

Abstract
The 1918 “Spanish flu” was the fastest spreading and most deadly influenza pandemic in recorded history. Hypotheses of its origin have been based on a limited collection of case and outbreak reports from before its recognized European emergence in the summer of 1918. These anecdotal accounts, however, remain insufficient for determining the early diffusion and impact of the pandemic virus. Using routinely collected monthly age-stratified mortality data, we show that an unmistakable shift in the age distribution of epidemic deaths occurred during the 1917/1918 influenza season in New York City. The timing, magnitude, and age distribution of this mortality shift provide strong evidence that an early wave of the pandemic virus was present in New York City during February-April 1918.
Keywords: age-specific mortality, epidemic, herald wave, Spanish flu
The origin of the 1918 influenza pandemic remains elusive. The causes for its transmissibility, virulence, and unique age pattern remain inadequately understood. In less than 2 years, the pandemic killed >675,000 people in the United States (1) and 40-100 million worldwide (2, 3), with the majority of deaths occurring among those <45 years old (4). Analysis of viral RNA recovered from preserved lung-tissue samples from geographically distant regions confirms that the pandemic wave in the northern hemisphere in the autumn of 1918 and its recrudescence in winter 1919 were caused by an A/H1N1 influenza virus with a highly conserved hemagglutinin gene identified in two antigenic configurations (5-7). Whether the epidemic recognized as “Spanish flu” in Europe in the summer of 1918 (4) was caused by a similarly conserved virus is unknown, and whether anecdotal cases and outbreaks reported from Europe in 1916 and 1917 (8) and the United States in early 1918 (9-11) were caused by a virus related to the pandemic virus remains speculative.
At the peak of the catastrophic wave in the autumn of 1918, an influenza-like epidemic the preceding spring was retrospectively described as an early wave of the pandemic (9). At the end of the 1918/1919 season, an epidemiological study suggested that this early precursor wave had caused epidemic mortality in Atlantic seaboard cities before central and western U.S. cities (10). A limited collection of eyewitness case and outbreak reports, however, led to the consensus view that the pandemic emerged in a mild wave in the spring of 1918 from an isolated rural region in the central United States (11). This view became widely accepted (12-15), without rigorous reevaluation of the original evidence.
Reanalysis of contemporary studies and routinely collected data may provide insight into the epidemiology of the 1918 pandemic (16-18). A characteristic feature of influenza epidemiology has historically been that the burden of excess mortality in interpandemic seasons occurs primarily in older age groups, whereas the burden in pandemic seasons shifts disproportionately to younger ages (19). During and following each of the three major 20th-century pandemics in which this pattern occurred, in subsequent epidemic seasons, the burden of mortality shifted proportionally back to older age groups (19, 20). In this study, we compare age-specific mortality patterns from influenza epidemics in New York City from 1911 to 1921. Although absolute confirmation of a precursor wave depends on recovery of viral RNA, we present here an epidemiological analysis showing that the pattern of age-specific epidemic excess mortality shifted strongly toward younger age groups in the winter preceding the pandemic wave in the autumn of 1918. This result provides evidence that a prepandemic herald wave of mortality occurred during February-April 1918 in New York City.
Materials and Methods
We obtained historical monthly mortality statistics from New York City Health Department records. All-cause and pneumonia and influenza (P&I) deaths from 1907 to 1921 were used in our all-ages analyses. All-cause deaths during 1911-1921, stratified into six age groups (<5, 5-14, 15-24, 25-44, 45-64, and ≥65), were used in our age-specific analyses. We standardized observed deaths by length of month and calculated rates by using New York City population estimates linearly interpolated from 1910 and 1920 U.S. census reports.
The model approach we used was originally developed for measuring the impact of influenza epidemics by estimating an expected baseline level to which observed epidemic period deaths were compared (21-23). Other studies have similarly estimated baseline mortality from weekly or monthly deaths, exclusive of epidemic periods, by using least-squares regression with sinusoidal terms. Although alternatives have been developed (24-26) and variations on this model have continued to be widely used (27-30), excess mortality remains an insensitive measure of severity, particularly in pediatric populations (1). We used the same monthly baseline equation with annual and 6-month-cycle terms for total P&I and for total and age-specific all-cause mortality analyses:
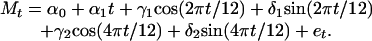 |
Our estimate of expected deaths at time t (Mt) was determined by the constant α0, secular trend α1, annual predictors γ1 and δ1, semiannual predictors γ2 and δ2, and an error term et, derived from linear regression of nonepidemic monthly mortality data. We purged months that the New York City Health Department reported increased respiratory disease activity or a major mortality event such as summer diarrheal epidemics, the 1915/1916 influenza epidemic, the 1916 polio epidemic, or the 1917 heat wave. For each available 5-year period of mortality data, we projected out the expectation and an arbitrarily defined 95% confidence limit and purged months exceeding this threshold. Each 5-year series and annual forecast was for August through July.
To focus on the most severe seasons, we arbitrarily defined an influenza season epidemic as two or more consecutive months when observed P&I mortality exceeded the upper 95% confidence limit of the baseline. Excess deaths were calculated for each epidemic season as the difference between expected and observed mortality for the consecutive epidemic months. Total and age-specific estimates of excess deaths were determined for the same months. Seasonal excess deaths from all-cause and P&I data for the entire population were compared, along with all-cause <45 and ≥45 excess death estimates. Due to the characteristic 1918 pattern of mortality and the age groupings available, we calculated the risk ratio (20) of <45 to ≥45 excess death rates to provide a single measure of relative risk of influenza-related death by age during the influenza periods identified by the model.
We obtained annual P&I mortality rates by age group for 1915-1917 to look for the typical “U-shaped” mortality curve (increased rates at the extremes of age) and for 1918 to look for the “W-shaped” curve characteristic of the pandemic (highest annual mortality rates among young children, young adults, and the elderly). To provide an alternative measure of the age-specific impact on interpandemic and pandemic influenza-related deaths, excess mortality rates were plotted by age and compared for the severe 1915/1916 influenza season, the March and April 1918 epidemic months, and the 1918/1919 pandemic season. As done recently to characterize mortality age patterns for the 1968 pandemic (31), we calculated the age-specific ratio of pandemic to interpandemic excess rates to estimate age-specific relative risk by using 45 years of age as the cutoff.
Results
Excess Mortality. Severe epidemic mortality occurred in 5 of the 10 influenza seasons during 1911-1921. The 1911/1912 influenza season was reported by the health department as distinctly free of epidemic influenza, and the 1915/1916 influenza epidemic was noted at the time as the worst to date that century (4). During the 1915-1920 epidemic seasons, the temporal pattern and peaks in all-cause and P&I deaths coincided (Fig. 1 A and B). Epidemic period estimates for all ages in the 1915/1916 and 1916/1917 seasons were 2,100 and 2,800 excess all-cause deaths, respectively (Table 1). Excess all-cause deaths in the 1918/1919 pandemic and 1919/1920 recrudescence were 29,200 and 8,200, respectively (Table 1). An estimated 4,600 excess all-cause deaths occurred during the 1917/1918 influenza season (Table 1); epidemic mortality in this season occurred in two peaks, in January and March 1918 (Fig. 1 A and B, arrowheads).
Fig. 1.

All-cause and P&I monthly mortality rates for all ages (A and B) and all-cause mortality rates by age group (C-H) are calculated per 10,000 population. Observed rates are days-per-month adjusted. Expected model baselines (solid lines) are derived from each series of nonepidemic months. Epidemic thresholds (dashed lines) are the upper 95% confidence limit above each baseline. The major epidemic influenza season months are indicated (shaded). Two 1917/1918 influenza season peaks (arrowheads) show excess mortality primarily confined to the ≥65-years age group in January (C), and to the groups <45 years old in March 1918 (E-H). Other severe mortality events are evident: summer diarrheal disease epidemics were confined to young children (H), the 1916 polio epidemic to all children (G and H), and the summer 1917 heat wave and diarrheal disease epidemic among the youngest and oldest age groups (C and H).
Table 1. Influenza season excess deaths.
| Number of excess deaths (rate per 10,000 population)†
|
Risk ratio‡ <45:≥45
|
||||
|---|---|---|---|---|---|
| Epidemic period* | AC | P&I | AC<45 | AC≥45 | |
| December 1915-February 1916 | 2,100 (4.0) | 1,600 (3.0) | 600 (1.3) | 1,400 (14.9) | 0.1 |
| December 1916-February 1917 | 2,800 (5.3) | 1,600 (3.0) | 800 (1.8) | 2,100 (21.2) | 0.1 |
| December 1917 and January 1918 | 1,500 (2.8) | 600 (1.1) | 1,000 (2.3) | 400 (3.9) | 0.6 |
| February 1918 | 400 (0.7) | 200 (0.5) | 400 (0.8) | 100 (0.6) | 1.3 |
| March and April 1918 | 2,700 (4.9) | 1,900 (3.5) | 2,500 (5.6) | 200 (2.1) | 2.6 |
| September 1918-April 1919 | 29,200 (53.0) | 28,600 (51.8) | 26,200 (58.7) | 2,100 (20.3) | 2.9 |
| January-March 1920 | 8,200 (14.6) | 6,000 (10.7) | 5,400 (11.8) | 1,800 (16.5) | 0.7 |
Epidemic periods are consecutive months when P&I mortality exceeded the arbitrarily defined threshold. The 1917/1918 influenza season is broken down by period to illustrate the changing age distribution.
Excess total all-cause (AC) and P&I mortality estimates are derived from the total population data. Excess AC<45 and AC≥45 mortality estimates are derived from the <45 and the ≥45 age groups. The difference between total all-cause and the sum of age-specificall-cause values was due to rounding and variation between the total and age-stratified analyses.
The risk ratio is derived from the AC<45 and AC≥45 excess mortality rates.
Age-Specific Analyses. The age pattern of mortality shifted profoundly during the 1917/1918 season. In the 1915/1916 and 1916/1917 influenza seasons and in January 1918, excess mortality incidence was greatest in those ≥65 years old (Fig. 1 C). During February-April 1918 and the 1918/1919 season, those ≥65 years old experienced little or no excess mortality (Fig. 1C), whereas those aged 15-24 and 25-44 years experienced sharply elevated death rates (Fig. 1 E and F). Mortality data for children <5 and 5-14 years old did not exhibit winter peaks before the 1917/1918 season (Fig. 1 G and H). Before 1918, children experienced mortality peaks from summer diarrheal disease epidemics, a severe polio epidemic in the summer of 1916 (Fig. 1 G and H), and a coincident diarrheal epidemic and heat wave in the summer of 1917 (Fig. 1H).
Because of the striking contrast in mortality impact by age during the 1918/1919 pandemic season, we calculated the ratio of <45 to ≥45 years epidemic period excess death rates (Table 1). The ratio of age-specific epidemic deaths shifted abruptly in February 1918 and reached values >20-fold higher in March and April 1918 and in the 1918/1919 season than in the 1915/1916 and 1916/1917 epidemic seasons (Table 1). The age-specific ratio in the 1919/1920 season was closer to prepandemic levels (Table 1); however, the total burden of excess deaths among people <45 years old remained extremely high, compared with the interpandemic period (Fig. 1 E-H).
The W-Shaped Curve. Average annual P&I death rates by age for 1915-1917 showed the classic U-shaped mortality pattern characteristic of influenza epidemic years (Fig. 2A). For calendar year 1918, the age-specific mortality pattern showed the classic W-shaped distribution of the pandemic (Fig. 2 A). In contrast, when we plotted seasonal age-specific excess mortality rates (specifically representing influenza-related mortality), a strikingly different pattern emerged. For the 1915/1916 interpandemic season, the U pattern is flattened on one end because children <5 years had little excess epidemic-period impact, whereas the U pattern was elevated on the other, because the greatest increase in epidemic period mortality was among the elderly (Fig. 2B). For the 1918/1919 pandemic season, mortality rates in young children and young adults were sharply higher than for the 1915/1916 epidemic, whereas mortality rates were lowest among the oldest age group, presenting an age pattern resembling an attenuated W shape (Fig. 2B).
Fig. 2.

Annual and epidemic period mortality rates by age group. (A) Average age-specific calendar year P&I death rates are plotted in a U-shaped age distribution for 1915-1917 (○), and in the characteristic W-shaped distribution for the pandemic year 1918 (▪). (B) Influenza-season-attributable excess deaths are plotted for the 1915/1916 epidemic influenza season (○), the epidemic months March and April 1918 (▴), and the pandemic season from September 1918 through April 1919 (▪). (C) The excess rates are plotted on a log10 scale. (D) Relative risk of death is plotted by age group on a log10 scale for the March and April 1918 epidemic period (▴) and the pandemic from September 1918 through April 1919 (▪) relative to the severe 1915/1916 epidemic season.
The age pattern of excess epidemic death rates in early 1918, when the risk ratio shifted toward younger ages (Table 1), is not immediately apparent as plotted (Fig. 2B). However, on a logarithmic scale, the relative pattern appears strikingly similar to the 1918/1919 pandemic period, and the 1915/1916 epidemic pattern clearly was different from both the early 1918 epidemic and the 1918/1919 season (Fig. 2C). To further illustrate the age pattern as a measure of mortality risk, the ratio of the early 1918 epidemic and 1918/1919 pandemic periods relative to the 1915/1916 epidemic are plotted (relative risk, Fig. 2D); this graph underscores that the oldest age groups fared better during these periods than during a typical severe influenza epidemic season such as 1915/1916.
Discussion
In the absence of virological evidence of an early wave of the 1918 pandemic, we have alternatively pursued an epidemiological approach based on previously unexploited age-detailed mortality data from New York City, which at the time comprised >5% of the entire U.S. population. In this study, we have characterized the age-specific impact of influenza epidemics on mortality for 1911-1921 to identify and determine the timing of a “signature” age shift previously shown to distinguish pandemic from epidemic influenza periods (19). We have demonstrated that a distinct shift in the age distribution of excess deaths occurred during the 1917/1918 season in a pattern suggestive of a herald wave of pandemic mortality. A prevailing hypothesis of the 1918 pandemic's origin has been that it emerged in a mild wave in the spring of 1918 from the central United States (11-15); however, the original data supporting this argument have never been rigorously evaluated. The data we present for New York City suggest that prepandemic activity was occurring before spring 1918, and that the impact was far from mild. These findings are inconsistent with the prevailing hypothesis of a spring 1918 Kansas origin, and they reopen the possibility that the virus had spread from Europe to New York City in the context of troop movement during World War I.
The severe wave of mortality from February to April 1918 in New York City caused more than one-tenth the number of excess all-cause deaths as occurred during the subsequent pandemic season. The impact of the early wave on young adults was extreme, with a peak associated with a doubling of all-cause deaths among 15- to 24-year-olds (Fig. 1F), and the impact on the oldest age group was typical of nonepidemic period winter mortality (Fig. 1C). If the 1918/1919 pandemic season had never occurred, the age-specific impact of the early 1918 epidemic would remain an unprecedented event in the history of New York City municipal mortality statistics. With the exception of the 1918/1919 pandemic season, no reported influenza-like respiratory disease epidemic, other than the proposed herald wave that we report, has ever caused such an extreme increase in young adult deaths while causing little or no impact among older adults. The relative impact on the young and the lack of impact on the oldest age group present similar patterns for both the herald and pandemic waves, compared with surrounding epidemic seasons (Fig. 2D).
Based on calendar-year data, the age pattern of the 1918 pandemic has typically been described as W-shaped, with high mortality rates in three age groups: very young children, young adults, and the elderly (Fig. 2A). Our study, which attempts to provide an estimate of epidemic influenza-attributable deaths, specifically shows that during pandemic months, the oldest age group experienced little increase in mortality, thus attenuating the older end of the W curve (Fig. 2B). Other studies have similarly shown this lack of impact in older age groups, based on quarterly (32) and annual (33) data. And other studies have shown a similar pandemic shift in age-specific relative risk, in the case of the 1968 A/H3N2 pandemic (31), consistent with serological evidence of previous exposure to a similar antigenic subtype virus among those ≥77 years old (34). The relative sparing of the oldest age groups in our study is consistent with the hypothesis of protection due to the recycling of a similar virus subtype a half-century earlier (34) and also with the hypothesis that antibody-dependent enhancement led to extreme virulence, specifically in younger adults (35). We could not address these possibilities further with the data at hand, and we did not have the age detail to further characterize the age cutoff among the middle age groups that would separate those affected from those not affected by this influenza pandemic. Furthermore, we did not have the spatial and temporal detail necessary to further characterize precisely when and where the pandemic mortality herald wave began. A shift in the relative distribution of mortality toward younger ages occurred in each of the three major pandemics of the 20th century (19, 20). Whether and to what degree early evidence of the 1918 pandemic shift can be identified in other cities remains an important test of our hypothesis. Although our approach can detect only a significant mortality impact and would miss any sporadic pandemic activity, the magnitude and age specificity of the early 1918 epidemic that we describe suggest that such an early wave may be identifiable in much smaller regional data sets. However, the recovery of A/H1N1 influenza virus RNA from human tissue from this period remains the gold standard for identifying a precursor epidemic wave.
Since the original confirmation that an A/H1N1 influenza virus caused the 1918 pandemic (5), further phylogenetic characterization of the virus (6, 7, 36) and elucidation of its hemagglutinin structure (37, 38) have presented a complex and inconclusive evolutionary story of an avian-like mammalian-adapted virus, more like the H5N1 avian influenza viruses that have been isolated since 1997 than the pandemic reassortments of either 1957 or 1968 (39, 40). Our epidemiological approach cannot provide insight into the specific genetic characteristics of the virus that caused the observed early 1918 epidemic. However, the most parsimonious explanation is that the virus that caused the early wave was similar to that which caused the catastrophic wave that exploded globally in the autumn of 1918. We believe that the magnitude of the increase in young-adult deaths accompanied by the lack of impact among older age groups provides compelling evidence of an early phase or herald wave of the 1918 pandemic.
Further analysis of age-detailed historical mortality data from other regions and periods may continue to provide insight into the epidemiology of past influenza pandemics and may help to inform current preparedness planning efforts for future pandemics (41). The historical-epidemiological evidence of the precursor wave that we present, along with a recent study of transmissibility in the pandemic wave of autumn 1918 (42), suggest that if events similar to those preceding autumn 1918 were to occur again, strengthened surveillance and increased capacity for rapid public health intervention could possibly prove critical in limiting the emergence and impact of the next pandemic. The speed with which human-to-human transmissibility of a highly pathogenic mammalian adapted avian influenza virus develops cannot currently be predicted. Nevertheless, the historical precedent of 20th-century influenza pandemics suggests that theoretically controllable transmissibility and a herald wave may occur before the onset of a catastrophic pandemic wave, leaving a critical window of opportunity for production and distribution of pandemic vaccines and antivirals.
Acknowledgments
We thank D. S. Fedson, J. K. Taubenberger, and C. Viboud for their critical input and stimulating discussions during the development of this study, and R. J. Taylor for help in editing the manuscript. S.S.M. is supported by Centers for Disease Control and Prevention Cooperative Agreements A1013-21/21 and U90/CCU224241 and by the Arts and Letters and Achelis Foundations.
Author contributions: D.R.O., L.S., and S.S.M. designed research; D.R.O. performed research; and D.R.O., L.S., P.J.E., and S.S.M. analyzed data and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: P&I, pneumonia and influenza.
References
- 1.Glezen, W. P. (1996) Epidemiol. Rev. 18, 64-76. [DOI] [PubMed] [Google Scholar]
- 2.Johnson, N. P. A. S. & Müller, J. (2002) Bull. Hist. Med. 76, 105-115. [DOI] [PubMed] [Google Scholar]
- 3.Reid, A. H. & Taubenberger, J. K. (2003) J. Gen. Virol. 84, 2285-2292. [DOI] [PubMed] [Google Scholar]
- 4.Jordan, E. O. (1927) Epidemic Influenza: A Survey (Am. Med. Assoc., Chicago).
- 5.Taubenberger, J. K., Reid, A. H., Krafft, A. E., Bijwaard, K. E. & Fanning, T. G. (1997) Science 275, 1793-1796. [DOI] [PubMed] [Google Scholar]
- 6.Reid, A. H., Fanning, T. G., Hultin, J. V. & Taubenberger, J. K. (1999) Proc. Natl. Acad. Sci. USA 96, 1651-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reid, A. H., Janczewski, T. A., Lourens, R. M., Elliot, A. J., Daniels, R. S., Berry, C. L., Oxford, J. S. & Taubenberger, J. K. (2003) Emerg. Infect. Dis. 9, 1249-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oxford, J. S., Lambkin, R., Sefton, A., Daniels, R., Elliot, A., Brown, R. & Gill, D. (2005) Vaccine 23, 940-945. [DOI] [PubMed] [Google Scholar]
- 9.Soper, G. A. (1918) Science 48, 451-456. [DOI] [PubMed] [Google Scholar]
- 10.Frost, W. H. (1919) J. Am. Med. Assoc. 73, 313-318. [Google Scholar]
- 11.Vaughn, W. T. (1921) Influenza: An Epidemiological Study (Am. J. Hyg., Baltimore), Monograph 1.
- 12.Crosby, A. W. (1989) America's Forgotten Pandemic: The Influenza of 1918 (Cambridge Univ. Press, New York).
- 13.Patterson, K. D. & Pyle, G. F. (1991) Bull. Hist. Med. 65, 4-21. [PubMed] [Google Scholar]
- 14.Phillips, H. & Killingray, D. (2003) in The Spanish Influenza Pandemic of 1918-1919: New Perspectives, eds. Phillips, H. & Killingray, D. (Routledge, New York), pp. 1-25.
- 15.Barry, J. M. (2004) The Great Influenza: The Epic Story of the Deadliest Plague in History (Viking, New York).
- 16.Kilbourne, E. D. (2003) in The Spanish Influenza Pandemic of 1918-1919: New Perspectives, eds. Phillips, H. & Killingray, D. (Routledge, New York), pp. 29-38.
- 17.Schoenbaum, S. C. (2003) in The Spanish Influenza Pandemic of 1918-1919: New Perspectives, eds. Phillips, H. & Killingray, D. (Routledge, New York), pp. 241-251.
- 18.Knobler, S. L., Mack, A., Mahmoud, A. & Lemon, S. M., eds. (2005) The Threat of Pandemic Influenza: Are We Ready? A Workshop Summary (Natl. Acad. Press, Washington, DC). [PubMed]
- 19.Simonsen, L., Clarke, M. J., Schonberger, L. B., Arden, N. H., Cox, N. J. & Fukuda, K. (1998) J. Infect. Dis. 178, 53-60. [DOI] [PubMed] [Google Scholar]
- 20.Dauer, C. C. & Serfling, R. E. (1961) Am. Rev. Resp. Dis. 83, 15-28. [Google Scholar]
- 21.Eickhoff, T. C., Sherman, I. L. & Serfling, R. E. (1961) J. Am. Med. Assoc. 176, 776-782. [DOI] [PubMed] [Google Scholar]
- 22.Serfling R. E. (1963) Public Health Rep. 78, 494-506. [PMC free article] [PubMed] [Google Scholar]
- 23.Serfling, R. E., Sherman, I. L. & Houseworth, W. J. (1967) Am. J. Epidemiol. 86, 433-441. [DOI] [PubMed] [Google Scholar]
- 24.Choi, K. & Thacker, S. B. (1981) Am. J. Epidemiol. 113, 215-226. [DOI] [PubMed] [Google Scholar]
- 25.Thompson, W. W., Shay, D. K., Weintraub, E., Brammer, L., Cox, N., Anderson, L. J. & Fukuda, K. (2003) J. Am. Med. Assoc. 289, 179-186. [DOI] [PubMed] [Google Scholar]
- 26.Reichert, T. A., Simonsen, L., Sharma, A., Pardo, S.A., Fedson, D.S. & Miller, M.A. (2004) Am. J. Epidemiol. 160, 492-502. [DOI] [PubMed] [Google Scholar]
- 27.Housworth, J. & Langmuir, A. D. (1974) Am. J. Epidemiol. 100, 40-48. [DOI] [PubMed] [Google Scholar]
- 28.Lui, K. J. & Kendal, A. P. (1987) Am. J. Pub. Health 77, 712-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simonsen, L., Clarke, M. J., Williamson, G. D., Stroup, D. F., Arden, N. H. & Schonberger, L. B. (1997) Am. J. Pub. Health 87, 1944-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viboud, C., Boelle, P. Y., Pakdaman, K., Carrat, F., Valleron, A. J. & Flahault, A. (2004) Emerg. Infect. Dis. 10, 32-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simonsen, L., Reichert, T. A. & Miller, M. (2004) in Options for the Control of Influenza V, International Congress Series 1263, ed. Kawaoka, Y. (Elsevier, Okinawa, Japan), Vol. ICS 1263, pp. 791-794. [Google Scholar]
- 32.Nguyen-Van-Tam, J. S. & Hampson, A. W. (2003) Vaccine 21, 1762-1768. [DOI] [PubMed] [Google Scholar]
- 33.Luk, J., Gross, P. & Thompson, W. W. (2001) Clin. Infect. Dis. 33, 1375-1378. [DOI] [PubMed] [Google Scholar]
- 34.Schoenbaum, S. C., Coleman, M. T., Dowdle, W. R. & Mostow, S. R. (1976) Am. J. Epidemiol. 103, 166-173. [DOI] [PubMed] [Google Scholar]
- 35.Morens, D. M. (1994) Clin. Infect. Dis. 19, 500-512. [DOI] [PubMed] [Google Scholar]
- 36.Reid, A. H., Fanning, T. G., Janczewski, T. A., Lourens, R. M. & Taubenberger, J. K. (2004) J. Virol. 78, 12462-12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamblin, S. J., Haire, L. F., Russell, R. J., Stevens, D. J., Xiao, B., Ha, Y., Vasisht, N., Steinhauer, D. A., Daniels, R. S., Elliot, A., et al. (2004) Science 303, 1838-1842. [DOI] [PubMed] [Google Scholar]
- 38.Stevens, J., Corper, A. L., Basler, C. F., Taubenberger, J. K., Palese, P. & Wilson, I. A. (2004) Science 303, 1866-1870. [DOI] [PubMed] [Google Scholar]
- 39.Webster, R. G. (2001) Science 293, 1773-1775. [DOI] [PubMed] [Google Scholar]
- 40.Reid, A. H., Taubenberger, J. K. & Fanning, T. G. (2004) Nat. Rev. Microbiol. 2, 909-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simonsen, L., Olson, D. R., Viboud, C. Heiman, E., Taylor, R. J., Miller, M. A. & Reichert, T. A. (2005) in The Threat of Pandemic Influenza: Are We Ready? A Workshop Summary, eds. Knobler, S. L., Mack, A., Mahmoud, A. & Lemon, S. M. (Natl. Acad. Press, Washington, DC), pp. 89-114.
- 42.Mills, C. E., Robins, J. M. & Lipsitch, M. (2004) Nature 432, 904-906. [DOI] [PMC free article] [PubMed] [Google Scholar]