

Abstract
We have disabled TNF receptor (TNFR) function by inducing allosteric modulation of tryptophan-107 (W107) in the receptor. The allosteric effect operates by means of an allosteric cavity found a short distance from a previously identified loop involved in ligand binding. Occupying this cavity by small molecules leads to perturbation of distal W107 and disables functions of the TNFR, a molecule not known to undergo conformational change upon binding TNF-α. TNF-α-induced NF-κB and p38 kinase activities and clinical symptoms of collagen-induced arthritis in mice were all diminished. Thus, disabling receptor function by induced conformational changes of active binding surfaces represents an innovative paradigm in structure-based drug design.
Keywords: allosteric, inhibitor, drug design, structure, arthritis
Conformational flexibility is often essential for a protein to function. Structural changes in proteins can be induced by various physical factors, including pH, solvents, ligand binding, and oligomerization. Conformational changes can occur at a defined local site or, as observed in multimeric proteins, at a distance from the ligand-binding site (allosterism) (1-3). The basis and functional importance of propagation of structural allosteric changes are poorly understood.
To our knowledge, altering a protein's function by perturbing a defined conformational state (i.e., not known to undergo conformational change on activation) using rational structure-based design has not been achieved. Although surface cavities on nonenzymatic classes of proteins have been largely unexplored, inactivation of enzymes has been accomplished by designing competitive or substrate analog inhibitors that bind at active sites and noncompetitive inhibitors identified generally by using high-throughput screening. For example, extensive studies have been reported on exosites (allosteric sites) of the thrombin-hirudin complex (4, 5). Also, allosteric inhibitors of G protein-coupled receptors (6) and muscarinic (7) and adenosine receptors (8) have been found. Bodian et al. (9) have used the knowledge that fusion-induced conformational change of influenza hemagglutinin is required for virus entry and by a structure-based approach developed an “inhibitor” of the conformational change by targeting the trimeric interface of the target protein.
Elements responsible for regulating conformational movements in proteins are not fully understood. Little information exists concerning the role of surface cavities/clefts in proteins that are not part of an active site or binding site or located at the protein-protein interface. The cavities' relationship to inherent flexibility of secondary structural elements in proteins has not been explored. We believe that cavities (internal and external) near critical secondary elements may represent a significant feature related to protein flexibility and function.
Based on these notions, we hypothesized that cavities and clefts on the surface of proteins distal to regulatory sites, such as ligand-binding sites or catalytic sites, that were hitherto unknown to have any discrete function might be used to modulate the function of proteins/receptors by inducing conformational changes from their native state. Induced allosterism would occur as a consequence of lodging small molecules into the cavities. The mode of inhibition is expected to share some features with that of allosteric inhibitors, and hence the small molecules are referred to as “pseudoallosteric” inhibitors, the cavity is termed a “pseudoallosteric cavity,” and the method is described as “cavity-induced allosteric modification” (”CIAM”). However, in our studies, the pseudoallosteric sites are not known to be natural allosteric sites.
We have tried to define the basic principles by which predictable conformational perturbation can be induced by selected compounds. Our efforts have used well characterized receptor molecules, which bind ligand by a structurally resolved mechanism but are not known to have any allosteric site or allosterically mediated function or undergo any conformational change on ligand binding. The studies have focused on a class of previously unappreciated surface cavities on the receptor ectodomain that are at defined distances from the ligand-binding sites.
The TNF receptor (TNFR) is one of the central mediators of inflammation (10). The three-dimensional structure of the TNFR1 complex has been determined with and without its ligand (11-14). Active TNFR1 and TNF-α form a trimeric complex. The crystal structure analysis of the TNFR complex with and without ligands did not reveal any changes consistent with ligand-induced fit (13). Hence, the structural role of the ligand was postulated to bring the receptor together and facilitate receptor activation. A flexible hinge (G81 and G97) was also identified from the crystal structure analysis (13), and it was predicted that this flexible hinge might provide ligand-induced conformational changes. But, contrary to that expectation, no significant conformational changes were observed in the crystallographic complex (13). Here, we describe the activity of a set of small molecules designed to bind to a discrete surface cavity that disable ligand-induced TNFR functions as a consequence of the defined conformational perturbation of tryptophan-107 (W107) on the receptor. The conformational perturbation approach identifies surface sites that are relevant for TNFR's biological activity in vitro and in vivo.
Experimental Protocol
Materials and Cell Cultures. All chemicals were obtained from Sigma. L929, THP1, and NE91 cells were all from the American Type Culture Collection. L929 cells were cultured in RPMI medium 1640 (Invitrogen) containing 5% FBS. THP1 cells were cultured in RPMI medium 1640 with the supplement of 50 mM Hepes/1 mM Na pyruvate/50 μM 2-mercaptoethanol/2.5 mg/ml glucose/50 μg/ml gentamicin/10% FBS. The cells were cultured in a 12-well plate at a density of 6 × 105 cells per well with a supplement of 100 ng/ml phorbol 12-myristate 13-acetate. The cells were plated for 72 h to differentiate. NE91 cells were cultured in RPMI medium 1640 containing 10% FBS.
WT and Mutant Human TNFR1 Cloning, Expression, and Purification. The ectodomain of WT TNFR was obtained by PCR from pKP13 (15) (a gift from B. Beutler, UT Southwestern Medical Center, Dallas) with 5′ primer AAA AAA CAT ATG TAC CCC TCA GGG GTT ATT GG and 3′ primer CCG CTC GAG TCA ATG ATG ATG ATGATG ATG TGT GGT GCC TGA GTC CTC AG and constructed into PET21 (Novagen) between NdeI and XhoT, verified by sequencing. Mutant TNFR1 was obtained from site-directed mutagenesis by using the QuikChange mutagenesis kit (Stratagene). The plasmid was then transformed into Origami (DE3) (Novagen). The WT and mutant TNFR1 were all expressed in the inclusion bodies of the cells and were extracted and refolded as described in ref. 16. The refolded protein solution was centrifuged at 14,000 × g for 30 min at 4°C to remove the aggregation. The supernatant was mixed with Talon metal affinity resin (QIAexpressonst, Qiagen, Valencia, CA), rocked for 2 h at 4°C, and then washed three times with 50 mM NaH2PO4 containing 300 mM NaCl and 20 mM imidazole. Finally, the purified TNFR were eluted with 50 mM NaH2PO4 containing 300 mM NaCl and 150 mM imidazole (pH 8.0).
Design of TNFR Binding Pseudoallosteric Ligand. A stepwise procedure to identify pseudoallosteric sites and cavities used to induce allosteric modification (a procedure termed CIAM) has been developed (see Supporting Text, which is published as supporting information on the PNAS web site).
Effect on TNF-α-Induced Cell Cytolysis. The murine fibroblast cell line L929 was maintained in RPMI medium 1640 supplemented with 5% FBS. L929 cells were seeded onto 96-well plates at a density of 3 × 104 cells per well and incubated for 20 h at 37°C under 5% CO2. The cells were then preincubated with designed small molecules at different concentrations as indicated for 2 h, TNF-α was added into the well at a final concentration of 100 pg/ml, and actinomycin D was added at 2 μg/ml. After 16 h, the cells were stained with crystal violet blue and lysed with 100 μl of extraction buffer (1% SDS). The optical density was then measured at 570 nm.
Effect on Regulation of Downstream Molecules of TNF-α Signaling. To evaluate the effect of the designed small molecules on the regulation of downstream molecules of TNF-α signaling, L929 cells (1 × 106 per well) were cultured in six-well plates for 12 h, treated with or without small molecules for 2 h, and then stimulated with TNF-α at 20 ng/ml for the indicated periods. Cells were then washed with ice-cold PBS and lysed with lysis buffer. Cell lysates (15-30 μg) were separated by 12% SDS/PAGE, electroblotted onto nitrocellulose membrane (Osmonics, Westborough, MA), and probed with anti-phospho-IκBα, anti-IκBα, anti-phospho-p38, anti-p38, and anti-β-actin antibodies (Cell Signaling Technology, Beverly, MA).
The membranes were then developed by using the enhanced chemiluminescence (ECL) system (Amersham Pharmacia Biosciences). For THP1 cells, differentiated THP1 cells (as described under the heading “Materials and Cell Cultures”) at day 3 were pretreated with or without small molecules for 2 h and then treated with TNF-α at 20 ng/ml at the indicated periods. Cells were then lysed the same as L929 cells and examined the same way by Western blotting. To verify whether the effect on TNF-α downstream signaling is specific for TNF-α signaling, NE91 cells were cultured in a six-well plate at a density of 1 × 106 per well for 12 h. After 2 h of treatment with or without designed small molecules, the cells were stimulated with EGF at 50 ng/ml for the indicated time and the cells were lysed and probed with the same antibodies as the TNF signaling.
Fluorescence Quenching Studies. Quenching experiments with acrylamide were performed in stirred cells at 25°C, titrating from a stock of 1 M acrylamide by adding 2.5 μl of acrylamide each time. Recombinant TNFR1 were at a concentration of 5 μM in 1% DMSO. Tryptophan emission, monitored at 340 nm, was observed by using 295-nm excitation. Intensity data after quencher additions were averaged over a 10-s collection and were corrected for background emission (paired control lacking of protein). Intensities, F, at given quencher concentration, [Q], were analyzed by using the Stern-Volmer equation,
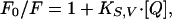 |
where F0 is the emission intensity of the protein in the absence of quencher and KSV is the Stern-Volmer constant for quenching, given by the slope when data are plotted as F0/F versus [Q].
For synthesized small molecules, F002 at 20 μM were preincubated with 5 μM TNFR1 in 1% DMSO for 30min and titrated with 1 M acrylamide the same way as TNFR1 alone. The F0/F was analyzed by using the Stern-Volmer equation, and two slopes from TNFR and from TNFR with F002 were compared.
Kinetic Binding Studies by Surface Plasmon Resonance. Recombinant TNFR WT and mutant were immobilized to the CM5 sensot chip with a surface density of 2,000 resonance units. The binding affinity of TNF-α to TNFR1 was estimated by using the Biacore 3000 instrument at 25°C. The apparent rate constants (kon and koff) and the equilibrium dissociation constant (Kd) for TNF/TNFR binding interaction were estimated from the kinetic analysis of sensorgrams by using biaevaluation 3.0 software (Biacore).
Isothermal Titration Calorimetry (ITC). The binding thermodynamics of inhibitors to the TNFR was measured by ITC using a high-precision VP-ITC titration calorimetric system (Microcal LLC, Northampton, MA). The calorimetric cell containing WT or mutant TNFR at a concentration of ≈1-6 μM dissolved in 5 mM Tris (pH 8.0) with 2% DMSO was titrated with the inhibitors dissolved in the same buffer. The concentration of inhibitor was 50-120 μM, depending on the solubility in buffer. Injection volumes were 10 μl. All solutions were properly degassed to avoid any formation of bubbles in the calorimeter during stirring. The heat evolved upon each injection of inhibitor was obtained from the integral of the calorimetric signal. The heat associated with the binding of the inhibitor to TNFR was obtained by subtracting the heat of dilution from the heat of reaction. The measurements were made at 25°C. Data were analyzed and fitted by using the data analysis software supplied by Microcal (origin 5.0).
Collagen-Induced Arthritis (CIA). Six- to 8-week-old male DBA/1 mice were purchased from The Jackson Laboratory and housed in the University of Pennsylvania Animal Care Facilities. Animals were maintained in accordance with guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. For CIA induction, mice were immunized by multiple intradermal injections of 100 μg of chicken type II collagen (Sigma) in 100 μl of 0.1 M acetic acid emulsified in an equal volume of complete Freund's adjuvant. Mice were challenged with the same antigen preparation i.p. on the 21st day. Mice were injected daily with vehicle or F002 at different dosages (2 and 4 mg/kg of body weight) from day 21. Mice were examined physically every other day in a blinded manner. Their paws were scored individually as follows: 0, normal; 1, erythema and mild swelling confined to the ankle joint or toes; 2, erythema and mild swelling extending from the ankle to the midfoot or ankle joint; 3, erythema and moderate swelling extending from the ankle to the metatarsal joints; 4, erythema and severe swelling encompassing the ankle, foot, and digits. The maximum disease score per foot is 4, and the maximum disease score per mouse is 16. For histological examination of the joint, mice were killed at different time points, and their paws were collected and fixed in 10% formalin. The paws were then decalcified in hydrochloric acid, embedded in paraffin, sectioned, and stained with hematoxylin/eosin.
Results
Identification of a Pseudoallosteric Cavities in TNFR. Earlier, in TNFR1 we identified three critical contact sites (WP5, WP8, and WP9) on the receptor needed for stable ligand complex formation and found that WP9 (105-113) is the most critical loop for this receptor's functional interaction with TNF-α (17). We hypothesized that if the configuration or disposition of the WP9 loop could be perturbed, then the receptor's structure would be such that it might lead to a defective complex formation.
A site to perturb the WP9 loop was located by using an internally developed a procedure called CIAM (cavity-induced allosteric modification) (see Supporting Text). Using this algorithm, we identified a pseudoallosteric cavity distal to the WP9 loop. The algorithm exploits several structural criteria that define flexibility of the receptor, including thermal parameters and the presence of internal cavities. To identify small molecule ligands that can be used to induce conformational perturbation at WP9, standard virtual screening procedures were used. Identified molecules were then further selected and tested. Subsequently, active molecules were further developed through synthetic modifications and evaluated based on their ability to induce conformational changes, using molecular simulation studies as a guide. These second-generation molecules were used in the studies described below. The target pseudoallosteric cavity in TNFR1 is shown in Fig. 1.
Fig. 1.

Molecular model of the TNFR-F002 complex. A molecular model of TNFR1 (Brookhaven Protein Data Bank ID code 1TNR) and the pseudoallosteric cavity is shown. TNFR1 is shown in a ribbon representation, and the allosteric cavity is shown as a dot surface model rendered with insightii. The small molecule, F002, is shown in a Corey-Pauling-Koltun (CPK) model to highlight the volume of the pseudoallosteric cavity. The functionally critical residue W107 (in the CPK model) is located in the loop WP9 (yellow), which represents the target of conformational perturbation by F002.
Binding Studies of Pseudoallosteric Inhibitors of TNFR. We evaluated whether the second-generation compound F002 (Fulcrum Pharmaceuticals, Las Vegas, NV) was able to bind to the isolated and purified TNFR. First, we used surface plasmon resonance to evaluate the kinetic binding of F002 to immobilized soluble TNFR. Because of the inadequate sensitivity of this approach, the binding results could not be interpreted unambiguously. ITC was used to deduce the binding characteristics, and the results are shown in Fig. 2. The compound F002 bound selectively to TNFR1 at one site with an affinity of 0.45 μM. Binding was characterized by small positive (unfavorable) enthalpy changes (ΔH = 2.1 kcal/mol), indicating that the interaction was entropically driven, as expected from a binding reaction dominated by hydrophobic interactions. No binding could be observed for F001, a control compound with similar mass and hydrophobicity to F002 but designed to be unable to interact with the cavity.
Fig. 2.

Binding of F002 to TNFR1 by ITC. Direct binding of F002 to the soluble TNFR1 was measured by using ITC. Isothermal titration microcalorimetry of F002 (55 μM) binding to recombinant WT (A) and mutant (B) human TNFR1 at 25°C in 50 mM Tris·HCl buffer containing 2% DMSO is shown. F002 exhibited binding affinity of 0.45 μM and positive enthalpy change of 2.1 kcal/mol to WT human TNFR1; no binding affinity to mutant TNFR1 was exhibited.
Next, we determined that F002 actually binds to the pseudoallosteric cavity, using a genetic approach. To address this question, we created mutants of the TNFR1 receptor in which the residues 82Q and 112F in the putative cavity were mutated to 82E and 112E, respectively. Hydrophilic isosteric changes were introduced because we considered that charge introduction might limit the entropic binding. We expected these changes to limit F002 access to the cavity and to induce a change in disposition of the WP9 loop. The structural integrity of the mutant receptor was verified by ligand binding in surface plasmon resonance studies. TNF-α bound to the WT TNFR1 (Kd = 3.79 × 10-10 M) and also to the recombinant mutant TNFR1 (Kd = 0.45 μM), suggesting that the mutations affected the ligand-binding sites to some extent as expected. Using ITC, we found that F002 no longer bound to the mutant receptor (Fig. 2B) at all. These studies confirm that F002 bound to a single and specific pseudoallosteric cavity as designed and that TNF-α bound to the WT and the mutant receptor to a lesser extent.
Conformational Perturbation of the WP9 Loop in TNFR. Subsequently, we evaluated whether the F002 binding involved conformational changes in the WP9 loop, which had been shown to be critical for TNF-α binding (17). Because there were no large detectable conformational changes on ligand binding (13), we expected that conformational alterations might be subtle, perhaps on the order of 2 Å. Small conformational changes can be observed by x-ray crystallography, but the technique requires high-resolution data. Alternatively, fluorescence quenching can identify small modulating changes in proteins (18). Fortunately, only one tryptophan residue exists in the TNFR1 ectodomain, located in the WP9 loop, and we examined whether the tryptophan (W107) in WP9 undergoes any change on F002 binding to the receptor. Fig. 3 shows the results from fluorescence quenching induced by acrylamide. The residue W107 in the WP9 loop fluoresces at ≈340 nm. In this set of experiments, the resultant concentration of quencher ranged up to 0.25 M, quenching 77.4% of the total intrinsic fluorescence of TNFR1. The Stern-Volmer constant for TNFR1 quenching by acrylamide calculated from the slope of the plot is 14.4 ± 0.2 M-1, compared with 11.6 ± 0.2 M-1 for TNFR1 plus F002, indicating that binding of FT002 to TNFR1 introduces conformational changes in the TNFR1, which partly protects W107 from the quencher. Some downward curvature in the Stern-Volmer plot observed in the presence of F002 can be attributed to the existence of two receptor populations, drug-bound and drug-free, with different degrees of tryptophan exposure to the solvent (19, 20). Thus, binding of F002 to the receptor changes the disposition of tryptophan-107 from partially exposed to more completely buried. We expected that this conformational change induced by F002 might lead to reduced affinity of ligand binding to the receptor and might also create a defective receptor-ligand complex disabled for signaling.
Fig. 3.

Binding of F002 to TNFR induced conformational change in the WP9 loop. Stern-Volmer plots for quenching of the intrinsic tryptophan fluorescence of TNFR1 by acrylamide are shown. Aliquots of acrylamide at 1 M were added to 5 μM TNFR1 in PBS containing 1% DMSO. Parallel experiments were carried out with 5 μM TNFR1 plus 20 μM F002 in PBS containing 1% DMSO. Florescence emission at 340 nm was recorded at 25°C after excitation at 295 nm. The resultant concentration of quencher is shown up to 0.25 M, quenching 77.4% of the total intrinsic fluorescence of TNFR1. The slope of TNFR1 is 14.4 ± 0.2 M-1; the slope of TNFR1 plus F002 is 11.6 ± 0.2 M-1.
In Vitro Effects of F002 on TNF-Mediated Signaling. Having established that F002 bound to the pseudoallosteric cavity and induced a conformational change, we next studied whether the F002-induced conformational perturbation was sufficient to alter TNFR1 function in vitro and in vivo. We examined changes in TNF-α-mediated cytotoxicity in murine L929 cells. In this assay, F002 rescued cells from TNF-α-induced cytotoxicity in a dose-dependent manner (Fig. 4A). Moreover, F002 binding inhibited TNF-α-mediated IκBα and p38 phosphorylation in both murine L929 and human monocyte cells, THP1 cells (Fig. 4 B and C), which are thought to be key intermediaries of TNF-α-induced inflammation in human cells.
Fig. 4.

F002 limits TNF-α-induced cytolysis of murine L929 cells and attenuates TNF-α-induced downstream signaling but does not affect EGF-induced signaling. (A) Inhibition of TNF-α-induced cytolysis. L929 cells were treated with human TNF-α and pseudoallosteric inhibitors at different concentrations. Absorbance obtained without human TNF-α and with 1 unit/ml human TNF-α was used as reference for 100% survival and 0% survival, respectively. F002 exhibited the inhibition dose-dependently; 10 μg/ml F002 (or 27.2 μM) shows ≈50% inhibition of cytolysis caused by 1 unit of human TNF-α. (B) F002 attenuates TNF-α-mediated proinflammatory signals. F002 inhibited TNF-α-induced phosphorylation of p38 in murine fibroblast L929 and in the human THP1 cells. The cells were pretreated with F002 at 20 μg/ml for 2 h and stimulated by human TNF-α at 20 ng/ml for the indicated time. F002 reduced the phosphorylation level of IκB and p38 at 5 min. Extracellular-regulated kinase 2 (Erk2) was analyzed as loading control. The relative phosphorylation intensity level of IκBα was 1.0, 2.2, 0.8, 0.3, 0.7, and 0.4, and the relative phosphorylation intensity level of P38 was 1, 4.5, 2.0, 1.3, 1.7, and 1.8, from left to right. The intensity was quantified by nih image software and normalized to the control Erk2 level. (C) Effect on the human promonocytic leukemia THP1. THP1 cells were pretreated with or without F002 for 1 h and stimulated by human TNF-α at 100 ng/ml for 1 h. The results showed that F002 inhibited TNF-α-induced phosphorylation of IκB and p38 but had no effect on LPS-induced activation. Lane 1, THP1 cells treated with vehicle; lane 2, LPS at 10 ng/ml; lane 3, TNF-α at 100 ng/ml; lane 4, F002 at 20 μg/ml; lane 5, LPS plus F002 at 20 μg/ml; lane 6, TNF-α plus F002 at 20 μg/ml. β-Actin was analyzed as loading control. The relative phosphorylation intensity of IκB normalized to the basal IκB levels is 1.0, 2.2, 1.2, 0.6, 1.4, and 0.5, and the phosphorylation intensity of p38 normalized to the basal p38 level is 1.0, 2.4, 2.2, 1.3, 2.3, and 0.9, from lane 1 to lane 6. The data were quantified by nih image software. (D) F002 showed no effect on EGF-induced p38 activation in NE91 cells. NE91 cells were cultured in DMEM (10%) and pretreated with or without F002 at 20 μg/ml for 2 h and stimulated by EGF at 50 ng/ml for 10 min. The cells were lysed and examined by Western blot. Lane 1, NE91 cells treated with vehicle; lane 2, EGF at 50 ng/ml; lane 3, EGF at 50 ng/ml plus F002 at 20 μg/ml. Erk2 was analyzed as loading control.
As a critical control, we asked whether F002 could affect EGF-stimulated cell signaling. EGF-mediated signals affect many of the same pathways, and we sought to exclude the possibility that the compound was acting through shared sites in the two signaling pathways. We did not observe any F002-induced changes in EGF signaling (Fig. 4D), supporting the notion that F002 specifically alters the TNFR1 signaling pathways by binding to the receptor ectodomain and inducing conformational changes. Alteration rather than complete abrogation of the receptor's function suggests that overall structural integrity of the trimeric complex is intact.
Effect of F002 on CIA in Mice. Finally, we examined whether the F002-induced conformational perturbation of W107 is important for in vivo biological activity. Activity of F002 was studied in the CIA system, which is a prototype animal model for human rheumatoid arthritis mediated by TNF-α. Six- to 8-week-old male DBA/1 mice were immunized by multiple intradermal injections of 100 μg of chicken type II collagen in 100 μl of 0.1 M acetic acid emulsified in an equal volume of complete Freund's adjuvant and were then challenged with the same antigen preparation i.p. on the 21st day. Animals were injected daily with F002 at different dosages (2-4 mg/kg of body weight per day) from day 21 on. In this model, disease typically develops 7-10 days after the second immunization, and the severity can be determined by both physical examination and joint histochemistry (21). Mice treated with F002 showed a dose-dependent decrease in the clinical symptoms of arthritis compared with the untreated or control group (Fig. 5). Histological analysis of ankles of the animals revealed that F002-treated mice have significantly reduced synovitis and mononuclear cell infiltration. Cartilage destruction was also prevented in the F002-treated group (Fig. 5B).
Fig. 5.

F002 suppresses CIA in DBA/1 mice. (A) Inhibition of CIA by F002. DBA/1 mice (n = 6) were immunized with type II collagen to induce arthritis. Twenty-eight days after immunization, mice were treated with vehicle and F002 at 50 μg/ml and 100 μg/ml and were scored every other day as described in Experimental Protocol. Data are expressed as mean ± SEM. *, P < 0.05; **, P < 0.01, by Student's t test as compared with vehicle-treated control group. (B) Joint histopathology at day 46. (a) The ankle joint from a normal DBA/1 mouse. (b) Severe inflammation and cartilage destruction occurred in a vehicle-treated CIA mouse. Arrow indicates severe synovitis and erosion of cartilage. (c) Animals treated with 100 μg/ml F002 show absence of synovitis in ankle joints. (d) High-power view of the inflamed synovium in b.
Discussion
Our previous efforts on targeted disabling of receptor ectodomains by monoclonal antibodies or mimetics established the paradigm of selectively inhibiting receptor function by creating misaligned receptors (22-25). Here, we have shown that targeted conformational perturbation using small molecules that lodge in surface cavities that reside distal to protein-protein interaction sites is an alternate way to modify receptor function.
Mutational, crystallographic, and computer simulation studies have suggested that a protein's activity can be affected by changes at the distal sites (4, 26, 27). In support of this hypothesis, myoglobin, a multimeric protein, undergoes structural modification and function due to physical changes in a distal cavity induced by a haem prosthetic group (28-31). Recently, tyrosine kinases and coagulation-relevant serine protease factors have been found to have exo sites (32, 33) similar to pseudoallosteric cavities defined in this study. Finally, White and colleagues (34) have found that it is possible to develop protein-interface binding molecules when the proteins are known to undergo conformational change on oligomerization. We have now demonstrated that in the absence of ligand-induced conformational changes, alteration of flexible regions by means of distal surface cavities is possible not only in protein enzymes, but also in oligomeric complex receptors such as TNFR.
The relationship between protein function and the extent of conformational change is unknown, and, more practically, the issue of whether inherent conformational plasticity can be exploited to alter protein function is untested. Several crystallographic analyses of protein complexes (35) reveal ligand-induced conformational changes in the range of 2 Å or less, which are often unappreciated due to the limited resolution of typical diffraction studies. Koshland and colleagues (36, 37) have recently suggested that small (<2 Å) induced conformational changes in the aspartate receptor complex may cause profound effects on receptor function. These types of notions suggest that a small conformational change might be sufficient to alter protein function. Our studies confirm these observations. Small changes in the conformation of W107 are sufficient to disable TNFR1 functions. Conformational changes clearly can be induced either by point mutations or by small molecules that are lodged near the WP9 loop. This study highlights the role of inherent flexibility at the WP9 loop and the critical nature of its disposition for TNFR function and suggests that small molecules can be used effectively to perturb conformational elements critical for protein function and thereby disable function.
The binding of F002 to allosteric cavities induces conformational changes, and this evident from small positive (unfavorable) enthalpy changes (ΔH = 2.1 kcal/mol), indicating that the interaction was entropically driven, as expected from a binding reaction dominated by hydrophobic interactions. Thus, binding of F002 to the receptor cavity might involve displacing water molecules. Earlier studies with the glucose binding yeast hexokinase implied that entropic binding correlated with conformational changes in the active site (38).
The F002 binding cavity is located away from the ligand interaction site. Thus, F002 may not prevent ligand binding to the TNFR, but F002 and the subsequent movement of the WP9 loop does affect ligand affinity for the receptor and limits effective ligand-mediated signaling. Our view is that the F002-bound receptor is in essence a defective complex that is less able to transduce signals in an efficient manner. We suggest that F002 is not a competitive inhibitor. It preferentially binds to the receptor before ligand binding and hinders the formation of a stable complex. F002 might also diffuse through the trimeric receptor complex and weaken ligand interactions with the receptor. This latter scenario is indeed possible, because the crystal structure of the complex reveals that W107 is partially exposed to solvents (13).
Small conformational changes mediate receptor function in vivo. It is not known whether small conformational changes in proteins have any biological effect in mice. Point mutations can change biological functions through small conformational rearrangements at the location. Now, our studies suggest that small molecules can also mimic point mutation in terms of conformational changes in the receptor and alter protein function. Results from the CIA model analyses reveal that small conformational perturbations can limit the progression of inflammatory responses in mice, suggesting that the receptor's conformational integrity is critical for biological function. These studies show that disabling receptor function by conformational perturbation is effective for inhibiting arthritis in mice.
In summary, these studies also indicate that certain but not all receptor functions may be altered either positively or negatively by discrete protein clefts and cavities located distant from the ligand-binding sites. Further understanding of the relationship between the clefts and cavities on the protein surface and inherent plasticity of secondary structural elements can be exploited, and thus conformation-induced inhibition of protein function can be considered an innovative component in structure-based rational drug design.
Supplementary Material
Acknowledgments
We thank Prof. Ray Ottenbrite and Dr. Ben Xiao (Fulcrum Pharmaceuticals) for providing F002 and Hongtao Zhang and Yuan Shen for critical reading and helpful suggestions. This work was supported in part by grants from the National Cancer Institute (to R.M. and M.I.G.) and the National Institutes of Health (to M.I.G.) and by the Abramson Family Cancer Research Institute (M.I.G.) and Fulcrum Pharmaceuticals (R.M.).
Author contributions: R.M. and M.I.G. designed research; R.M., X.C., A.B., X.D., A.S., and Y.H.C. performed research; R.M., E.F., X.X., and Y.H.C. analyzed data; and R.M. and M.I.G. wrote the paper.
Abbreviations: CIA, collagen-induced arthritis; ITC, isothermal titration calorimetry; TNFR, TNF receptor.
References
- 1.Stanfield, R. L. & Wilson, I. A. (1995) Curr. Opin. Struct. Biol. 5, 103-113. [DOI] [PubMed] [Google Scholar]
- 2.Roux, K. H., Strelets, L. & Michaelsen, T. E. (1997) J. Immunol. 159, 3372-3382. [PubMed] [Google Scholar]
- 3.Hayward, S. (1999) Proteins 36, 425-435. [PubMed] [Google Scholar]
- 4.Vijayalakshmi, J., Padmanabhan, K. P., Mann, K. G. & Tulinsky, A. (1994) Protein Sci. 3, 2254-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dennis, M. S., Eigenbrot, C., Skelton, N. J., Ultsch, M. H., Santell, L., Dwyer, M. A., O'Connell, M. P. & Lazarus, R. A. (2000) Nature 404, 465-470. [DOI] [PubMed] [Google Scholar]
- 6.Kenakin, T. (2004) Recept. Channels 10, 51-60. [DOI] [PubMed] [Google Scholar]
- 7.Ellis, J. (1997) Drug Dev. Res. 40, 193-204. [Google Scholar]
- 8.Kolliasbaker, C. A., Ruble, J., Jacobson, M., Harrison, J. K., Ozeck, M., Shryock, J. C. & Belardinelli, L. (1997) J. Pharmacol. Exp. Ther. 281, 761-768. [PubMed] [Google Scholar]
- 9.Bodian, D. L., Yamasaki, R. B., Buswell, R. L., Stearns, J. F., White, J. M. & Kuntz, I. D. (1993) Biochemistry 32, 2967-2978. [DOI] [PubMed] [Google Scholar]
- 10.Beutler, B. & Cerami, A. (1988) Annu. Rev. Biochem. 57, 505-518. [DOI] [PubMed] [Google Scholar]
- 11.Eck, M. J. & Sprang, S. R. (1989) J. Biol. Chem. 264, 17595-17605. [DOI] [PubMed] [Google Scholar]
- 12.Eck, M. J., Ultsch, M., Rinderknecht, E., de Vos, A. M. & Sprang, S. R. (1992) J. Biol. Chem. 267, 2119-2122. [PubMed] [Google Scholar]
- 13.Banner, D. W., D'Arcy, A., Janes, W., Gentz, R., Schoenfeld, H. J., Broger, C., Loetscher, H. & Lesslauer, W. (1993) Cell 73, 431-445. [DOI] [PubMed] [Google Scholar]
- 14.Naismith, J. H., Devine, T. Q., Brandhuber, B. J. & Sprang, S. R. (1995) J. Biol. Chem. 270, 13303-13307. [DOI] [PubMed] [Google Scholar]
- 15.Peppel, K., Crawford, D. & Beutler, B. (1991) J. Exp. Med. 174, 1483-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin, K. H. & Cheng, S. Y. (1991) BioTechniques 11, 748, 750, 752, 753. [PubMed] [Google Scholar]
- 17.Takasaki, W., Kajino, Y., Kajino, K., Murali, R. & Greene, M. I. (1997) Nat. Biotechnol. 15, 1266-1270. [DOI] [PubMed] [Google Scholar]
- 18.Strambini, G. B., Cioni, P. & Felicioli, R. A. (1987) Biochemistry 26, 4968-4975. [DOI] [PubMed] [Google Scholar]
- 19.Lehrer, S. S. (1971) Biochemistry 10, 3254-3263. [DOI] [PubMed] [Google Scholar]
- 20.Lakowicz, J. R. (1983) Principles of Fluorescent Spectrosopy (Plenum, New York).
- 21.Song, K., Chen, Y., Goke, R., Wilmen, A., Seidel, C., Goke, A. & Hilliard, B. (2000) J. Exp. Med. 191, 1095-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drebin, J. A., Link, V. C., Stern, D. F., Weinberg, R. A. & Greene, M. I. (1985) Cell 41, 697-706. [DOI] [PubMed] [Google Scholar]
- 23.Cheng, X., Kinosaki, M., Takami, M., Choi, Y., Zhang, H. & Murali, R. (2004) J. Biol. Chem. 279, 8269-8277. [DOI] [PubMed] [Google Scholar]
- 24.Park, B. W., Zhang, H. T., Wu, C., Berezov, A., Zhang, X., Dua, R., Wang, Q., Kao, G., O'Rourke, D. M., Greene, M. I. & Murali, R. (2000) Nat. Biotechnol. 18, 194-198. [DOI] [PubMed] [Google Scholar]
- 25.Hasegawa, A., Cheng, X., Kajino, K., Berezov, A., Murata, K., Nakayama, T., Yagita, H., Murali, R. & Greene, M. I. (2004) Proc. Natl. Acad. Sci. USA 101, 6599-6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph, D., Petsko, G. A. & Karplus, M. (1990) Science 249, 1425-1428. [DOI] [PubMed] [Google Scholar]
- 27.Weinglass, A. B. & Kaback, H. R. (1999) Proc. Natl. Acad. Sci. USA 96, 11178-11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hubbard, S. J. & Argos, P. (1994) Protein Sci. 3, 2194-2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson, M. L., Regan, R. M. & Gibson, Q. H. (1996) Biochemistry 35, 1125-1136. [DOI] [PubMed] [Google Scholar]
- 30.Fitzgerald, M. M., Musah, R. A., McRee, D. E. & Goodin, D. B. (1996) Nat. Struct. Biol. 3, 626-631. [DOI] [PubMed] [Google Scholar]
- 31.Towler, E. M., Thompson, S. K., Tomaszek, T. & Debouck, C. (1997) Biochemistry 36, 5128-5133. [DOI] [PubMed] [Google Scholar]
- 32.Pargellis, C., Tong, L., Churchill, L., Cirillo, P. F., Gilmore, T., Graham, A. G., Grob, P. M., Hickey, E. R., Moss, N., Pav, S. & Regan, J. (2002) Nat. Struct. Biol. 9, 268-272. [DOI] [PubMed] [Google Scholar]
- 33.Krishnaswamy, S. (2005) J. Thromb. Haemost. 3, 54-67. [DOI] [PubMed] [Google Scholar]
- 34.Hoffman, L. R., Kuntz, I. D. & White, J. M. (1997) J. Virol. 71, 8808-8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ottemann, K. M., Thorgeirsson, T. E., Kolodziej, A. F., Shin, Y. K. & Koshland, D. E., Jr. (1998) Biochemistry 37, 7062-7069. [DOI] [PubMed] [Google Scholar]
- 37.Koshland, D. E., Jr. (1998) Nat. Med. 4, 1112-1114. [DOI] [PubMed] [Google Scholar]
- 38.Bennett, W. S., Jr., & Steitz, T. A. (1978) Proc. Natl. Acad. Sci. USA 75, 4848-4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.