

Abstract
The unphosphorylated regulatory (R) domain of the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) is often viewed as an inhibitor that is released by phosphorylation. To test this notion, we studied domain interactions using CFTR channels assembled from three polypeptides. Nucleotides encoding the R domain (aa 635–836) were replaced with an internal ribosome entry sequence so that amino- and carboxyl-terminal half-molecules would be translated from the same mRNA transcript. Although only core glycosylation was detected on SplitΔR, biotinylation, immunostaining, and functional studies clearly demonstrated its trafficking to the plasma membrane. SplitΔR generated a constitutive halide permeability, which became responsive to cAMP when the missing R domain was coexpressed. Each half-molecule was co-precipitated by antibody against the other half. Contrary to expectations, GST-R domain was pulled down only if prephosphorylated by protein kinase A, and coexpressed R domain was precipitated with SplitΔR much more efficiently when cells were stimulated with cAMP. These results indicate that phosphorylation regulates CFTR by promoting association of the R domain with other domains rather than by causing its dissociation from an inhibitory site.
Keywords: cystic fibrosis, chloride channel, domain–domain interactions, ion channel regulation, protein kinase
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel mediates transepithelial anion transport when stimulated by secretagogues that raise [cAMP]. It belongs to the ATP binding cassette (ABC) superfamily of membrane proteins and has transmembrane domains (TMD1 and TMD2), nucleotide binding domains (NBD1 and NBD2), and a regulatory (R) domain, which confers most, if not all, control by phosphorylation (Hanrahan et al, 2003). The R domain is unique to CFTR and is mostly unstructured when studied as a soluble polypeptide (Dulhanty and Riordan, 1994; Ostedgaard et al, 2000). Consequently, there are few clues into the mechanism by which its phosphorylation activates gating (Ostedgaard et al, 2001).
A popular hypothesis for CFTR regulation considers the R domain as an inhibitory particle, which becomes released upon phosphorylation by protein kinase A (PKA). This scheme was inferred from the obvious domain structure of CFTR, and by analogy to the ‘ball-and-chain' model for fast (N-type) inactivation of voltage-gated Shaker B potassium channels (Hoshi et al, 1990). Shaker K+ channels have an amino-terminal region, which occludes the pore through sequential binding steps, and which is released by membrane hyperpolarization (Zhou et al, 2001). Shaker K+ channels are inhibited by exogenously applied amino-terminal peptides (Zagotta et al, 1990), and CFTR channels are similarly inhibited by adding unphosphorylated R domain to the cytoplasmic aspect (Ma et al, 1997). CFTR channels with large deletions in the R domain have slightly elevated open probability, further evidence that it normally functions as an inhibitor, although stimulatory effects of phosphorylated R domain have also been observed (Winter and Welsh, 1997; Ma, 2000). PKA can activate mutant channels in which the R domain is transplanted to the carboxyl-terminus (Baldursson et al, 2001); thus, regulation is apparently preserved if the carboxy-terminal ‘chain' is long enough to allow the R domain ‘ball' to reach its inhibitory site. The phosphorylation-regulated ball-and-chain model remains the framework for interpreting electrophysiological studies and can explain most findings, although phosphorylation-dependent binding (or more accurately, unbinding) of the R domain has not been demonstrated.
Many domain–domain interactions have been reported within CFTR, for example, between TMD1 and TMD2 (Ostedgaard et al, 1997), NBD1 and NBD2 (Lu and Pedersen, 2000; Kidd et al, 2004), and R domain and NBD (Wang et al, 2002). Interactions between NBD1 and NBD2 form nucleotide binding sites from apposed ABC signature and Walker A motifs (Jones and George, 1999; Smith et al, 2002; Vergani et al, 2005) although it is not known whether this dimerization is enhanced by phosphorylation. Recombinant R domain can bind fusion proteins comprising the amino-terminus of CFTR and glutathione-S-transferase (GST), but these interactions reportedly had little dependence on phosphorylation and were implicated in the regulation of CFTR by syntaxin 1A (Naren et al, 1999). The R domain also associates with the carboxy-terminal half of CFTR independently of cAMP stimulation, and this binding was proposed to facilitate phosphorylation of the R domain (King and Sorscher, 2000). Finally, unphosphorylated GST-R domain fusion protein can bind short peptides (20-mers) from the amino-terminus, cytoplasmic loops 1 and 4, and both NBDs; however regulation of these interactions was not examined (Wang et al, 2002). Thus, despite much evidence for domain–domain interactions within CFTR, there is little information concerning their dependence on phosphorylation.
In this paper, we use cell lines expressing a novel bicistronic ‘split' construct to investigate intramolecular interactions in CFTR channels assembled from three polypeptides. We expected phosphorylation of the R domain to inhibit its association with the rest of CFTR but found the opposite; that is, PKA phosphorylation caused a striking increase in R domain binding both in vitro and in vivo. Rather than causing dissociation of the R domain from an inhibitory site, we propose that phosphorylation induces binding of the R domain to another domain and the resulting intramolecular rearrangements cause channel activation. A preliminary account of this work has been published (Irvine et al, 2003).
Results
Stable expression of SplitΔR CFTR channels with or without R domain polypeptide
Stable transfection of a bicistronic SplitΔR construct encoding amino-terminal (TMD1-NBD1; aa 1–634) and carboxy-terminal (TMD2-NBD2; aa 837–1480) fragments of CFTR yielded polypeptides of Mr=62–65 kDa that were recognized by monoclonal antibodies against the ‘front half' (L12B4 and MM13-4, epitopes at aa 386–412 and 24–35, respectively) and by M3A7 against the ‘back half' (aa 1365–1395; Figure 1A). There are two consensus sequences for N-linked glycosylation in the fourth extracellular loop, but only core oligosaccharide chains were detected on the back half of SplitΔR when its mobility in SDS–PAGE gels was compared before and after incubation with endoglycosidase H or peptidyl N-glycosidase F (Figure 1B). This suggests most back-half molecules had only high-mannose oligosaccharide chains corresponding to the immature ‘Band B' form of full-length CFTR. Densitometry revealed lower expression of SplitΔ compared to full-length CFTR (17.7±1.9%, n=8 for front half and 14.8±1.7%, n=6 for back half; difference between front and back significant, P=0.035). Nevertheless, SplitΔR levels in BHK cells were still higher than those observed for full-length protein in most other mammalian cell lines (unpublished observations). Cells were treated with 10% glycerol overnight at 26°C, conditions that partially correct misfolding of some CFTR mutants (Denning et al, 1992; Brown et al, 1996), but these maneuvers did not increase SplitΔR levels noticeably or induce a high molecular weight form corresponding to ‘Band C' (Figure 1C). Nevertheless, cell surface biotinylation, immunostaining, and functional studies revealed significant amounts of core-glycosylated SplitΔR in the plasma membrane (see below).
Figure 1.

Expression of bicistronic SplitΔRpNUT, SplitΔRpIND, and SplitΔRpIND+RDpNUT CFTR in BHK cells. (A) Western blots of cell lysates from six BHK variants that had been transfected with SplitΔRpNUT CFTR and selected using 500 μM methotrexate. Note the expression of both amino-terminal (aa 1–634; top) and carboxy-terminal (aa 837–1480) halves of CFTR in colonies 1, 2, 4, and 5, as detected using the monoclonal antibodies L12B4 or M3A7, respectively. (B) Effect of glycerol treatment on maturation of full-length wild type (WT) or SplitΔR CFTR. Mature CFTR was sensitive to PNGase but not EndoH, consistent with the complex glycosylation (Band ‘C'). The back half of SplitΔR was sensitive to EndoH, indicating the presence of core oligosaccharide chains, which are characteristic of immature (Band ‘B') CFTR. (C) Western blot probed with antibody against front (L12B4) and back (M3A7) half-molecules showing the effect of incubating cells overnight with 10% glycerol at 26°C on expression of SplitΔRpNUT and wild-type CFTR. (D) Inducible expression of SplitΔRpIND with or without coexpression of RDpNUT. Western blot showing the front, back, and R domain (450) with (+) or without (−) induction by 10 μM Ponasterone A (Pon. A) for 24, 48, or 72 h. (E) Covalent crosslinking of coexpressed R domain and SplitΔR fragments into a complex. Cells were incubated with 2 mM dithiobis (succinimidyl propionate) (DSP) for 30 min, lysed, and immunoprecipitated using the anti-R domain. After crosslinking, most R domain (23 kDa) migrates in a high-molecular-weight complex of 175–200 kDa that also contains the front and back halves of SplitΔR. (F) Cell surface biotinylation of SplitΔRpIND, SplitΔRpIND+RDpNUT, or wild-type CFTR. Both halves were pulled down on streptavidin beads after cell surface biotinylation using sulfo-NHS-SS-biotin (left and right panels, respectively). SplitΔR was not co-precipitated if biotin was omitted from the reaction mix (‘no biotin').
SplitΔRpNUT expression declined after a few passages despite continued selection with 500 μM methotrexate. To generate a more stable cell line, bicistronic SplitΔR was subcloned into pIND and transfected into BHK cells. With this system, SplitΔR was detected only after cells were exposed to Ponasterone A (10 μM; Figure 1D), and SplitΔR levels were two- to three-fold higher than in pNUT-transfected cells constitutively expressing the same construct. Expression of the front and back half-molecules did not change significantly during 1–3 days induction (front/back ratios=1.02±0.28, 1.34±0.64, and 1.04±0.22 by densitometry, respectively; P>0.5; open bars in Supplementary Figure S1A) and did not decline with passage number; therefore, SplitΔRpIND-transfected cells were used in most subsequent experiments.
Western blots were probed for each CFTR fragment before or after SplitΔR induction with Ponasterone A. RDpNUT expression driven by the metallothionein promoter was robust and constitutive as expected. The steady-state level of R domain was not altered by coexpressing SplitΔR (Figure 1D), and conversely, SplitΔR levels were not affected by coexpression of R domain polypeptide (compare SplitΔRpIND versus SplitΔRpIND+RDpNUT). Interestingly, relative expression of the back half of SplitΔRpIND declined somewhat at 72 h (i.e. the increase in ratio in Supplementary Figure S1A; n=4, P<0.022), but only in cells coexpressing the R domain.
SplitΔR and R domain colocalize at the plasma membrane
To obtain biochemical evidence that the three fragments of CFTR can assemble into a complex in vivo, cells coexpressing SplitΔR and RD were stimulated with cAMP, exposed to chemical crosslinkers, and immunoprecipitated using anti-R domain antibody. Precipitates were subjected to Western blot analysis using antibodies against each fragment (i.e. R domain, front half, and back half, respectively). Figure 1E shows one such experiment with 2 mM DSP (dithiobis (succinimidyl propionate)), which is representative of three experiments with DSP and one with lysates exposed to DTSSP (3,3′-dithiobis (sulfosuccinimidyl propionate)) disuccinimyl tartrate (DST). After crosslinking, most R domain appeared in a high-molecular-weight complex of 175–200 kDa rather than at its predicted Mr=23 kDa (compare Figure 1E with Figures 1D and 6E). When aliquots of the same precipitates were analyzed for the front half by Western blotting with MM13-4, the front half was also detected in the complex (Figure 1E, center panel; predicted Mr=62 kDa as in Figure 1A). Stripping the blot and reprobing with M3A7 revealed that the back half of CFTR was also co-precipitated in the 175–200 kDa complex (predicted Mr=64 kDa). Thus, CFTR fragments assemble in vivo, and crosslinkers apparently stabilize the complex so that it migrates at a molecular weight similar to that of full-length CFTR (Figure 1E). This argues against significant binding to large scaffolding proteins, which would have further increased its mass. The R domain was readily pulled down without crosslinking when the front and back halves were immunoprecipitated (see below).
Figure 6.

Interactions between SplitΔR fragments and R domain. (A) Co-immunoprecipitation of the front and back halves of CFTR: lane 1, in the absence of GST-R; lane 2, with unphosphorylated GST-R added to the lysate; lane 3, after addition of phosphorylated GST-R without phosphatase inhibitors; and lane 4, with phosphorylated GST-R added to lysates along with excess PKA, ATP, and phosphatase inhibitors (see Materials and methods). (B) Effect of PKA phosphorylation on electrophoretic mobility of GST-R. (a) Autoradiogram of GST after preincubation with PKA and [γ-32P]ATP. (b–d) Western blots probed using anti-R domain mAb 25 (b), anti-R domain mAb 450 (c), or anti-GST antibody (d). (e) Unphosphorylated and phosphorylated GST-R in a Coomassie-stained gel. (C) Immunoprecipitates obtained with antibodies against the front or back half of SplitΔR after preincubation of lysates as described in (A). Note that GST-R was only detected when prephosphorylated. Electrophoretic mobility shifts confirmed that elevated phosphorylation promotes the interaction of GST-R, particularly with the amino-terminal half-molecule. (D) Co-immunoprecipitation of the front and back halves of CFTR from cells coexpressing SplitΔRpIND+RDpNUT CFTR. Phosphorylation was varied by pretreating cells for 3 h with H7 (lane 1), control medium (lane 2), or medium containing 150 μM cpt-cAMP+1 mM IBMX (lane 3). (E) Co-immunoprecipitation of endogenously expressed R domain with front or back halves of CFTR under the three conditions described in (D) except for lane 1 of the right panel, in which cells were pretreated with the PKA inhibitor H89. More R domain was pulled down with both half-molecules under phosphorylating conditions.
Biochemical evidence that the core-glycosylated SplitΔR complex is trafficked to the plasma membrane was obtained by exposing cells to sulfo-NHS-SS-Biotin on ice and pulling down biotinylated cell surface proteins on streptavidin beads (Figure 1F). Both halves of SplitΔR CFTR were detected after cell surface biotinylation, but not in negative controls (‘no biotin') performed to exclude nonspecific binding to the beads. Confocal microscopy was used to further test if core-glycosylated SplitΔR and coexpressed R domain reach the cell periphery (Figure 2). Intracellular distributions of full-length wild-type and ΔF508 CFTR (the most common disease mutation) were assessed by immunostaining and then the same protocols were applied to cells expressing SplitΔR±R domain polypeptide. Transfected cells were fixed, permeabilized, and blocked in solutions containing paraformaldehyde and Triton X-100/BSA and then exposed to L12B4, M3A7, or 450 monoclonal antibodies directed against the front half, back half, or R domain of CFTR, respectively. CFTR was visualized using goat anti-mouse IgG secondary antibody that had been conjugated with the fluorophores Cy3 (L12B4) and Cy5 (M3A7 or 450; see Materials and methods).
Figure 2.

Localization of full-length WT and ΔF508 CFTR (rows A and B, respectively), and amino- and carboxy-terminal fragments in cells expressing SplitΔRpIND or SplitΔRpIND+RDpNUT (rows C and D). Cells were fixed using paraformaldehyde and permeabilized with 0.1% Triton X-100 and then incubated with anti-CFTR antibodies L12B4, M3A7, or 450 (1:1000). Signals were detected using a secondary Cy3-conjugated goat anti-mouse antibody (1:100, in red, column i) or Cy5-conjugated goat anti-mouse antibody (1:200, in green for M3A7 (ii) or in light blue for 450 (iii)). Colocalization is shown for front half and back half (column iv) and for the front half and R domain (column v).
Sharply defined cell margins and strong peripheral immunostaining were obtained with all three antibodies when cells expressed full-length, wild-type CFTR (Figure 2A, panels i–iii). By contrast, only weak immunofluorescence was detected in cells expressing ΔF508 CFTR, and it was mostly perinuclear, consistent with the reduced half-life of this mutant and its retention in the endoplasmic reticulum (Figure 2B, panels i–iii). Similar distributions were observed using antibodies directed against the front or back halves, or the R domain. In some experiments, cells were incubated sequentially with two different antibodies that recognize distinct domains so that each could be visualized with a different fluorophore (i.e. L12B4/Cy3-anti-mouse followed by M3A7/Cy5-anti-mouse, or L12B4/Cy3-anti-mouse followed by 450/Cy5-anti-mouse; see Materials and methods for details). As expected, dual labelling of full-length, wild-type CFTR yielded overlapping membrane localization (Figure 2A, panels iv and v), and overlapping perinuclear staining was obtained with ΔF508 CFTR as indicated by yellow and mauve in panels iv and v, respectively (Figure 2B). Thus, antibodies recognizing distinct amino- and carboxy-terminal epitopes in full-length CFTR gave equivalent results when the protein was expressed in different cellular compartments, validating the immunolocalization.
The same protocols were used to localize the front and back halves of SplitΔR (Figure 2C, panels i and ii). SplitΔR immunofluorescence resembled that of wild-type rather than ΔF508 CFTR (compare panels i and ii in Figure 2C with those in Figure 2A and B), and Cy3 and Cy5 signals could be superimposed in ∼1/3 of cells that had been stained sequentially using L12B4 (front half) and M3A7 (back half). R domain antibody also caused peripheral staining of cells coexpressing the R domain with SplitΔR (Figure 2D, panel iii), consistent with the crosslinking results. Immunofluorescence was not detected if cells were incubated with Cy3- or Cy5-conjugated goat anti-mouse IgG without exposure to primary antibody (Supplementary Figure S2A). Fluorescence signals from Cy3 and Cy5 were well resolved in cells that had been incubated sequentially with two pairs of antibodies, that is, with a 1° mAb against one epitope followed by saturation with Cy3-conjugated 2° antibody, then application of a different 1° mAb and Cy5-conjugated 2° antibody (Supplementary Figure S2B). Finally, immunofluorescence was not detected when control cells that had been mock transfected with empty pIND vector were stained using M3A7 or 450 (Supplementary Figure S2C), and nonspecific staining with L12B4 was predominantly nuclear and therefore easily distinguished from CFTR at the periphery.
In summary, chemical crosslinking, biotinylation, and immunostaining indicate that core-glycosylated SplitΔR and coexpressed R domain can assemble and reach the cell surface.
SplitΔR expression generates constitutively active channels in BHK cells
To investigate if surface-expressed SplitΔR is functional in intact cells, we compared halide efflux after stably transfecting cells with SplitΔR, full-length CFTR, or empty vector. Consistent with the tight regulation of CFTR by phosphorylation, iodide release from unstimulated cells expressing full-length CFTR was low, and similar to control cells transfected with empty vector (Figure 3A). Unstimulated iodide efflux was also low in uninduced SplitΔRpIND cells, but increased dramatically (∼6-fold) when SplitΔR expression was induced using Ponasterone A. This elevated efflux was not responsive to cAMP agonists, in contrast to cells expressing full-length CFTR (Figure 3B). Thus, expression of SplitΔR generates an unregulated halide efflux pathway in BHK cells.
Figure 3.

Constitutive activity of SplitΔRpIND and restoration of PKA regulation by R domain. Iodide effluxes were measured from BHK cell monolayers at 1 min intervals. (A) Initial effluxes from BHK cells stably transfected with SplitΔRpIND with or without induction by 10 μM Ponasterone A for 48 h. Initial iodide effluxes from cells constitutively expressing full-length wild-type (WT) CFTR are shown for comparison. (B) Effect of the cAMP agonists 150 μM cpt-cAMP+1 mM IBMX added at t=0 min on iodide efflux from cells induced to express SplitΔRpIND or SplitΔRpIND+RDpNUT. Control cells were not induced. (C) Effect of R domain coexpression on basal halide permeability assayed as initial iodide efflux. (D) Restoration of iodide efflux response to cAMP agonists when R domain is coexpressed with SplitΔR CFTR. Means±s.e., n=6.
Single SplitΔR channels were studied after 2 days induction by sequentially exposing inside-out membrane patches to control solution, 1 mM MgATP, and 1 mM MgATP+100 nM PKA catalytic subunit. Only one channel having conductance in the 5–10 pS range was detected in 204 patches bathed with nominally ATP-free solution, and its open probability was low (NPo=0.019±0.015). By contrast, 1–3 active channels appeared in 32 of those patches upon addition of 1 mM MgATP, and the patches had a mean NPo=0.54±0.55; P<0.0085 (Figure 4A and C). PKA did not further increase SplitΔR channel activity (NPo=0.54±0.213, P>0.15, n=6; Figure 4A and C), consistent with the inability of cAMP to stimulate iodide efflux from cells expressing SplitΔRpIND. Their current–voltage relationships were linear and mean unitary conductance was 7.2 pS in symmetric 150 mM chloride solutions (Figure 5A and B), as shown previously for fibroblasts expressing full-length CFTR (Tabcharani et al, 1991). In summary, iodide efflux and patch-clamp experiments indicate that SplitΔR channels are functional at the plasma membrane of mammalian cells. Moreover, they are constitutively active and do not respond to PKA activation as reported previously in Xenopus oocytes (Csanády et al, 2000).
Figure 4.
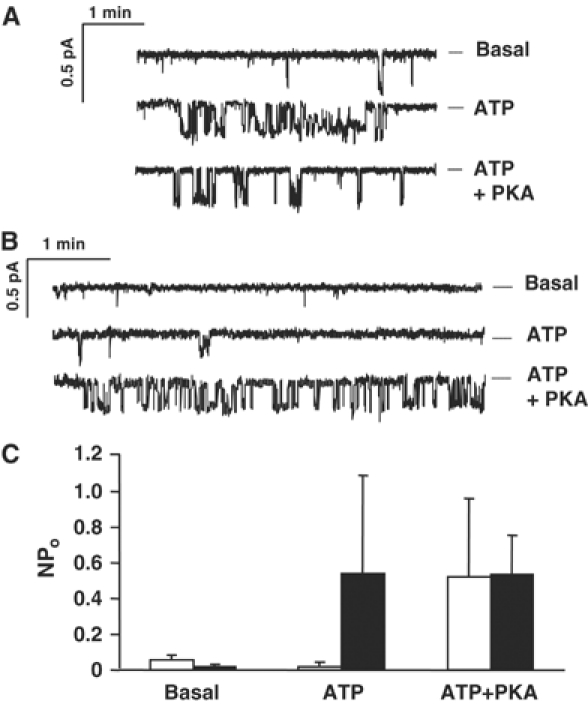
Single channel recordings from cells expressing (A) SplitΔRpIND or (B) SplitΔRpIND+RDpNUT in control solution (‘Basal') and after sequential addition of 1 mM ATP and 100 nM PKA to the bath. Dashed lines indicate closed level. (C) Mean number of channels in the open state in control solution (basal) and after addition of 1 mM Mg-ATP or 1 mM Mg-ATP and 100 nM PKA. Solid and open bars indicate cells expressing SplitΔRpIND and SplitΔRpIND+RDpNUT, respectively. Means±s.e., n=12 and 8, respectively.
Figure 5.
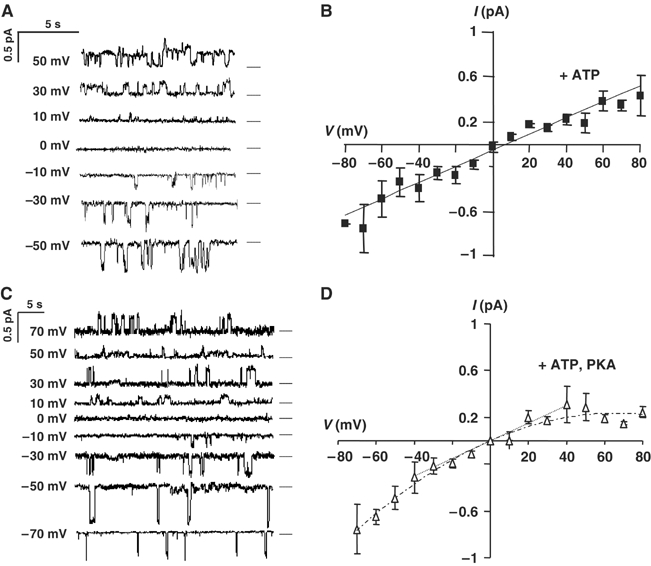
Current–voltage relations for SplitΔR channels in inside-out membrane patches. Representative activity in patches from cells expressing (A) SplitΔRpIND or (C) SplitΔRpIND+RDpNUT. Dashed lines indicate the current level when closed. Best-fitted current–voltage relationships are shown for (B) SplitΔRpIND and (D) SplitΔRpIND+RDpNUT channels in the presence of 1 mM Mg-ATP. Mean values±s.e. are for SplitΔRpIND (n=12) and SplitΔRpIND+RDpNUT (n=8).
Coexpressing the R domain inhibits spontaneous channel activity and confers PKA regulation
Unstimulated iodide efflux from cells coexpressing SplitΔR+RD was reduced by 32% compared to those expressing only SplitΔR (P<0.032, n=4; Figure 3C), although still higher than full-length CFTR despite lower expression. SplitΔR did not respond to cAMP, but when cells were stably cotransfected with RDpNUT, their responsiveness was partially restored, albeit with a delay of 2–3 min (Figure 3D). cAMP-stimulated iodide efflux from cells expressing SplitΔR+R domain reached a maximum of 32±3.9 nmol/min, as compared to 41±9.0 nmol/min (mean±s.e., P<0.02, n=6 monolayers) for full-length CFTR. cAMP responses were only observed after cells were induced with Ponasterone A (Figure 3D). The reason for the delayed efflux response may reflect altered gating by reassembled channels. Regardless, it is clear that coexpressing the R domain partially restores PKA regulation of SplitΔR channels in vivo.
Control of SplitΔR channels by the R domain was also studied in vitro by recording channels in membrane patches excised from cells expressing SplitΔR+R domain. Channels were detected only after Ponasterone A induction, and had low activity in 21/51 patches bathed with 1 mM MgATP (mean NPo for those patches with active channels was 0.02±0.023). Significantly, channel activity in cells expressing SplitΔR+R domain increased to NPo=0.52±0.44 (P<0.015) in the presence of MgATP+PKA (Figure 4B), similar to the value obtained when SplitΔR channels lacking the R domain were exposed to MgATP (Figure 4C). Thus, SplitΔR channels are functional, and PKA phosphorylation in the absence of the R domain has little, if any, effect on their activity. The current–voltage relationship of single channels was linear and indicated a unitary conductance of 7.6 pS between −40 and +40 mV, although some inward rectification was observed at large positive potentials (Figure 5C and D). We conclude that coexpressing the R domain with SplitΔR partially inhibits constitutive channel activity and restores regulation by PKA.
Phosphorylation enhances binding of GST-R domain to SplitΔR in vitro
The hypothesis that phosphorylation releases the R domain was tested by assessing the ability of monoclonal antibodies against the front and back halves of SplitΔR to co-precipitate the R domain. First, interaction between the front and back half was examined by immunoprecipitating each fragment and immunoblotting precipitates for the other half (Figure 6A). Lane 1 in both panels shows that each half-molecule was pulled down by the other, as reported previously (Ostedgaard et al, 1997; Chan et al, 2000). Co-precipitation was observed in the absence of exogenous GST-R (lane 1), after addition of unphosphorylated GST-R to lysates (lane 2), in the presence of weakly phosphorylated GST-R that had been pretreated with PKA but exposed to phosphatases in the lysates (lane 3), and when prephosphorylated GST-R was added to lysates along with excess PKA, ATP, and phosphatase inhibitors to ensure that it remained strongly phosphorylated (lane 4). We conclude that association between CFTR half-molecules is independent of the R domain and its phosphorylation state, consistent with interactions via the membrane (Ostedgaard et al, 1997) and/or NBDs (Kidd et al, 2004).
The phosphorylation dependence of R domain binding was explored by incubating prephosphorylated GST-R with SplitΔR, immunoprecipitating the front or back half of CFTR, and probing Western blots for R domain. Figure 6B demonstrates the ability of R domain antibodies to recognize the fusion protein and the effect of phosphorylation on its electrophoretic mobility in SDS–PAGE gels. An autoradiogram of GST-R that had been prephosphorylated using PKA and [γ-32P]ATP in panel a serves as a reference for the Western blots (panels b–d) of samples before (lane 1) and after (lane 2) phosphorylation. Probing Western blots with the anti-R domain antibodies 25 or 450 (panels b and c, respectively) or anti-GST antibody (panel d) produced similar results. Antibody 25 was phosphorylation independent whereas mAb 450 had somewhat higher affinity for dephospho-GST-R than for the phosphorylated form, although it still recognized both at 1:5000 dilution. A Coomassie-stained protein gel containing GST-R domain before and after phosphorylation is shown in panel e (lanes 1 and 2, respectively). GST-R was partially dephosphorylated by the lysate, as evidenced by increased mobility in Western blots. The mobility shift served as a convenient indicator of phosphorylation state during the pull-downs. Dephosphorylation was prevented by adding PKA and protein phosphatase inhibitors to cell lysates.
Interactions between domains were studied by incubating lysates with GST-R under various conditions. Figure 6C shows a typical experiment in which the front or back half of CFTR was precipitated by antibody MM13-4 (left) or M3A7 (right) followed by SDS–PAGE and immunoblotting for GST-R. Lane 1 shows a negative control precipitate from a sample incubated without GST-R. Unphosphorylated R domain was not co-precipitated with the front or back half (lane 2). By contrast, GST-R was consistently co-precipitated with both half-molecules if it was prephosphorylated using PKA before addition to the lysate (lane 3). The pull-down was further enhanced if PKA (400 nM), ATP (2 mM), and the phosphatase inhibitors cyclosporin A (5 μM) and calyculin A (100 nM) were added to the lysate with prephosphorylated GST-R to maintain its phosphorylation. Association of GST-R with the front appeared stronger and more dependent on phosphorylation, but more front half was usually expressed and GST-R binding might be influenced by GST; therefore, no conclusions were drawn concerning the site of R domain binding. Regardless, the results indicate phosphorylation-dependent association of GST-R with SplitΔR in vitro.
cAMP stimulates intracellular binding of R domain polypeptide to SplitΔR
To study R domain interactions under more physiological conditions and control for possible artifacts that might arise from use of a fusion protein, cells containing both SplitΔRpIND and RDpNUT were immunoprecipitated under conditions paralleling those used in vitro (Figure 6D and E). Cells were either exposed to the broad-spectrum kinase inhibitor H7 or the more specific PKA inhibitor H89 (10 μM) for 3 h to minimize phosphorylation (lane 1), left untreated (lane 2), or incubated with 150 μM cpt-cAMP+1 mM IBMX to stimulate PKA phosphorylation (lane 3). When kinase inhibitors were used, they were also added to the lysates.
MM13-4 against the front half of CFTR antibody co-precipitated the back half irrespective of kinase inhibition or activation (Figure 6D). Likewise, Western blots confirmed that the carboxy-terminal half co-precipitated the front half. More importantly, coexpressed R domain polypeptide was pulled down by antibody against either half-molecule, and these associations became progressively stronger under conditions that would increase phosphorylation (Figure 6E). Preferential binding to the front half was observed under control conditions (P<0.026, n=4) and during cAMP stimulation (P<0.031, n=4), but not with the kinase inhibitor H7 (P>0.837). Taken together, these results demonstrate that phosphorylation promotes binding of the R domain (no GST) to SplitΔR in live cells.
Discussion
The goal of this work was to investigate the role of R domain interactions in the regulation of CFTR by PKA. CFTR has many conserved consensus sequences for phosphorylation by protein kinases (Gadsby and Nairn, 1999; Dahan et al, 2001), and mutating them decreases PKA responsiveness primarily by reducing the opening rate (Mathews et al, 1998). Unphosphorylated R domain functions as an inhibitor (Rich et al, 1994; Csanády et al, 2000; Baldursson et al, 2001) but it remains unclear how phosphorylation relieves this inhibition. Electrostatic repulsion was suggested when mutagenesis of eight consensus PKA sites to negatively charged aspartates induced some constitutive activity (Rich et al, 1993), and when prephosphorylation of in vitro-translated R domain abolished its inhibition of single channels (Ma et al, 1996). However, phosphorylation-induced release of the R domain has not been demonstrated, and other mechanisms such as alterations in R domain conformation have also been suggested based on channel stimulation by the cis–trans peptidyl-prolyl isomerase cyclophilin A (Xie et al, 2000).
By studying binding of the R domain to SplitΔR in vivo and in vitro, we unexpectedly found that PKA phosphorylation strongly enhances its association with the rest of the molecule. Since PKA is the primary stimulus for CFTR gating, this phosphorylation-induced binding is likely to play a role in channel activation, perhaps by causing intramolecular rearrangements that allow nucleotide-induced conformational changes to be transmitted from the NBDs to the TMDs. Further studies are needed to establish the mechanism and identify the site where phosphorylated R domain binds; nevertheless, the present results strongly suggest that phosphorylation acts by enhancing association of the R domain rather than by causing its release. This is shown conceptually in Figure 7, although it should be noted that the receptor for phosphorylated R domain is speculative and there may be several binding sites in the front or back half.
Figure 7.
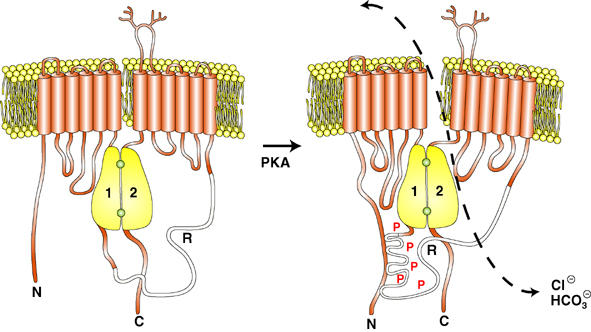
Cartoon summarizing the effect of CFTR phosphorylation on R domain interactions. Phosphorylated R domain is shown interacting with the amino-terminus; however, this is highly speculative and the R domain receptor remains to be identified.
SplitΔR was weakly expressed compared to full-length CFTR, and only endo-H-sensitive core glycosylation was detected. Attempts to improve maturation by exposing cells to low temperature and glycerol, procedures that partially correct misprocessing of ΔF508 CFTR and some other mutants (Denning et al, 1992; Brown et al, 1996; Sato et al, 1996), were unsuccessful, presumably because the efficacy of these treatments depends on the particular mutation and its associated folding defect. Immunofluorescence localization, cell surface biotinylation, iodide efflux assays, and patch clamping all confirmed that SplitΔR is functionally expressed at the plasma membrane. This was surprising given that core glycosylation on ΔF508 signifies retention in the endoplasmic reticulum, although it is well established that complex glycosylation is not required for CFTR trafficking or channel activity (Cheng et al, 1990; Kartner et al, 1991; Morris et al, 1993) and there is growing evidence for nonconventional trafficking of immature CFTR from the cis-Golgi to early endosomes (Yoo et al, 2002; Okiyoneda et al, 2004). It is conceivable that functional, immature SplitΔR can be diverted from endosomes to the plasma membrane instead of returning to the trans-Golgi.
Reconstitution of regulated CFTR channels from three separate protein fragments is consistent with the multidomain structure of CFTR and the fact that many ABC transporters in lower organisms are complexes comprising separate polypeptide domains. We confirmed a strong association between front and back half-molecules (Ostedgaard et al, 1997; Chan et al, 2000) and further showed that phosphorylated R domain can be co-precipitated by antibody against either half. The higher apparent efficiency of R domain pull-down with the front half was probably due to elevated expression of the sequence upstream of the internal ribosome entry sequence (IRES), as is commonly observed with bicistronic vectors. In Western blots, the predominant R domain band, which also contained the front and back halves, had an apparent Mr of 175–200 kDa after cells were exposed to crosslinkers rather than the predicted 23 kDa. This apparent mass is similar to that of full-length CFTR, which confirms assembly and also argues against R domain binding to large scaffolding proteins, although further studies are needed to exclude this definitively. Unphosphorylated R domain must associate weakly with SplitΔR since R domain coexpression inhibited constitutive iodide efflux and single channel activity, although this interaction is apparently too weak to survive our immunoprecipitation protocol. cAMP only activated iodide efflux from cells coexpressing SplitΔR and R domain; thus, phosphorylation-induced associations that we detect biochemically are probably relevant to physiological channel activation by PKA. Channel activity was not observed in control cells transfected with empty vector, and single channel current–voltage relations of reassembled channels were similar to those of full-length wild-type CFTR except for smaller currents at large positive voltages. Interestingly, rectification was only seen when cells coexpressed the R domain. Slight inward rectification has been reported previously for full-length CFTR channels and shown to be an intrinsic property of the pore (Cai et al, 2003).
Associations between CFTR domains have been observed previously using GST fusion proteins (Naren et al, 1999). The R domain interacted with four acidic residues near the amino-terminus but not with intracellular loops 1, 2, or 3. Another study showed that CFTR truncated after the R domain induces halide permeability in COS cells if coexpressed with the back half (King and Sorscher, 2000). The truncated fragment only became phosphorylated when coexpressed with the back half, leading the authors to suggest that R domain phosphorylation depends on interaction with the carboxyl-terminus. This differs from our conclusion that phosphorylation promotes R domain binding, but the interpretations are not mutually exclusive. Indeed, the present finding that binding is greatly enhanced by phosphorylation would explain why only phosphorylated R domain was detected previously in immunoprecipitates (King and Sorscher, 2000). High constitutive halide permeability was observed previously despite coexpression of the R domain, but this might have been due to use of a different expression system. Constitutive halide permeability in that study was not further stimulated by cAMP yet was sensitive to the PKA inhibitor Rp-8-cpt-cAMP, implying high basal PKA activity in Vaccinia-infected COS cells.
Recent Fourier transform infrared spectroscopy and tryptophan fluorescence studies of CFTR indicate that phosphorylation increases the solvent accessibility of the cytoplasmic domains and TMDs of CFTR (Grimard et al, 2004). These alterations could reflect long-range conformational changes transmitted from the phosphorylation sites, or from domain rearrangements resulting from enhanced R domain binding, as suggested by the present results. A helix between aa 817 and 838 called NEG2 may mediate binding of the R domain to other regions of CFTR (Xie et al, 2002), and a model has been proposed in which NEG2 interacts with hypothetical stimulatory or inhibitory binding sites according to its phosphorylation state (Xie et al, 2002). The present results are compatible with such a model and provide the first direct evidence for phosphorylation-dependent binding of the R domain and its role in CFTR activation.
Materials and methods
SplitΔR cDNA was constructed by replacing the nucleotides that encode the R domain of wild-type CFTR (aa 635–836) with an IRES while introducing a stop codon at the 3′ end of the front half and a Kozak consensus for translation initiation of the back half. The final construct was confirmed by sequencing and ligated into pNUT and pIND. Stable baby hamster kidney (BHK-21) cell lines were selected using methotrexate (SplitΔRpNUT), G418+Zeocin (pVgRXR+SplitΔRpIND), or all three drugs (pVgRXR+SplitΔRpIND+RDpNUT) and induced for 2 days using Ponasterone A. A fusion protein with aa 635–836 of CFTR attached to the carboxyl-terminus of GST was prepared as described previously (Zhu et al, 1999). Purified GST-R was phosphorylated by incubation in phosphorylation buffer (140 mM NaCl, 4 mM KCl, 2 mM MgCl2, 0.5 mM CaCl2, 10 mM Tris–HCl pH 7.4) containing 10 μM Na2-ATP and 400 nM PKA catalytic subunit (kindly prepared by the laboratory of Dr MP Walsh, Univ. Calgary, AB) for 30 min at 30°C. These conditions were also used to radiolabel the fusion protein, except that Na2-ATP was replaced by 20 μCi [γ-32P]ATP (see Supplementary data for details).
Immunoprecipitation, crosslinking, and biotinylation
Anti-CFTR antibodies were bound to Protein G using the SeizeX immunoprecipitation kit (Pierce Laboratories Inc., Rockford, IL). In vitro association of GST-R domain with SplitΔR was assessed by incubating lysates with GST-R under one of the following conditions: (1) control, without any manipulation that would cause phosphorylation, (2) low phosphorylation: after preincubation with PKA and ATP but susceptible to phosphatases in the lysate, or (3) high phosphorylation, prephosphorylated and added with PKA, ATP, and the phosphatase inhibitors cyclosporin A and calyculin A. In vivo association of endogenously expressed R domain with SplitΔR was studied using cells stably expressing both SplitΔRpIND and RDpNUT. Cells were induced, treated for 3 h with either cpt-cAMP+1 mM IBMX or 10 μM H7 or H89 to increase or reduce PKA phosphorylation, respectively, and lysed for immunoprecipitation as described above. When cells were pretreated with H7 or H89, they were also added to the lysates to maintain inhibition in vitro.
To crosslink CFTR fragments, cells coexpressing SplitΔRpIND and RDpIND were induced and stimulated with cpt-cAMP+IBMX and then treated with the membrane-permeable crosslinker DSP (2 mM; Pierce) for 30 min at 22°C. The reaction was stopped using Tris, cells were washed, lysed in PBS/1% Triton X-100, and immunoprecipitated using R domain antibody (450) on IgIP beads for SDS–PAGE and Western blotting. Blots were probed with 450 and M3A7 to identify the R domain and back half of CFTR, respectively, and then stripped and reprobed with MM13-4 against the front half. To biotinylate SplitΔR at the cell surface, cells expressing full-length CFTR, SplitΔRpIND, or SplitΔRpIND+RDpNUT were cultured at high density, induced, and washed 3 × with ice-cold PBS and once with ice-cold borate buffer. After incubating cells with 0.5 mg/ml sulfo-NHS-SS-biotin, the reaction was quenched and they were washed, harvested by scraping, lysed in RIPA buffer, centrifuged, and incubated with streptavidin-coated beads on a rotator at 4°C for 2 h. Unbound proteins were removed by washing the beads five times with RIPA buffer and biotinylated proteins were eluted with 5 × sample buffer and subjected to Western blot analysis as described previously (Chappe et al, 2003) (see Supplementary data). Protein expression levels were compared by densitometry of scanned Western blots using ImageJ software from Wayne Rasband, NIH (http://rsb.info.nih.gov/ij/). Densities were normalized to full-length CFTR run on the same gel to correct for differences in antibody affinity.
Immunolocalization
Cells stably expressing wild-type or ΔF508 CFTR (both in pNUT), SplitΔRpIND (in pIND), or SplitΔRpIND/RDpNUT (i.e. both plasmids) were plated at low density on glass coverslips, induced with Ponasterone A, and fixed with 2% paraformaldehyde. After permeabilization and blocking with 0.1% Triton X-100/2% BSA in PBS, they were incubated in fresh solution containing 0.1% Triton X-100, 0.2% BSA, and the anti-CFTR antibodies L12B4 or M3A7 diluted 1:1000 in PBS for 1 h at room temperature, and then washed and incubated with a secondary antibody (Cy3-conjugated goat anti-mouse at 1:100 diluted in PBS/0.1% Triton X-100/0.2% BSA; Jackson ImmunoResearch Lab., West Grove, PA). In one set of experiments, cells were washed and incubated with anti-CFTR antibody (M3A7 or 450, 1:1000) and then with Cy5-conjugated goat anti-mouse secondary antibody (1:200) in PBS/0.1% Triton X-100/0.2% BSA. After the final wash, slides were mounted, dried, and viewed using a Zeiss LSM 510 confocal microscope. Cells expressing full-length wild-type or ΔF508 CFTR served as positive controls. Protocol details and negative controls (Supplementary Figure S2A–C) are in Supplementary data.
Functional studies
Cell lines expressing full-length were cultured at high density in six-well plates, induced for 2 days, and assayed for cAMP-stimulated iodide efflux as described previously (Chappe et al, 2003). For patch-clamp studies, cells containing SplitΔRpIND or SplitΔRpIND/RDpNUT were plated at low density on glass coverslips, induced, and single channels recorded as described previously (Chappe et al, 2004).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S2
Supplementary Materials 3
Supplementary Materials 2
Supplementary Materials 1
Acknowledgments
We thank JR Riordan (Mayo Foundation and SC Johnson Medical Research Center, Mayo Clinic, Scottsdale, AZ) for providing anti-CFTR antibodies, F Chappe for technical assistance, and Julie Wang for preparing Figure 7. This work was supported by a Canadian CF Foundation (CCFF) fellowship to VC, CCFF studentship to TI, Canadian Institutes of Health Research (CIHR) senior scientist award to JWH, and grants from the Canadian CF Foundation, CIHR, CF Foundation (USA), and National Institutes of Health (NIDDK).
References
- Baldursson O, Ostedgaard LS, Rokhlina T, Cotten JF, Welsh MJ (2001) Cystic fibrosis transmembrane conductance regulator Cl− channels with R domain deletions and translocations show phosphorylation-dependent and -independent activity. J Biol Chem 276: 1904–1910 [DOI] [PubMed] [Google Scholar]
- Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal Biochem 72: 248–254 [DOI] [PubMed] [Google Scholar]
- Brown CR, Hong-Brown LQ, Biwersi J, Verkman AS, Welch WJ (1996) Chemical chaperones correct the mutant phenotype of the ΔF508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1: 117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Scott-Ward TS, Sheppard DN (2003) Voltage-dependent gating of the cystic fibrosis transmembrane conductance regulator Cl− channel. J Gen Physiol 122: 605–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KW, Csanády L, Seto-Young D, Nairn AC, Gadsby DC (2000) Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH2-terminal nucleotide binding domain. J Gen Physiol 116: 163–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappe V, Hinkson DA, Howell LD, Evagelidis A, Liao J, Chang X-B, Riordan JR, Hanrahan JW (2004) Stimulatory and inhibitory protein kinase C consensus sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc Natl Acad Sci USA 101: 390–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappe V, Hinkson DAR, Zhu T, Chang X-B, Riordan JR, Hanrahan JW (2003) Phosphorylation of protein kinase C sites in NBD1 and the R domain control CFTR channel activation by PKA. J Physiol 548: 39–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834 [DOI] [PubMed] [Google Scholar]
- Csanády L, Chan KW, Seto-Young D, Kopsco DC, Nairn AC, Gadsby DC (2000) Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J Gen Physiol 116: 477–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan D, Evagelidis A, Hanrahan JW, Hinkson DAR, Jia Y, Luo J, Zhu T (2001) Regulation of the CFTR channel by phosphorylation. Pflügers Arch 443: S92–S96 [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358: 761–764 [DOI] [PubMed] [Google Scholar]
- Dulhanty AM, Riordan JR (1994) Phosphorylation by cAMP-dependent protein kinase causes a conformational change in the R domain of the cystic fibrosis transmembrane conductance regulator. Biochemistry 33: 4072–4079 [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Nairn AC (1999) Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 79: S77–S107 [DOI] [PubMed] [Google Scholar]
- Grimard V, Li C, Ramjeesingh M, Bear CE, Goormaghtigh E, Ruysschaert J-M (2004) Phosphorylation-induced conformational changes of cystic fibrosis transmembrane conductance regulator monitored by attenuated total reflection-Fourier transform IR spectroscopy and fluorescence spectroscopy. J Biol Chem 279: 5528–5536 [DOI] [PubMed] [Google Scholar]
- Hanrahan JW, Gentzsch M, Riordan JR (2003) The cystic fibrosis transmembrane conductance regulator (ABCC7). In ABC Proteins: From Bacteria to Man, Holland B, Higgins CF, Kuchler K, Cole SPC (eds) pp 589–618. New York: Elsevier Sci. Ltd [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW (1990) Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250: 533–538 [DOI] [PubMed] [Google Scholar]
- Irvine T, Chappe V, Evaglelidis A, Hanrahan JW (2003) Role of phosphorylation in domain–domain interactions studied using split ΔR-channels stably expressed in mammalian cells. Pediatric Pulmonol S25: 187 [Google Scholar]
- Jones PM, George AM (1999) Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol Lett 179: 187–202 [DOI] [PubMed] [Google Scholar]
- Kartner N, Hanrahan JW, Jensen TJ, Naismith AL, Sun S, Ackerley CA, Reyes EF, Tsui L-C, Rommens JM, Bear CE, Riordan JR (1991) Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell 64: 681–691 [DOI] [PubMed] [Google Scholar]
- Kidd JF, Ramjeesingh M, Stratford F, Huan L-J, Bear CE (2004) A heteromeric complex of the two nucleotide binding domains of CFTR mediates ATPase activity. J Biol Chem 279: 41664–41669 [DOI] [PubMed] [Google Scholar]
- King SA, Sorscher EJ (2000) R-domain interactions with distal regions of CFTR lead to phosphorylation and activation. Biochemistry 39: 9868–9875 [DOI] [PubMed] [Google Scholar]
- Lu NT, Pedersen PL (2000) Cystic fibrosis transmembrane conductance regulator: the purified NBF1+R protein interacts with the purified NBF2 domain to form a stable NBF1+R/NBF2 complex while inducing a conformational change transmitted to the C-terminal region. Arch Biochem Biophys 375: 7–20 [DOI] [PubMed] [Google Scholar]
- Ma J (2000) Stimulatory and inhibitory functions of the R domain on CFTR chloride channel. News Physiol Sci 15: 154–158 [DOI] [PubMed] [Google Scholar]
- Ma J, Tasch JE, Tao T, Zhao J, Xie J, Drumm ML, Davis PB (1996) Phosphorylation-dependent block of cystic fibrosis transmembrane conductance regulator chloride channel by exogenous R domain protein. J Biol Chem 271: 7351–7356 [DOI] [PubMed] [Google Scholar]
- Ma J, Zhao J, Drumm ML, Xie J, Davis PB (1997) Function of the R domain in the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem 272: 28133–28141 [DOI] [PubMed] [Google Scholar]
- Mathews CJ, Tabcharani JA, Chang X-B, Jensen TJ, Riordan JR, Hanrahan JW (1998) Dibasic protein kinase A sites regulate bursting rate and nucleotide sensitivity of the cystic fibrosis transmembrane conductance regulator chloride channel. J Physiol 508: 365–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AP, Cunningham SA, Benos D, Frizzell RA (1993) Glycosylation status of endogenous CFTR does not affect cAMP-stimulated Cl− secretion in epithelial cells. Am J Physiol Cell Physiol 265: C688–C694 [DOI] [PubMed] [Google Scholar]
- Naren AP, Cormet-Boyaka E, Fu J, Villain M, Blalock JE, Quick MW, Kirk KL (1999) CFTR chloride channel regulation by an interdomain interaction. Science 286: 544–548 [DOI] [PubMed] [Google Scholar]
- Okiyoneda T, Harada K, Yamahira K, Wada I, Hashimoto Y, Ueno H, Suico MA, Shuto T, Kai H (2004) Characterization of the trafficking pathway of cystic fibrosis transmembrane conductance regulator in baby hamster kidney cells. J Pharmacol Sci 95: 471–475 [DOI] [PubMed] [Google Scholar]
- Ostedgaard LS, Baldursson O, Vermeer DW, Welsh MJ, Robertson AD (2000) A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc Natl Acad Sci USA 97: 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard LS, Baldursson O, Welsh MJ (2001) Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by its R domain. J Biol Chem 276: 7689–7692 [DOI] [PubMed] [Google Scholar]
- Ostedgaard LS, Rich DP, DeBerg LG, Welsh MJ (1997) Association of domains within the cystic fibrosis transmembrane conductance regulator. Biochemistry 36: 1287–1294 [DOI] [PubMed] [Google Scholar]
- Rich DP, Berger HA, Cheng SH, Travis SM, Saxena M, Smith AE, Welsh MJ (1993) Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by negative charge in the R domain. J Biol Chem 268: 20259–20267 [PubMed] [Google Scholar]
- Rich DP, Gregory RJ, Cheng SH, Smith AE, Welsh MJ (1994) Effect of deletion mutations on the function of CFTR chloride channels. Receptors Channels 1: 221–232 [PubMed] [Google Scholar]
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR (1996) Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 271: 635–638 [DOI] [PubMed] [Google Scholar]
- Smith PC, Karpowich N, Millen L, Moody JE, Rosen J, Thomas PJ, Hunt JF (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell 10: 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani JA, Chang X-B, Riordan JR, Hanrahan JW (1991) Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature 352: 628–631 [DOI] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC, Gadsby DC (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433: 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, He Z, O'Shaughnessy TJ, Rux J, Reenstra WW (2002) Domain–domain associations in cystic fibrosis transmembrane conductance regulator. Am J Physiol Cell Physiol 282: C1170–C1180 [DOI] [PubMed] [Google Scholar]
- Winter MC, Welsh MJ (1997) Stimulation of CFTR activity by its phosphorylated R domain. Nature 389: 294–296 [DOI] [PubMed] [Google Scholar]
- Xie J, Adams LM, Zhao J, Gerken TA, Davis PB, Ma J (2002) A short segment of the R domain of CFTR contains channel stimulatory and inhibitory activities that are separable by sequence modification. J Biol Chem 277: 23019–23027 [DOI] [PubMed] [Google Scholar]
- Xie J, Zhao J, Davis PB, Ma J (2000) Conformation, independent of charge, in the R domain affects cystic fibrosis transmembrane conductance regulator channel openings. Biophys J 78: 1293–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JS, Moyer BD, Bannykh S, Yoo HM, Riordan JR, Balch WE (2002) Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J Biol Chem 277: 11401–11409 [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW (1990) Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science 250: 568–571 [DOI] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R (2001) Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 411: 657–661 [DOI] [PubMed] [Google Scholar]
- Zhu T, Dahan D, Evaglelidis A, Zheng S-X, Luo J, Hanrahan JW (1999) Association of cystic fibrosis transmembrane conductance regulator and protein phosphatase 2C. J Biol Chem 274: 29102–29107 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S2
Supplementary Materials 3
Supplementary Materials 2
Supplementary Materials 1