

Abstract
Skipping of mammalian exons during pre-mRNA splicing is commonly mediated by the activity of exonic splicing silencers (ESSs). We have recently identified a regulated ESS within variable exon 4 of the CD45 gene, named ESS1, that is necessary and sufficient for partial exon repression in resting T cells and has additional silencing activity upon T-cell activation. In this study, we identify three heterogeneous nuclear ribonucleoproteins (hnRNPs) that bind specifically to ESS1. The binding of one of these proteins, hnRNP-L, is significantly decreased by mutations that disrupt both the basal and induced activities of ESS1. Recombinant hnRNP-L functions to repress exon inclusion in vitro in an ESS1-dependent manner. Moreover, depletion of hnRNP-L, either in vitro or in vivo, leads to increased exon inclusion. In contrast, the other ESS1-binding proteins, PTB and hnRNP E2, do not discriminate between wild-type and mutant ESS1 in binding studies, and do not specifically alter ESS1-dependent splicing in vitro. Together, these studies demonstrate that hnRNP-L is the primary protein through which CD45 exon 4 silencing is mediated by the regulatory sequence ESS1.
Keywords: alternative splicing, CD45, ESS, hnRNP
Introduction
The recent effort in genome sequencing has demonstrated that gene number does not necessarily correlate with organism complexity. Therefore, generation of proteome and functional complexity must rely heavily on mechanisms that amplify the informational content of the genome. One such mechanism for increasing proteome complexity is alternative pre-mRNA splicing, which is estimated to occur in 60–75% of human genes (Modrek et al, 2001; Black, 2003; Johnson et al, 2003). Alternative pre-mRNA splicing is defined as the differential inclusion or exclusion of exons in mature mRNA, such that a single gene can encode for multiple distinct proteins. In addition to amplifying genomic diversity, alternative splicing is emerging as a common mechanism for regulating protein expression in a cell-specific manner or in response to precise environmental cues.
During pre-mRNA processing, excision of noncoding intron sequence and ligation of exonic coding sequence is directed by sequence-specific splice sites at the exon and intron boundaries. Splicing is catalyzed by the spliceosome, a large complex of small nuclear ribonucleoproteins (snRNPs) U1, U2, U4, U5, and U6 and associated proteins, which assemble on pre-mRNA in a precise and coordinated manner (Black, 2003). In higher eukaryotes, pre-mRNA splicing is further under the control of positive and negative regulatory elements, located in exon or intron sequence. These regulatory elements help determine if a particular exon is included in mature mRNA, by either promoting or preventing spliceosome assembly on the exon. Depending on their physical location and activity, these elements are called intronic or exonic splicing enhancers (ESEs) or silencers (Black, 2003).
Numerous studies regarding ESEs have led to the conclusion that these motifs are present in most mammalian exons and promote exon inclusion primarily by serving as binding sites for the SR family of splicing activator proteins (Schaal and Maniatis, 1999; Blencowe, 2000). In contrast to ESEs, exonic splicing silencers (ESS) have been less well studied, although recent estimates suggest that these sequences are likely to be as abundant and important as ESEs (Fairbrother and Chasin, 2000; Fu, 2004; Wang et al, 2004). Specifically, ESS motifs are thought to play a critical role in repressing the use of pseudo-exons and controlling the efficiency of alternative exon inclusion (Fu, 2004; Zhang and Chasin, 2004).
Most ESSs studied have been shown to function by binding to members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family (Black, 2003; Zheng, 2004). HnRNP proteins are a structurally diverse family of proteins that were first identified by virtue of their interaction with nascent pre-mRNA (Pinol-Roma et al, 1988). More recently, individual members of this family have been shown to have sequence-specific effects on numerous steps in RNA processing and function, including alternative splicing (Krecic and Swanson, 1999; Dreyfuss et al, 2002). The hnRNP proteins most commonly associated with ESS function are hnRNP A/B, hnRNP I (also known as PTB) and hnRNP H (Caputi et al, 1999; Chen et al, 1999; Del Gatto-Konczak et al, 1999; Wagner and Garcia-Blanco, 2001; Zheng, 2004). These proteins have been shown to repress the splicing of many exons by binding to UAGG, UCUC, or polyG motifs, respectively, and are presumed to function by inhibiting the earliest steps of spliceosomal recognition of the surrounding splice sites (Burd and Dreyfuss, 1994; Chan and Black, 1997; Caputi and Zahler, 2001; Zhu et al, 2001). Other hnRNP proteins, such as hnRNP L, K, E1 and E2, have traditionally been thought to regulate the cytoplasmic export, stability, and translation of specific mRNA, with no indication that these proteins influence pre-mRNA splicing (Liu and Mertz, 1995; Hahm et al, 1998; Shih and Claffey, 1999; Makeyev and Liebhaber, 2002; Perrotti and Calabretta, 2002). However, each of these four proteins is found in the nucleus as well as the cytoplasm (Pinol-Roma and Dreyfuss, 1992), and a recent study revealed a novel role for hnRNP L in stimulating the removal of CA repeat-containing intron in the eNOS gene (Hui et al, 2003). Thus, while hnRNP L, K, E1, and E2 have yet to be implicated in the function of ESS motifs, it is certainly possible that these proteins may be involved in repressing the inclusion of specific exons.
The human CD45 gene encodes a haematopoietic-specific transmembrane protein tyrosine phosphatase. Three of the exons (exons 4, 5, and 6) that encode a portion of the extracellular domain of the CD45 protein are alternatively spliced. These variable exons are excluded from a portion of the transcripts that are generated in resting cells, and from the majority of the transcripts that are expressed in activated cells (Trowbridge and Thomas, 1994; Lynch and Weiss, 2000; see Figure 1A). The extent of inclusion of these variable exons has been shown to alter CD45 homodimerization and consequently to modulate the threshold for T-cell activation (Xu and Weiss, 2002). Thus, identifying the factors responsible for silencing the CD45 variable exons is of significant importance to understanding the finely tuned control of the immune system. Previously, we identified a 60-nucleotide (nt) ESS within CD45 variable exon 4 that is responsible for the partial repression of exon 4 in resting cells (Lynch and Weiss, 2001). We further demonstrated that this silencer element, termed ESS1, is necessary and sufficient for the regulated increase in skipping of CD45 exon 4 that is observed upon cell stimulation (Rothrock et al, 2003). Therefore, we concluded that ESS1 is the primary determinant of CD45 exon 4 inclusion, and that ESS1 may be classified as a ‘regulated ESS' as its activity is augmented in response to extracellular signals.
Figure 1.
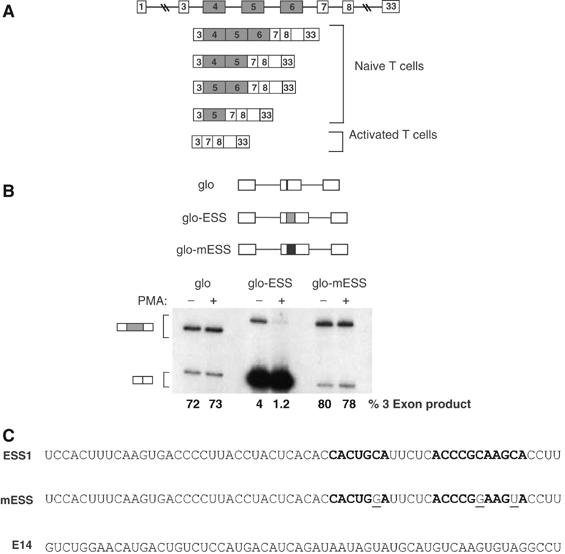
ESS1 controls both basal and activation-induced regulation of CD45 exon 4. (A) A schematic of the CD45 gene and the five spliced isoforms of CD45 that are translated in humans, demonstrating the variable level of exon 4 inclusion in resting and activated T cells. (B) RT–PCR analysis of the splicing of three minigenes stably expressed in JSL1 cells under resting (−PMA) or stimulated (+PMA) conditions (see Materials and methods). These minigenes consist of introns and exons from the human β-globin gene (lines and white boxes), and either no insert (glo) or insertion of a WT (grey box, glo-ESS) or mutant (black box, glo-mESS) version of the ESS1-regulated silencer. The reactions for glo-ESS were overloaded on the gel as compared to glo and glo-mESS to allow for visualization of the three-exon product in these samples. Quantitation of inclusion of the central exon (% 3 exon) was achieved by averaging results from at least four independent clones and two independent experiments. The standard deviation for the averages shown is <10% in each case. (C) Sequence of the WT ESS1 RNA, functionally mutant ESS1 (mESS), and nonspecific (E14) RNAs used in all the experiments in this study. Bold nt's refer to the ARS consensus motif. Mutations in mESS relative to ESS1 are within the ARS motif and are indicated by plain underlined text.
In the present study, we identify three hnRNP proteins, hnRNP L, PTB, and hnRNP E2, that specifically associate with ESS1. Notably, the binding of one of these proteins, hnRNP L, is markedly decreased by mutations that abolish both the basal and stimulated activity of ESS1. In addition, we show that hnRNP L represses exon usage in an ESS1-dependent manner in vitro and in vivo. HnRNP E2 can also repress exon usage when present at high levels in vitro; however, this protein is normally much less abundant in nuclear extract than hnRNP L. Therefore, we conclude that hnRNP L is the primary protein responsible for exonic silencing mediated by ESS1, although the activity of hnRNP L is perhaps facilitated by the presence of other proteins such as hnRNP E2.
Results
Mutations that disrupt ESS1 function inhibit the association of a complex required for exon silencing
The three variable exons of the human CD45 gene are partially repressed in resting T lymphocytes and more completely repressed upon T-cell activation (Lynch and Weiss, 2000; see Figure 1A). Previous mapping of the exonic splicing regulatory sequences of CD45 variable exon 4 led to the identification of a 60-nt regulated ESS that we named ESS1 (see Figure 1C). This ESS1 sequence is necessary and sufficient for both basal and activation-induced exonic silencing, and contains a core motif (the ‘ARS consensus', bolded in Figure 1C) that is also found within variable exons 5 and 6 (Lynch and Weiss, 2001; Rothrock et al, 2003). As we have shown previously (Rothrock et al, 2003), insertion of ESS1 into a heterologous exon reduces the inclusion of that exon, as revealed by RT–PCR analysis of minigene-derived mRNA expressed in unstimulated cells (Figure 1B, glo versus glo-ESS(−)PMA). Moreover, the presence of the ESS1 element is sufficient to confer further exon repression upon phorbol ester treatment of cells (Figure 1B, glo-ESS(−) versus (+)PMA). Importantly, both the basal and induced activity of ESS1 are completely abolished by the presence of three point mutations within the conserved ARS motif, demonstrating that this sequence is critical for exon silencing (Figure 1B, glo-ESS versus glo-mESS, and C). We note that, interestingly, the ARS motif does not match any of the predicted constitutive ESS motifs identified by others (Wang et al, 2004; Zhang and Chasin, 2004), nor does ESS1 contain any other matches to functional silencers in the FAS-hex3 (http://genes.mit.edu/fas-ess/) or PESS (http://cubweb.biology.columbia.edu/pesx) ESS databases.
The vast majority of splicing enhancer and silencer sequences characterized function as binding sites for regulatory proteins (Black, 2003). To determine if potential trans-acting factors stably interact with the ESS1 sequence, binding reactions were performed with ESS1 RNA and nuclear extracts prepared from JSL1 cells, a cell line that faithfully recapitulates all aspects of CD45 splicing in peripheral human T cells. As shown by native RNA mobility shift assays, at least two slower migrating complexes are visible upon incubation of 32P-labeled ESS1 RNA with nuclear extract derived from unstimulated JSL1 cells. These apparent protein–RNA complexes are stable up to 500 mM KCl, are independent of ATP, and appear to behave identically in all competition studies (data not shown and see below). This native gel shift analysis, as well as all other binding assays described in this study, were also conducted with nuclear extract prepared from stimulated JSL1 cells; however, no difference in complex formation or associated proteins was observed (data not shown), suggesting that the increase in ESS1-mediated repression upon stimulation is not simply due to a conspicuous change in protein binding. Therefore, in this study, we have focused on characterizing the proteins involved in the activity of ESS1 under unstimulated conditions as a critical first step toward understanding the interplay between basal and induced exon silencing via ESS1 (see Discussion).
To determine the specificity of the native gel shift complex, we analyzed the ability of various unlabeled RNAs to compete for complex formation. As shown in Figure 2A, assembly of a complex on the wild-type (WT) ESS1 RNA probe is almost completely abolished by addition of an ∼10-fold molar excess (1 pmol) of unlabeled ESS1 RNA, but is not inhibited by addition of up to 100-fold molar excess (10 pmol) of nonspecific RNA (‘E14'; see also Figure 1C). Moreover, the native gel complex showed a marked binding preference for WT ESS1 RNA versus mESS RNA that contains the three mutations which abolish function in vivo, as demonstrated by the fact that approximately 10-fold more mESS RNA than ESS1 RNA is required to achieve an equivalent level of competition (Figure 2A). Therefore, we conclude that a complex associates specifically with the active form of ESS1.
Figure 2.
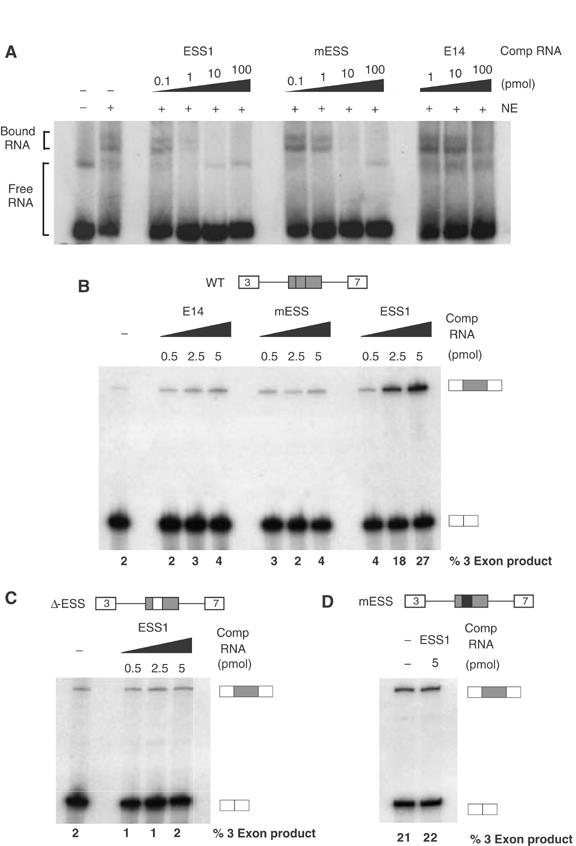
An exon-repression complex specifically associates with ESS1. (A) 32P-labeled ESS1 RNA (0.1 pmol) was incubated in the presence (+) or absence (−) of nuclear extract from unstimulated JSL1 cells and resolved on a native gel to observe free and bound RNA species. Unlabeled competitor RNA was also added to the reactions as indicated. (B) In all, 1 fmol unlabeled, capped RNA derived from a minigene containing WT CD45 variable exon 4, flanked by constitutive exons 3 and 7, was incubated in nuclear extract from unstimulated JSL1 cells in the presence or absence of exogenous competitor RNA. The resulting spliced products were then assayed by RT–PCR, as done for RNA derived from cells. Quantitation of the % 3-exon product is the average of at least four experiments with a standard deviation of <20% for all values given. (C) RNA derived from a CD45 exon 4 minigene in which the ESS1 has been deleted and replaced with heterologous sequence was spliced in the same assay as used for (B). (D) In vitro splicing as in panel B of RNA derived from a CD45 exon 4 minigene in which the ESS1 has been mutated to the ‘mESS' sequence shown in Figure 1C.
The specificity of the native gel complex suggests that the binding of trans-acting factors to ESS1 is functionally relevant. However, to more directly confirm the functional role of trans-acting factors in ESS1-mediated exon silencing, we examined the effect of addition of exogenous ESS1 RNA to an in vitro splicing assay. Splicing of a standard CD45 exon 4 minigene in the nuclear extract derived from resting JSL1 cells shows a low level of exon 4 inclusion or ‘three-exon product' (Figure 2B), consistent with the partial repression of exon 4 that we have previously observed in resting cells (Lynch and Weiss, 2001). Addition of exogenous ESS1 RNA to the splicing reaction greatly alleviates exon 4 silencing (up to a 10-fold increase in three-exon product; Figure 2B). Significantly, no effect on exon 4 inclusion is observed upon addition of either the mutant ESS1 (mESS) or nonspecific RNA (E14) (Figure 2B). To determine the specificity of ESS addition, we tested the effect of titration of exogenous ESS1 RNA on the splicing of a minigene in which the ESS1 element has been removed and replaced with heterologous sequence (ΔESS; Figure 2C). The heterologous sequence used in ΔESS has inherent silencer activity and represses three-exon splicing in JSL1 cells (Rothrock et al, 2003) and nuclear extract (Figure 2C). However, the silencer activity in the ΔESS construct is clearly distinct from that of ESS1 in that repression is not stringently observed in all JSL1 extract preparations (see Figure 6B) and is not active in 293 cells (see Figure 6C). Importantly, this heterologous silencer activity is unaffected by the presence of competitor ESS1 sequence (Figure 2C). Additionally, ESS1 RNA does not increase inclusion of an exon that contains the functionally dead element mESS (Figure 2D). Taken together, the specificity of complex binding and functional competition strongly indicate that ESS1 mediates splicing silencing by recruiting trans-acting factors to the repressed exon.
Figure 6.
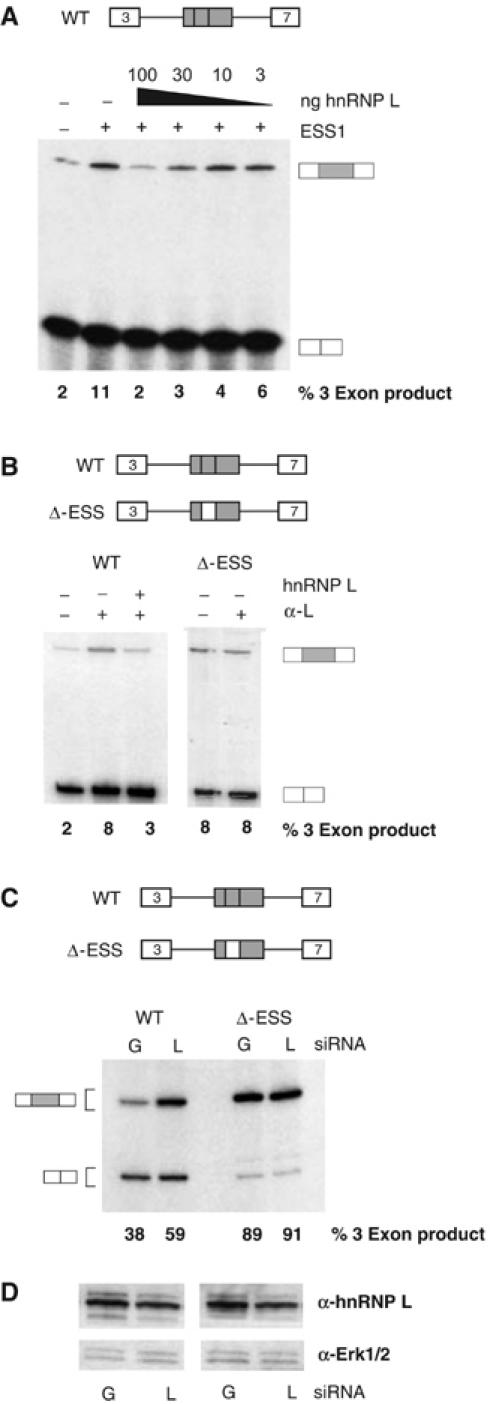
Depletion of hnRNP L in vitro or in vivo leads to increased inclusion of an ESS1-containing exon. (A) In vitro splicing assays lacking (−ESS) or containing (+ESS) exogenous ESS1 RNA similar to experiments shown in Figure 2B, in which purified recombinant GST-hnRNP L is also added to splicing reactions at the concentrations indicated. Quantitation is from at least four independent experiments, with a standard deviation of <20% of value. (B) In vitro splicing assays in the presence (+) or absence (−) of 2 μl 4D11 antibody and/or 100 ng recombinant GST-hnRNP L. Quantitation is derived from at least two independent experiments with standard deviation <10% of value. (C) RT–PCR analysis of WT or ΔESS minigene-derived RNA harvested from 293 cells transiently cotransfected with a minigene encoding vector and siRNAs against either GFP (G) or hnRNP L (L). Quantitation of three-exon product is derived from four independent transfections with standard deviation of <5%. (D) Western blot analysis of hnRNP L, or Erk1/2 as internal control, from transfections corresponding to those shown in panel C. The average reduction of hnRNP L in transfections with hnRNP L siRNAs versus GFP siRNAs was 50±10%.
HnRNPs L, E2, and PTB specifically associate with ESS1
To identify the protein components of the ESS1-specific complex, we employed an RNA affinity approach. WT ESS1 RNA, mutant ESS RNA, or nonspecific RNA (identical to ESS1, mESS, and E14 in Figure 1C) was chemically synthesized to contain a 5′ terminal biotin group. The biotinylated RNAs were individually incubated in JSL1 nuclear extracts under binding conditions similar to those used for the native RNA mobility shift assays. Following binding, the RNAs were coupled to streptavidin agarose beads, precipitated, and washed. Finally, RNA-associated proteins were eluted with SDS sample buffer and analyzed by SDS–PAGE and silver stain (Figure 3A). Comparison of the proteins precipitated with ESS1 RNA versus nonspecific (E14) RNA reveals several bands that associate specifically with ESS1 (indicated by arrows in Figure 3A). The most prominent of these ESS1-specific bands, which migrates at approximately 65 kDa, also appears to have a significantly stronger interaction with ESS1 RNA than with the mutant mESS RNA (Figure 3A). Each of the ESS1-specific bands was excised from the gel and analyzed by mass spectrometry (see Materials and methods for detail). Multiple peptides corresponding to hnRNP L, hnRNP I (PTB) and hnRNP E2 were identified for each band as indicated (Figure 3A). No peptides to other proteins were reproducibly present in any of the excised bands, including closely related proteins such as hnRNP E1, hnRNP K, YB-1, or KSRP.
Figure 3.
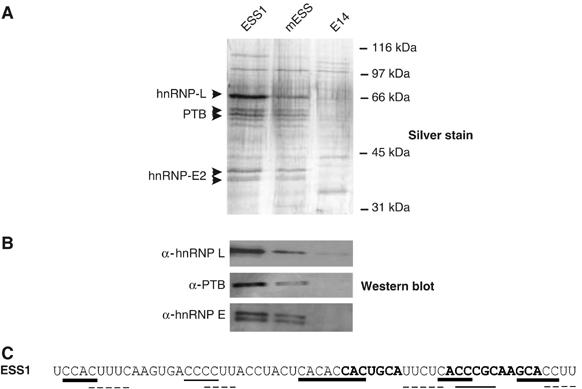
HnRNPs L, E2, and PTB are identified as ESS1-associated proteins by RNA affinity purification. (A) Affinity purification of proteins associated with ESS1 RNA. Silver stained SDS–PAGE gel of proteins isolated from JSL1 nuclear extract by virtue of association with chemically synthesized, 5′-biotinylated ESS1, mESS, and E14 RNAs (see Materials and methods for details). (B) Western blot of SDS–PAGE gel from panel A with specified antibodies. (C) Sequence of ESS1 with predicted high-affinity binding sites indicated for hnRNP L (bold lines), PTB (dotted lines), and hnRNP E (plain lines).
As further confirmation of the mass spectrometry results, hnRNP L, PTB, and hnRNP E2 were all detected using specific antibodies against these proteins in Western blot analysis of the ESS or mESS-affinity-purified proteins, but not in lanes corresponding to E14-associated proteins (Figure 3B). In contrast, no signal was detected when the RNA-associated proteins were probed with antibodies against hnRNP U, hnRNP K, and YB-1 (data not shown). Moreover, addition of hnRNP L, PTB, or E2 antibodies to the RNA mobility shift assays induces formation of a slower mobility complex, while antibodies against U2AF65 or hnRNP U have no effect (see Supplementary Figure S1). Thus, although we cannot fully rule out the presence or importance of other proteins, RNA-affinity purification clearly identifies the hnRNP family members L, E2, and PTB as primary proteins associated with the ESS1-regulated exonic silencer. Previous studies have identified high-affinity binding sites for hnRNP L, PTB, and hnRNP E2 as ACAC, UCUC, and Poly(C), respectively (Leffers et al, 1995; Chan and Black, 1997; Hui et al, 2005). Although the specificity of RNA-binding proteins can be highly context dependent (Lynch and Maniatis, 1996), the presence of predicted binding motifs for hnRNPs L, PTB, and E2 within ESS1 (shown in Figure 3C) is consistent with our identification of these three hnRNPs as major ESS1-binding proteins.
The ability of hnRNP L, PTB, and E2 to bind to the ESS1 regulatory sequence of CD45 exon 4 was also independently confirmed using UV crosslinking assays (Figure 4). 32P-labeled ESS1 RNA was incubated with nuclear extract or recombinant proteins under standard binding conditions. Following treatment with UV light to induce crosslinks between RNA and bound protein, the RNA was degraded with RNases such that ESS1-associated proteins could be visualized by SDS–PAGE and autoradiography by virtue of being covalently bound to one or more 32P-labeled nt's. UV crosslinking of ESS1 RNA to JSL1 nuclear extract reveals the presence of one major crosslinked species (Figure 4A, NE). This predominant crosslinked species could be specifically immunoprecipitated with antibodies against hnRNP L, thus identifying hnRNP L as the protein in nuclear extract most strongly associated with ESS1 (Figure 4A). In addition, several weaker crosslinked species between ESS1 and nuclear extract could be detected with variable clarity and intensity. One of these weaker species, a doublet migrating slightly smaller than hnRNP L on the SDS–PAGE gel, was selectively precipitated with antibodies specific for PTB, while antibodies against U2AF65, which is similar in size and binding specificity to PTB, did not precipitate any significant crosslinked products (Figure 4A). We also often detect a weak doublet at approximately 35–40 kDa, which we predict to be hnRNP E2. However, we cannot confirm this species to correspond to endogenous hnRNP E2 since our antibodies are not competent for immunoprecipitation.
Figure 4.
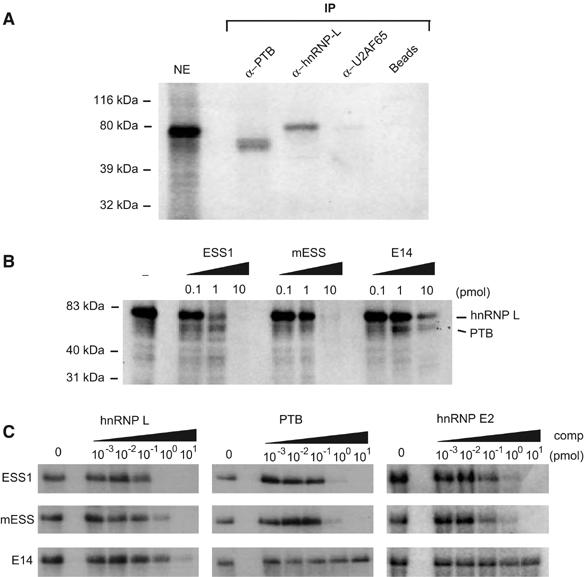
Mutations that disrupt the function of ESS1 inhibit binding of HnRNP L. (A) HnRNP L and PTB crosslink to ESS1 RNA from nuclear extract. Nuclear extract was incubated under splicing conditions with approximately 0.1 pmol uniformly 32P-labeled ESS1 RNA, treated with UV light, digested with RNAses, and then either directly resolved on a 10% SDS–PAGE gel (NE lane) or immunoprecipitated with antibodies as specified (IP lanes), and the precipitates were resolved on the gel. (B) UV crosslinking experiment as in panel A, except that no immunoprecipitation was carried out and unlabeled competitor RNAs as indicated were added during the binding reaction. The indicated identities of the crosslink species are based on experiments as shown in panel A. (C) UV crosslinking experiments using approximately 0.05 pmol purified recombinant GST-hnRNP L, GST-PTB or MBP-hnRNP E, and 0.1 pmol 32P-labeled ESS1 either alone (0) or in the presence of additional unlabeled ESS, mESS, or E14 competitor, as indicated.
Binding of hnRNP L to ESS1 is specifically inhibited by mutations that disrupt exon silencing
To more explicitly correlate the binding of hnRNP L, PTB, and E2 with the function of ESS1, we used UV crosslinking assays to determine how binding of these proteins is individually affected by the mutations that disrupt ESS1 function. The addition of 1 pmol of unlabeled competitor ESS1 RNA to the crosslinking of JSL1 nuclear extract with 32P-labeled ESS1 results in almost complete inhibition of the interaction between hnRNP L and probe, whereas approximately 10-fold more mESS or E14 RNA is required to achieve a similar inhibition of hnRNP L binding (Figure 4B). This ability of hnRNP L to discriminate between functional and nonfunctional ESS1 is consistent with the preliminary suggestion of specificity from the RNA-affinity experiments (Figure 3A) and with the observation that at least some of the mutations in mESS fall within putative hnRNP L-binding sites (Figure 3C). In contrast, crosslinking of PTB to the ESS1 probe, although resistant to competition by E14 RNA, was inhibited equally by ESS1 and mESS competitors (Figure 4B), suggesting that the binding of PTB to ESS1 is not altered by mutations that disrupt exonic silencing.
In the UV crosslinking experiments with JSL1 nuclear extract, we were not able to detect sufficient signal for the putative hnRNP E2 species to determine binding specificity. Furthermore, we also wanted to determine if the observed specificity of hnRNP L and PTB binding in nuclear extract was inherent to each protein or conferred by accessory proteins. Therefore, we repeated the UV crosslinking competition experiments using recombinant GST-tagged hnRNP L, PTB purified from Sf9 cells, and recombinant MBP-hnRNP E2 purified from bacteria. As shown in Figure 4C, recombinant hnRNP L binds with approximately 10-fold greater affinity to ESS1 than mESS, as indicated by the requirement for 10 versus 1 pmol of competitor to achieve complete inhibition of crosslinking. Therefore, the binding specificity of hnRNP L we observe in nuclear extract is an inherent feature of hnRNP L and not induced by cooperative interactions with other protein(s). By contrast, crosslinking of recombinant PTB and hnRNP E2 is inhibited equally by ESS1 and mESS RNA, although these proteins appear to have no ability to bind the unrelated E14 RNA (Figure 4C). Thus, the association of PTB and hnRNP E2 with ESS1, while demonstrating specificity with respect to the E14 sequence, does not correlate with the exonic silencer activity of this regulatory element.
Recombinant hnRNP-L induces ESS1-dependent exon silencing in vitro
The correlation of sequence requirements for the binding of hnRNP L in vitro, and the function of ESS1 in vivo, suggests that the exonic silencing activity of ESS1 is mediated predominantly by the association of hnRNP L. To test this prediction, we first used our in vitro splicing assays to determine the functional effect of hnRNP-L, PTB, and E2 on repression of CD45 exon 4. Titration of purified recombinant hnRNP L into in vitro splicing reactions using our standard WT CD45 exon 4 minigene results in a dose-dependent decrease in exon 4 inclusion (Figure 5A and C). Importantly, this activity of hnRNP L is dependent on the presence of the ESS1 sequence within the repressed exon, since only the highest concentration of hnRNP L shows even a modest silencing effect on the ΔESS substrate (Figure 5B and C). By contrast, addition of purified, recombinant PTB has no effect on exon 4 splicing, either on its own or in the presence of low levels of additional recombinant hnRNP L (Figure 5A and C). Addition of an unrelated hnRNP, hnRNP A1, also does not alter the splicing of exon 4 (data not shown).
Figure 5.
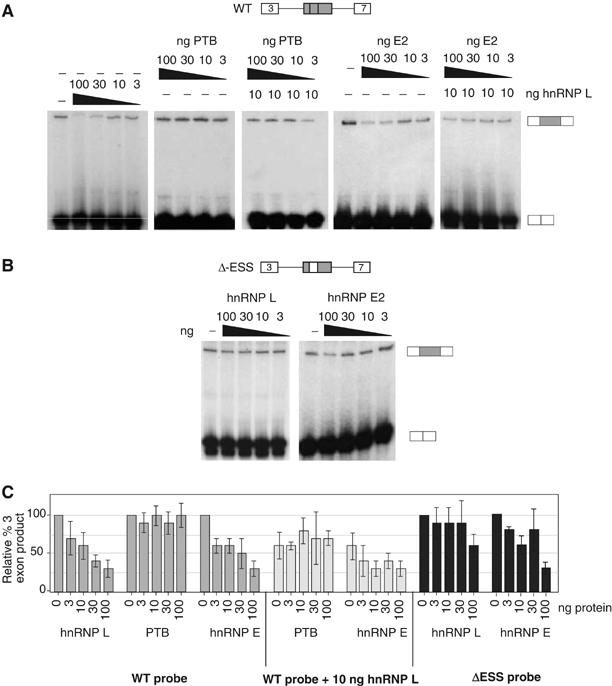
HnRNP L induces exon repression in in vitro splicing assays. (A) In vitro splicing assays with WT CD45 exon 4 minigene as described in Figure 2B. Splicing reactions contained either nuclear extract alone (−) or supplemented with purified recombinant GST-hnRNP L, GST-PTB, and/or MBP-hnRNP E2, as indicated. Quantitation of replicate experiments is given in panel C. (B) In vitro splicing as in panel A, but using the ΔESS splicing substrate that lacks the ESS1 regulatory sequence. Quantitation of replicate experiments is given in panel C. (C) Graphical representation of the three-exon product from at least four independent experiments, identical to those shown in panels A and B, and similar experiments with purified recombinant hnRNP A1. Numbers are given as % of three-exon product as compared to NE alone (set at 100%).
Addition of recombinant hnRNP E2 into in vitro splicing reactions does result in repression of exon 4 inclusion and shows a roughly additive silencing effect when added in conjunction with hnRNP L (Figure 5A and C). The ability of hnRNP E2 to repress splicing in vitro was initially surprising given that the mESS sequence is not sufficient to mediate repression, despite the fact that hnRNP E2 binds efficiently to this mutant element. However, comparison of the abundance of hnRNPs L and E2 in nuclear extract suggests that hnRNP L is present in approximately a 30-fold molar excess over hnRNP E2 (300 fmol versus 10 fmol per μg total protein based on comparison to known quantities of recombinant protein; data not shown). Therefore, the standard concentration of hnRNP E2 in the nucleus may not be sufficient to mediate exon repression when the binding of hnRNP L is decreased. Moreover, recombinant hnRNP E2 also partially represses the exon 4 variant lacking the ESS1 motif (Figure 5B and C). Thus, hnRNP E2-mediated exon repression in vitro may be due to mechanisms separate from its ESS1-binding activity.
Depletion of hnRNP L relieves exon repression in vitro and in vivo
As a further test of the function of hnRNP L, we asked whether this protein could complement an ESS1-depleted extract. As shown in Figure 2B, we can functionally deplete ESS1-repressing activity by the addition of exogenous ESS1 RNA into in vitro splicing assays. Importantly, in such a functionally depleted background, we observe robust rescue of exonic repression upon the addition of recombinant hnRNP L (Figure 6A), but not PTB (data not shown). Blocking of hnRNP L by addition of anti-hnRNP L antibody to the in vitro splicing reactions also relieves repression of exon 4, and is reversed by supplementing the reactions with recombinant hnRNP L (WT; Figure 6B). In contrast, no change in the inclusion of exon 4 was observed upon addition of antibody specific for hnRNP A1 (data not shown), and the anti-hnRNP L antibody has no effect on the splicing of an exon lacking ESS1 (ΔESS; Figure 6B).
While the above experiments demonstrate a role of hnRNP L in ESS1-mediated silencing in vitro, we wanted to confirm that hnRNP L is also critical for ESS1 function in vivo. Due to the high level of hnRNP L expression in the JSL1 cell line and the fact that these cells do not support RNAi, we have been unable to modulate hnRNP L expression levels in the JSL1 cells. While other cell lines do not support activation-induced alternative splicing of CD45, and may differ from JSL1 cells in the relative expression of hnRNP L and other splicing regulatory factors, we have found that the 293 cell line does recapitulate the splicing pattern of the CD45 exon 4 minigene observed resting JSL1 cells. HnRNP L is highly abundant in 293 cells; however, using a pool of siRNAs against hnRNP L, we were able to achieve about a 50% decrease in hnRNP L protein levels (Figure 6D). Strikingly, this 50% reduction in hnRNP L is sufficient to induce a significant increase in exon 4 inclusion from a cotransfected WT minigene (38±4 to 59±5%), consistent with decreased activity of the ESS1 silencer. In contrast, parallel transfections with the ΔESS minigene show no effect of hnRNP L depletion (Figure 6C). These data strongly suggest that hnRNP L is required for ESS1-dependent exon silencing in vivo as well as in vitro.
Discussion
Previously, we demonstrated that the 60-nt ESS1 sequence in human CD45 exon 4 is a regulated ESS that is necessary and sufficient both for basal exon repression and for increased exon repression in response to PMA treatment (Lynch and Weiss, 2001; Rothrock et al, 2003; see Figure 1). Here we identify three hnRNPs, L, PTB, and E2, as the primary proteins that bind to the ESS1-regulated ESS. Several lines of evidence indicate that hnRNP L is the predominant protein through which the function of ESS1 is mediated. First, hnRNP L is the most prominent ESS1-binding protein as assayed by both RNA-affinity purification and UV crosslinking (Figures 3 and 4). Secondly, hnRNP L is the only one of the ESS1-binding proteins whose affinity is decreased by the introduction of mutations that disrupt both the basal and inducible activities of ESS1 (Figure 4). Finally, we have shown that modulating the levels of hnRNP L, both in vitro and in vivo, specifically alters the splicing of an ESS1-containing exon (Figures 5 and 6).
In contrast to hnRNP L, PTB and hnRNP E2 bind with indistinguishable affinity to ESS1 and the functionally inactive mutant mESS (Figure 4C). Therefore, the level of association of these proteins in cells with mESS, and by analogy ESS1, must not be sufficient to mediate exonic silencing. The inability of hnRNP E2 or PTB to functionally compensate for hnRNP L may be due, at least in part, to the fact that the binding of both of these proteins to ESS1 in nuclear extract is much less pronounced than that of hnRNP L. Indeed, when we increased the concentration of hnRNP E2 in in vitro splicing assays, this protein was able to promote exonic silencing, although this effect was not strictly dependent on the presence of the ESS1 element (Figure 5). Therefore, while we have no evidence for a functional role of PTB in ESS1 activity, we cannot rule out that hnRNP E2 might contribute in some way to the overall exonic silencing.
Interestingly, hnRNP L, PTB, and hnRNP E2 have previously been shown to interact with one another by both yeast two-hybrid and GST pulldown experiments (Kim et al, 2000). Using UV crosslinking assays with recombinant hnRNP L, PTB, and E2, we do not detect any evidence for cooperative binding of these proteins to ESS1 (data not shown). However, interaction between these proteins may occur once bound to the RNA and may increase the overall stability of the ESS1-bound regulatory complex and/or be important for mediating exon repression. Further mapping of the precise sites of association of hnRNP E2 and PTB within the ESS1 element will be required to better understand the potential contribution of these proteins to the overall activity of this silencer.
HnRNP L may have broad function as a splicing regulatory protein
PTB has been shown to bind ESSs in several genes, and to be essential for the silencing activity of many of these regulatory sequences (Wagner and Garcia-Blanco, 2001). However, neither hnRNP L nor hnRNP E2 has been previously implicated in ESS function. To our knowledge, hnRNP E2 has, in fact, never been shown to influence pre-mRNA splicing. However, hnRNP-L has recently been shown to bind to a CA-rich element within an intron of the eNOS gene and stimulate its removal (Hui et al, 2003). Other splicing regulatory proteins have been shown to have dual function to promote the removal of introns or skipping of exons depending whether they are bound within the intron or exon, respectively (Dredge et al, 2005). Thus, although the mechanisms by which hnRNP L stimulates either intron removal or exon repression have yet to be elucidated, these activities may be related to one another and may reflect a general ability of hnRNP L to mark RNA sequences for removal by the splicing machinery.
Previously, we have shown that the ARS core motif from ESS1 is also present in the other CD45 variable exons and in several signal-regulated exons from other genes (Rothrock et al, 2003). Interestingly, in a study parallel to those described here for exon 4, we have found that hnRNP L binds to the minimal ARS-containing silencing sequences from CD45 variable exons 5 and 6, and this binding of hnRNP L is abrogated by mutations within the ARS core of these exons (A Tong, J Nguyen and KW Lynch, in preparation). Therefore, it is possible that hnRNP L may influence the splicing of all of the CD45 variable exons, or even ARS-containing exons, from other genes. Similarly, recent studies from the Bindereif group have found that hnRNP L binds to CA-rich intronic enhancer and silencer elements in three other genes in addition to eNOS (Hui et al, 2005). Taken together, these studies suggest that hnRNP L may play a broad role as a master splicing regulatory protein that controls the isoform expression of a large number of genes.
Signal-induced alternative splicing in CD45
While the data presented in this study clearly demonstrate a role for hnRNP L, and perhaps hnRNP E2, in the ESS1-dependent repression of exons in resting cells, we have not addressed the important question of how the activity of ESS1 is enhanced upon cellular stimulation, as shown in Figure 1B. Extracts prepared from resting or activated cells show a robust and specific difference in inclusion of CD45 exon 4 in in vitro splicing assays (AE House and KW Lynch, unpublished data). However, despite this functional difference, no difference in the intensity or identity of the ESS1-binding proteins was observed between these two extracts (data not shown). Moreover, careful Western blot analysis of hnRNPs L, PTB, and E2 in extracts from resting and activated cells indicated no differences in the concentration of these proteins within our limits of detection (>1.5-fold difference, data not shown).
It is possible that relatively subtle changes in hnRNP concentration (i.e. <1.5-fold) do occur upon stimulation and are sufficient to induce the 3–5-fold increase in exon repression observed in vivo and in vitro. Consistent with this model, a two-fold decrease in protein level of hnRNP L was sufficient to cause a significant change in exon 4 inclusion in the RNAi experiment (Figure 6C). Furthermore, addition of approximately 1 pmol of recombinant hnRNP L significantly increased exon skipping in the in vitro splicing assays shown in Figure 5A, even though the background level of hnRNP L contributed by the nuclear extract in these experiments was approximately 7–10 pmol per reaction. Therefore, while we do not know the level of free or active hnRNP L in nuclear extract, 293 or JSL1 cells, it is possible that the regulation of CD45 exon 4 is exquisitely poised to respond to small variations in the level of hnRNP L. The fact that hnRNP L can interact with itself (Kim et al, 2000), and thus may bind cooperatively to an RNA substrate, may suggest a mechanism by which a slight change in protein concentration could result in a large functional outcome. However, we cannot exclude the possibility that the enhanced activity of ESS1 in activated cells is due to additional or alternate mechanisms such as changes in the post-translational modification or activity of the ESS1-binding proteins. Studies are currently in progress to more thoroughly and precisely assess the relative expression, activity, and RNA binding of hnRNP L, PTB, and E2 in resting and activated cells.
In conclusion, we show here that hnRNP L, PTB, and hnRNP E2 are the primary ESS1-binding proteins found in nuclear extract from a T-cell-derived cell line, and that hnRNP L is the key regulatory protein responsible for mediating ESS1-dependent exonic silencing under resting conditions. These studies not only broaden the known functions of hnRNP L by implicating it in ESS binding and function, but also lay a critical foundation for future studies aimed at understanding the mechanism and regulation of ESS1-dependent silencing activity and the control of CD45 splicing.
Materials and methods
Plasmids and RNAs
The Glo minigene was engineered by modifying the central exon of the three exon Dup175 construct (Xie and Black, 2001) to contain extraneous sequence from CD45 exon 14 to elongate the exon and an MluI restriction site to facitilate modification. Oligonucleotides encoding the ESS or mESS 60 nt were inserted into the MluI site of Glo to result in Glo-ESS and Glo-mESS. These same oligonucleotides, plus that encoding E14, were also cloned directly downstream of a T7 polymerase promoter to create pSPT7-ESS, -mESS, and -E14. Biotinylated ESS, mESS, and E14 RNAs for affinity purification were chemically synthesized by Dharmacon. All other RNA probes and competitor RNAs were transcribed with T7 polymerase (Promega), in the absence or presence of 32P-CTP added during transcription reactions to radioactively label probes. The WT and ΔESS minigenes used for in vitro splicing were also transcribed with T7 polymerase and from plasmids essentially identical to the CD4 and CD14 constructs described in Rothrock et al (2003).
RT–PCR assay
RT–PCR and analysis was carried out as described previously (Rothrock et al, 2003).
RNA mobility shift assays
Nuclear extract was purified from JSL1 cells with a previously described standard protocol (Lynch and Weiss, 2001). Probes were gel-purified and adjusted for to 104 c.p.m./μl specific activity. Standard binding reactions were carried out in 10 μl, with a final concentration of 3.2 mM MgCl2, 20 mM phosphocreatine, 1 mM ATP, 1.3% polyvinyl alcohol, 25 ng of yeast tRNA, 0.8 μg of BSA, 1 mM DTT, 0.1 μl Rnasin (Promega, 40 U/μl), 75 mM KCl, 10 mM Tris, pH 7.5, 0.1 mM EDTA, 10% glycerol, and 0–30% JSL1 nuclear extract. Binding reactions were preincubated for 5 min at 30°C before addition of 32P-labeled probe, and then incubated for 15 min at 30°C after addition of probe. After binding, heparin was added to a final concentration of 0.5 μg/μl; reactions were analyzed on a 4.5% native gel (Acrylamide/Bis 29:1, BioRad).
In vitro splicing
Approximately 1 fmol of unlabeled RNA substrate was incubated with 30% JSL1 nuclear extract in a total volume of 12.5 μl containing (final concentrations): 3.2 mM MgCl2, 20 mM phosphocreatine, 1 mM ATP, 3% polyvinyl alcohol, 1 mM DTT, 0.25 U RNasin (Promega), 90 mM KCl, 10 mM Tris, pH 7.5, 0.1 mM EDTA, 7% glycerol, and other proteins, as indicated in the figures. Reactions were incubated for 2 h at 30°C; then the RNA was recovered from the reactions by proteinase K treatment, phenol–chloroform extraction, and precipitation. The resulting RNA was then analyzed by RT–PCR as described above. For competition studies, the indicated amount of ESS, mESS, E14, or antibody was added to the splicing reactions immediately prior to addition of unlabeled substrate and reactions were processed as above.
RNA affinity purification
In all, 50 pmol of 5′-biointylated RNA (Dharmacon) was incubated with 100 μg of JSL1 nuclear extract in a 500 μl binding reaction similar to that described for RNA mobility shift assays, but omitting BSA. Binding reactions were incubated with gentle agitation for 30 min at 30°C. Streptavidin agarose beads (Pierce) were preblocked in GFB100 (100 mM KCl, 20 mM Tris–Cl, pH 7.5, and 0.2 mM EDTA, pH 8.0), and 1 μg/μl heparin and washed with GFB100. Preblocked strepavidin beads (25 μl) were added to the RNA-binding reactions, with additional heparin, to a final concentration of 0.5 μg/μl, 1.6 mM MgCl2, 100 μl BC400, in a final volume of 1 ml, and incubated for 60 min at 4°C with gentle agitation. RNA–protein-bead complexes were washed with GFB100, resuspended in 2 × SDS loading buffer, denatured for 5 min at 95°C, analyzed under denaturing conditions on an 8% gel (Acrylamide/Bis 37.5:1, BioRad), and detected by silver staining (BioRad).
Western blotting
Western blotting was carried out as previously described (Lynch and Weiss, 2000), with the antibodies specified below.
Antibodies
Antibodies for Western blots, splicing inhibition, and immunoprecipitation of crosslinked reactions were as follows: anti-hnRNP L (4D11, Abcam), anti-hnRNP E2 (rabbit polyclonal, kind gift of R Andino), anti-HA (HA.11, BabCo), anti-U2AF65 (rabbit polyclonal, kind gift of T Maniatis), anti-hnRNP U (IQ210, ImmunoQuest), anti-Erk1/2 (rabbit polyclonal, kind gift of M Cobb). Different antibodies against PTB were used for different applications, all of which were kind gifts of D Black. Immunoprecipitations were performed with BB7 (mouse monoclonal); Western blot was carried out using anti-PTB N-term (rabbit polyclonal).
Mass spectrometry
For identification of proteins by mass spectrometry, colloidal blue or silver-stained bands were excised from the gel and an in-gel digest was performed with porcine trypsin. Tryptic peptide mixtures were dissolved and injected into reversed-phase HPLC/ion trap with a nanospray source, using a ThermoFinnigan LCQ Deca XP MS instrument and Xcalibur 1.3 software. MS/MS files were searched against NCBI-nr protein sequence databases, using the Sonar database software (GenomicSolutions, Inc.).
UV crosslinking
JSL1 nuclear extract or purified recombinant hnRNP-L, PTB, or hnRNP E was incubated with 32P-labeled RNA under conditions described for RNA mobility shift assays, excluding RNasin. Reactions were incubated at 30°C for 20 min, crosslinked using UV light (254 nm) for 20 min at on ice, and digested with RNaseT1 and RNase A at a final concentration of 2 μg each for 20 min at 37°C. Reactions were resuspended in 2 × SDS loading buffer, denatured for 5 min at 95°C, analyzed under denaturing conditions on an 12% gel (Acrylamide/Bis 37.5:1, BioRad), and detected by autoradiography. Immunoprecipitation after crosslinking was carried out as described previously (Lynch and Maniatis, 1996), with the antibodies described above.
Recombinant proteins
HnRNP-L and PTB cDNAs were cloned into pFastBac (Invitrogen) and modified with an N-terminal GST tag. Expression plasmids were transformed into DH10Bac (Invitrogen) to generate recombinant bacmids which were then transfected into SF9 cells using cellfectin (Invitrogen). Viral stocks were harvested, amplified, and then used to infect SF9 cells for 60 h to generate recombinant proteins. SF9 cells containing GST-hnRNP L or GST-PTB were lysed by repeated freeze–thaw cycles, and recombinant proteins were purified with glutathione sepharose 4B resin (Amersham Biosciences) according to the manufacturer's protocol.
Recombinant HnRNP E2 was expressed as a MBP fusion protein in Escherichia coli. The expression plasmid pMALc2-PCBP2 was a kind gift of R Andino, and MBP-hnRNP E2 was expressed and purified as described previously (Gamarnik and Andino, 1997).
RNAi
3 × 105 293 cells were plated in each well of a six-well plate in 2 ml of high-glucose DMEM. At 24 h after seeding, the cells were transfected with 5 μl Lipofectamine 2000 (Invitrogen), 0.5 μg WT or ΔESS minigene, and 3 μl of a 200 μM stock of either GFP duplex siRNA (Dharmacon) or the SMARTpool of siRNAs against hnRNP L (Dharmacon). At 12 h after transfection, the lipid complexes were removed and replaced with fresh media. Cells were harvested for analysis of protein and RNA 48–60 h after transfection.
Supplementary Material
Supplementary Figure S1
Acknowledgments
We thank Raul Andino and Doug Black for the kind gift of reagents, and Chris Cox of the UTSW Protein Core Facility for the mass spectrometry analysis. We also thank Albrecht Bindereif for communicating results prior to publication and for helpful comments regarding our manuscript. This work was supported by NIH grant R01 GM067719 and NSF grant MCB0347104. AEH is supported by the Division of Cell and Molecular Biology Training Program grant at UT Southwestern Medical Center (T32 GM08203). KWL is an EE and Greer Garson Fogelson Scholar in Biomedical Research.
References
- Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Blencowe BJ (2000) Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseass. TIBS 25: 106–110 [DOI] [PubMed] [Google Scholar]
- Burd CG, Dreyfuss G (1994) RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J 13: 1197–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi M, Mayeda A, Krainer AR, Zahler AM (1999) hnRNP A/B protein are required for inhibition of HIV-1 pre-mRNA splicing. EMBO J 18: 4060–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputi M, Zahler AM (2001) Determination of the RNA binding specificity of the heterogeneous nuclear ribonucleoprotein (hnRNP) H/H′/F/2H9 family. J Biol Chem 276: 43850–43859 [DOI] [PubMed] [Google Scholar]
- Chan RC, Black DL (1997) The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol Cell Biol 17: 4667–4676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Kobayashi R, Helfman DM (1999) Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat beta-tropomyosin gene. Genes Dev 13: 593–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gatto-Konczak F, Olive M, Gensel M-C, Breathnack R (1999) hnRNP A1 recruited to an exon in vivo can function as an exon splicing silencer. Mol Cell Biol 19: 251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge BK, Stefani G, Engelhard CC, Darnell RB (2005) Nova autoregulation reveals dual functions in neuronal splicing. EMBO J 24: 1608–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N (2002) Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3: 195–205 [DOI] [PubMed] [Google Scholar]
- Fairbrother WG, Chasin LA (2000) Human genomic sequences that inhibit splicing. Mol Cell Biol 20: 6816–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD (2004) Towards a splicing code. Cell 119: 736–738 [DOI] [PubMed] [Google Scholar]
- Gamarnik AV, Andino R (1997) Two functional complexes formed by KH domain containing proteins with the 5′ noncoding region of poliovirus RNA. RNA 3: 882–892 [PMC free article] [PubMed] [Google Scholar]
- Hahm B, Kim YK, Kim JH, Kim TY, Jang SK (1998) Heterogeneous nuclear ribonucleoprotein L interacts with the 3′ border of the internal ribosomal entry site of hepatitis C virus. J Virol 72: 8782–8788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J, Hung LH, Heiner M, Schreiner S, Neumuller N, Reither G, Haas SA, Bindereif A (2005) Intronic CA-repeats and CA-rich elements: a new class of regulators of mammalian alternative splicing. EMBO J 24: 1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J, Stangl K, Lane WS, Bindereif A (2003) HnRNP L stimulates splicing of the eNOS gene by binding to variable-length CA repeats. Nat Struct Biol 10: 33–37 [DOI] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144 [DOI] [PubMed] [Google Scholar]
- Kim JH, Hahm B, Kim YK, Choi M, Jang SK (2000) Protein–protein interaction among hnRNPs shuttling between nucleus and cytoplasm. J Mol Biol 298: 395–405 [DOI] [PubMed] [Google Scholar]
- Krecic AM, Swanson MS (1999) hnRNP complexes: composition, structure, and function. Curr Opin Cell Biol 11: 363–371 [DOI] [PubMed] [Google Scholar]
- Leffers H, Dejgaard K, Celis JE (1995) Characterisation of two major cellular poly(rC)-binding human proteins, each containing three K-homologous (KH) domains. Eur J Biochem 230: 447–453 [PubMed] [Google Scholar]
- Liu X, Mertz JE (1995) HnRNP L binds a cis-acting RNA sequence element that enables intron-dependent gene expression. Genes Dev 9: 1766–1780 [DOI] [PubMed] [Google Scholar]
- Lynch KW, Maniatis T (1996) Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev 10: 2089–2101 [DOI] [PubMed] [Google Scholar]
- Lynch KW, Weiss A (2000) A model system for the activation-induced alternative-splicing of CD45 implicates protein kinase C and Ras. Mol Cell Biol 20: 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KW, Weiss A (2001) A CD45 polymorphism associated with multiple sclerosis disrupts an exonic splicng silencer. J Biol Chem 276: 24341–24347 [DOI] [PubMed] [Google Scholar]
- Makeyev AV, Liebhaber SA (2002) The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA 8: 265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Resch A, Grasso C, Lee C (2001) Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res 29: 2850–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti D, Calabretta B (2002) Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene 21: 8577–8583 [DOI] [PubMed] [Google Scholar]
- Pinol-Roma S, Choi YD, Matunis MJ, Dreyfuss G (1988) Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2: 215–227 [DOI] [PubMed] [Google Scholar]
- Pinol-Roma S, Dreyfuss G (1992) Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature 355: 730–732 [DOI] [PubMed] [Google Scholar]
- Rothrock C, Cannon B, Hahm B, Lynch KW (2003) A conserved signal-responsive sequence mediates activation-induced alternative splicing of CD45. Mol Cell 12: 1317–1324 [DOI] [PubMed] [Google Scholar]
- Schaal TD, Maniatis T (1999) Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Mol Cell Biol 19: 261–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih SC, Claffey KP (1999) Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J Biol Chem 274: 1359–1365 [DOI] [PubMed] [Google Scholar]
- Trowbridge IS, Thomas ML (1994) CD45: an emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu Rev Immunol 12: 85–116 [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Garcia-Blanco MA (2001) Polypyrimidine tract binding protein antagonizes exon definition. Mol Cell Biol 21: 3281–3288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB (2004) Systematic identification and analysis of exonic splicing silencers. Cell 119: 831–845 [DOI] [PubMed] [Google Scholar]
- Xie J, Black DL (2001) A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 410: 936–939 [DOI] [PubMed] [Google Scholar]
- Xu Z, Weiss A (2002) Negative regulation of CD45 by differential homodimerization of the alternatively spliced isoforms. Nat Immunol 3: 764–771 [DOI] [PubMed] [Google Scholar]
- Zhang XH, Chasin LA (2004) Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev 18: 1241–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZM (2004) Regulation of alternative RNA splicing by exon definition and exon sequences in viral and mammalian gene expression. J Biomed Sci 11: 278–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Mayeda A, Krainer AR (2001) Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell 8: 1351–1361 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1