

Abstract
Forward genetic screens have led to the isolation of several genes involved in secondary cell wall formation. A variety of evidence, however, suggests that the list of genes identified is not exhaustive. To address this problem, microarray data have been generated from tissue undergoing secondary cell wall formation and used to identify genes that exhibit a similar expression pattern to the secondary cell wall–specific cellulose synthase genes IRREGULAR XYLEM1 (IRX1) and IRX3. Cross-referencing this analysis with publicly available microarray data resulted in the selection of 16 genes for reverse genetic analysis. Lines containing an insertion in seven of these genes exhibited a clear irx phenotype characteristic of a secondary cell wall defect. Only one line, containing an insertion in a member of the COBRA gene family, exhibited a large decrease in cellulose content. Five of the genes identified as being essential for secondary cell wall biosynthesis have not been previously characterized. These genes are likely to define entirely novel processes in secondary cell wall formation and illustrate the success of combining expression data with reverse genetics to address gene function.
INTRODUCTION
The plant cell wall has many functions: it regulates cell expansion, contributes to cell adhesion, acts as a barrier to potential pests and pathogens, and determines the physical properties of the plant (Braam, 1999; Jones and Takemoto, 2004; Scheible and Pauly, 2004; Vorwerk et al., 2004) The differing functions of the cell wall are reflected in the large variation in cell wall composition between different cell types and during cell differentiation. One estimate suggests that as many as 15% of the genes in the genome may be concerned with cell wall synthesis, remodeling, or turnover (Carpita et al., 2001). The Arabidopsis thaliana genome contains >800 identifiable carbohydrate active enzymes. This figure represents a large proportion of the genome compared with nonplant organisms, and it is suggested that the overrepresentation of carbohydrate active enzymes is a requirement for synthesis, remodeling, and degradation of the plant cell wall (Coutinho et al., 2003). A large number of other genes are also required for synthesis of cell wall polymers, such as lignin, phenylpropanoids, structural proteins, and other cell wall components. Identifying and determining the function of genes involved in cell wall synthesis and modification remains a major challenge.
The deposition of a thick lignified secondary cell wall only occurs once cells have attained their final shape and size. As the major constituent of wood and plant fibers, understanding the synthesis of the secondary cell wall has important biological and economic implications. During inflorescence stem development in Arabidopsis, the xylem and interfascicular cells form a thick secondary cell wall that constitutes a large proportion of the dry weight of the stem (Turner and Somerville, 1997) and represents the predominant metabolic process during certain stages of stem development. Secondary cell wall formation is a complex process that requires the coordinate regulation of several diverse metabolic pathways. The wall is predominantly composed of cellulose, lignin, and xylan. It is unclear, however, what other components may be essential for cell wall function and integrity. Arabidopsis has proved an excellent model for secondary cell wall formation and has been used to identify genes involved in both the regulation of secondary cell wall synthesis as well as genes encoding individual steps in the lignin and cellulose biosynthetic pathways (Nieminen et al., 2004).
Defects in the secondary cell wall are characterized by a collapse of xylem vessels that are unable to withstand the negative pressure generated during water transport through the xylem. This phenotype, described as irregular xylem (irx) has been used to isolate Arabidopsis mutants defective in the biosynthesis of both cellulose (Turner and Somerville, 1997) and lignin (Jones et al., 2001). Similarly, defects in lignin biosynthesis (Piquemal et al., 1998) and in phenylpropanoid biosynthesis (Ranocha et al., 2002) in tobacco (Nicotiana tabacum) are all characterized by irregular or distorted xylem vessels. This would suggest that the irx phenotype will be indicative of any secondary cell wall mutation. Although this phenotype is a sensitive indicator of a secondary cell wall defect, it is not particularly suited to very large genetic screens. The original irx mutants were identified from stem sections, although subsequent lines were identified based on a resulting alteration in plant morphology (Taylor et al., 2003). Mutants containing xylem elements that only exhibit slight distortions are harder to discriminate; consequently, forward genetic screens have led to the isolation of quite severe phenotypes only (Turner and Somerville, 1997; Jones et al., 2001). Furthermore, very severe wall defects may result in reduced viability. This idea is confirmed by the fact that several potentially novel irx mutants have been isolated, but very low fertility has rendered them unsuitable for genetic analysis (S.R. Turner, unpublished data). Both of these points suggest that the original screen may not have identified all genes involved in secondary cell wall synthesis and that the irx phenotype is likely to be indicative of many more genes essential for proper secondary cell wall formation.
The irx1, irx3, and irx5 mutants are all caused by defects in members of the CesA gene family. The AtCesA4 (IRX5), AtCesA7 (IRX3), and AtCesA8 (IRX1) proteins all function in a nonredundant manner as part of a complex that is required to synthesize cellulose in the secondary cell wall (Gardiner et al., 2003; Taylor et al., 2003). The absence of any detectable primary cell wall phenotype together with promoter–β-glucuronidase fusions and RNA gel blot analysis suggest that these genes only function to synthesize cellulose in the secondary cell wall and as such represent specific markers for cells undergoing secondary cell wall formation (Holland et al., 2000; Taylor et al., 2000; Ha et al., 2002).
Recent annotation of the Arabidopsis genome suggests that of the 27,139 identified functional genes, 7592 (28%) are predicted as protein of either hypothetical or unknown function (http://arabidopsis.org/). Many of the remaining genes are annotated solely on the basis of a conserved domain or assigned to a gene family based on sequence homology that groups these genes into broad categories. Estimates suggest that only ∼10% of genes had any experimental data to support their function (Arabidopsis Genome Initiative, 2000). Consequently, to assign a role for a gene in a process such as cell wall biosynthesis requires further information. The use of expression data, coupled with reverse genetics, is a method that would offer the potential to rapidly and efficiently identify the function of particular genes. Using the expression data to identify genes expressed in particular cell types is also likely to help alleviate the problem associated with cell wall diversity described above.
In this study, microarray data generated from developing stems and hypocotyls have been used to identify those genes coregulated with IRX1, IRX3, and IRX5. By cross-referencing publicly available data, several genes were selected for reverse genetic analysis. The results of this analysis suggest that several, if not all, of the genes identified are essential for secondary cell wall formation. Only one mutant, however, exhibited a severe cellulose-deficient phenotype, suggesting that the other genes are involved in other, potentially novel, pathways required for secondary cell wall formation.
RESULTS
Expression of IRX1 and IRX3
At the onset of this study, few publicly available data sets exhibited high levels of expression of the secondary cell wall–specific genes IRX1 and IRX3. This presumably reflects the fact that few studies use tissues such as mature stems that contain a high proportion of cells undergoing secondary cell wall formation. To examine the expression of these genes in more detail, real-time RT-PCR was performed on RNA isolated from four parts of the stem, mature hypocotyls, and leaves. This material was selected on the basis that secondary cell wall formation had previously been shown to increase dramatically from the top to the base of the stem and to be high in hypocotyls and low in leaves (Turner and Somerville, 1997). The expression levels of IRX3 and IRX1 both mirrored this pattern. Their expression increased dramatically from the tip to the base of the stem and was high in hypocotyls and low in leaves (Figure 1). Although IRX3 was expressed consistently at a higher level than IRX1, they both exhibited a very similar pattern of expression that varied dramatically with >60-fold higher levels of expression in the hypocotyls compared with leaves.
Figure 1.

Analysis of IRX1 and IRX3 Expression in the Stem, Leaf, and Hypocotyl.
Expression of IRX1 (open bars) and IRX3 (closed bars) are represented as a percentage of actin expression. Standard error bars are shown (n = 3).
Expression Analysis of Secondary Cell Wall Formation
mRNA from the six stages described above were used for near genome-wide expression profiling using the Affymetrix ATH1 microarray chip (Santa Clara, CA). Experiments were performed using three biological replicates from each developmental stage. Principal component analysis (PCA) was used to analyze the variance within the data set. All the biological replicates for a particular developmental stage clustered closely together in this unsupervised multivariate analysis of all the genes on the microarrays. Similarly, the hypocotyls, leaf, and stem samples were all well separated from one another (Figure 2A). More importantly, the different stem samples separated on principal component 1 on the basis of their stage of development (i.e., from tip to base) (Figure 2A). This clear clustering of the microarray data in the PCA indicates highly reproducible and discrete mRNA expression patterns in the different stages of stem development that were collected.
Figure 2.

Interpretation and Analysis of Microarray Data.
(A) PCA of microarray data from leaves, hypocotyls, and four stages of stem development. The data include three biological replicates for each stage. The first two principal components are shown (component 1 accounts for 45% of the variance and component 2 accounts for 23%). Circles are drawn as a guide and have no statistical significance.
(B) Comparison of the gene expression between the base and tip of the stem. Changes in expression levels have been plotted against P values derived from a t test. Horizontal lines indicate a change in gene expression of twofold or more. The vertical line corresponds to a P value of 0.01. Shaded area shows genes that have a greater than twofold increase in gene expression between the tip and the base of the stem and a P value of <0.01 (n = 3). Crosses represent genes selected for further analysis.
(C) Slope profile analysis of microarray data showing the expression profile of the top 25 genes that most closely match that of IRX3 (thick line).
A comparison of changes in gene expression between the top and the bottom of the stem is shown in Figure 2B. This analysis identifies several hundred genes that apparently show an increase in expression at the base of the stem compared with the tip. This was considered as too many genes to examine efficiently. Consequently, to perform a detailed study, this set of genes was further refined (see below). For reference, the genes eventually selected are indicated in Figure 2B by crosses. This clearly shows that there is no clear relationship between fold change and those genes eventually selected.
Selection of Candidate Secondary Cell Wall Genes for Further Analysis
There are currently a large number of approaches to the analysis of microarray data. Two criteria were used to determine the best method of analyzing this data set. First, IRX1, IRX3, and IRX5 all form part of the same complex (Taylor et al., 2003) and are consequently likely to be coregulated. Secondly, although the pattern of expression of IRX1 and IRX3 is similar, their absolute levels of expression vary. IRX3 is always expressed more than twofold higher than IRX1 (Figure 1). The slope profile is generated by looking at the gradient of a line that connects the expression of a gene in different samples (Figure 2C). This profile filter was used to generate a ranking of genes whose score most closely matched that of IRX3 (Figure 2C, Table 1). This analysis selects genes that show a similar pattern of expression (covariance), rather than genes that exhibit similar absolute levels of expression (Figure 2C). Using this analysis, both IRX5 (position 4) and IRX1 (position 15) are closely matched to IRX3 (Table 1). This is a much closer ranking than methods based upon absolute expression levels (data not shown).
Table 1.
Summary of Genes, Insertion Lines, and Phenotypes
| Rankinga | Gene | MaxD Slope Valueb | Annotation | r2 Valuec | Insertion | Insertion Site | XIM Line Numberd (Mutant Name) | Phenotype |
|---|---|---|---|---|---|---|---|---|
| 1 | At5g17420 | 0.00 | Cellulose synthase (IRX3) | 1.00 | SALK_029940 | Intron | 59 (irx3-4) | Severe |
| 2 | At2g38080 | 0.52 | Laccase | 0.84 | SALK_051892 | 5′UTRe | 66 (irx12) | Mild |
| 3 | At2g37090 | 0.94 | Glycosyl transferase family 43 | 0.77 | SALK_058238 | Exon | 36 (irx9) | Severe |
| 4 | At5g44030 | 0.99 | Cellulose synthase (IRX5) | 0.88 | SALK_084627 | Exon | 55 (irx5-4) | Severe |
| 5 | At5g15630 | 1.20 | COBRA- like (COBL4) | 0.94 | FLAG_248B03 | Exon | 40 (irx6) | Moderate |
| 6 | At1g19940 | 1.28 | Glycosyl hydrolase family 9 | 0.50 | None selected | NAf | NA | NA |
| 7 | At1g09610 | 1.33 | Unknown protein (DUF579) | 0.69 | SALK_050883 | Exon | 54 | No |
| 8 | At3g16920 | 1.45 | Glycoside hydrolase family 19 | 0.90 | SALK_055713 | Exon | 28 | No |
| 9 | At1g62990 | 1.50 | Homeodomain containing protein | 0.60 | SALK_002098 | Intron | 47 (irx11) | Moderate |
| 10 | At4g33330 | 1.68 | Glycosyl transferase family 8 | 0.22 | None selected | NA | NA | NA |
| 11 | At5g54690 | 1.73 | Glycosyl transferase family 8 | 0.93 | SALK_008642 | Intron | 30 (irx8) | Severe |
| 12 | At4g27435 | 1.79 | Unknown protein | 0.90 | SALK_137109 | Intron | 41 | No |
| 13 | At2g45900 | 1.84 | Unknown protein | 0.07 | None selected | NA | NA | No |
| 14 | At1g27440 | 1.90 | Glycosyl transferase family 47 | 0.86 | SALK_055673 | 5′UTR | 29 (irx10) | Moderate |
| 15 | At4g18780 | 1.91 | Cellulose synthase (IRX1) | 0.80 | SALK_026812 | Intron | 58 (irx1-5) | Severe |
| 16 | At1g33800 | 2.14 | Unknown protein (DUF579) | 0.13 | None selected | NA | NA | NA |
| 17 | At3g12955 | 2.15 | Auxin-responsive protein | 0.05 | None selected | NA | NA | NA |
| 18 | At5g60720 | 2.15 | Unknown protein | 0.46 | SALK_048659 | Promoter | 43 | No |
| 19 | At1g72220 | 2.17 | Zinc finger protein | 0.35 | None selected | NA | NA | NA |
| 20 | At5g46340 | 2.22 | O-acetyltransferase protein | 0.72 | None selected | NA | NA | NA |
| 21 | At1g62800 | 2.23 | Aspartate aminotransferase4 | 0.07 | None selected | NA | NA | NA |
| 22 | At1g27380 | 2.26 | p21-rho binding protein | 0.87 | None selected | NA | NA | NA |
| 23 | At5g54160 | 2.28 | O-methyltransferase | 0.09 | None selected | NA | NA | NA |
| 24 | At3g59690 | 2.28 | Putative protein SF16 | 0.26 | None selected | NA | NA | NA |
| 25 | At2g28110 | 2.33 | Glycosyl transferase family 47 | 0.10 | SALK_120296 | 5′UTR | 27 (irx7) | Severe |
| 28 | At1g08340 | 2.39 | Rac GTPase activating protein | 0.71 | SALK_080212 | Exon | 60 | No |
| 38 | At3g18660 | 2.54 | Glycosyl transferase family 8 | 0.86 | SALK_063763 | Exon | 9 | No |
| 49 | At5g03170 | 2.80 | Fasciclin-like AGP | 0.95 | SALK_046976 | Exon | 10 | No |
| 79 | At2g29130 | 3.14 | Laccase | 0.83 | SALK_025690 | Exon | 1 | No |
| 87 | At5g16600 | 8.39 | Transcription factor (MYB43) | 0.27 | SALK_030146 | Exon | 12 | No |
Ranking based on MaxD slope profile value.
Value is derived from an algorithm from the MaxD software program.
r2 values generated from the pairwise comparison of IRX3 expression and other genes of interest using the program GeneCorrelator.
All Columbia except FLAG428B03, which is Ws.
UTR, untranslated region.
NA, not applicable.
The identification of two further genes that closely match the expression of IRX3 further increased the confidence in this method of analysis. At5g15630 (position 5) encodes a member of the COBRA family (COBL4) (Schindelman et al., 2001; Roudier et al., 2002). The ortholog of this gene in rice (Oryza sativa) is essential for cellulose deposition in the secondary cell wall (Li et al., 2003). Similarly, At3g16920 (position 8) encodes for a member of a family that exhibits homology to chitinase genes. A member of this family has been shown to be required for cellulose synthesis in the primary cell wall (Mouille et al., 2003). By contrast, expression of the endoglucanase KORRIGAN (KOR) (Nicol et al., 1998) does not closely match the pattern of IRX3 expression. Similarly, many genes required for lignin biosynthesis, such as IRX4 that encodes a cinnamoyl CoA reductase (Jones et al., 2001), do not exhibit a good match with IRX3 expression.
Many of the genes in Table 1 that exhibit a similar expression pattern to IRX3 are likely to be involved in carbohydrate metabolisms. Twelve of the 25 genes shown in Table 1 have been identified as belonging to various families of glycosyltransferase (GT) or glycosylhydrolase enzymes, including members of both the GT8 and GT47 families (http://afmb.cnrs-mrs.fr/CAZY/). Mutations in members of both of these gene families have been shown to result in cell wall defects in Arabidopsis (Bouton et al., 2002; Iwai et al., 2002; Madson et al., 2003), supporting the idea that genes identified in this analysis are likely to be involved in cell wall formation.
Cross-Referencing Publicly Accessible Data
One of the problems associated with looking at tissues such as the stem is the presence of many different cell types of which only some synthesize a secondary cell wall. One of the few studies in which cell type–specific expression has been analyzed in plants is the recent work by Birnbaum et al. (2003). In this study, individual cells from the Arabidopsis root were fractionated and analyzed to generate an expression map of the whole root. We have used this microarray data to examine the cell-specific expression of IRX1 and IRX3. The results are visualized in Figures 3A and 3B, respectively. There is little expression in the tip section containing the apical meristem, and high levels of expression are found only in the stele where secondary cell wall formation occurs during xylem development. Each of the top 200 genes from the slope ranking was checked for their cell-specific expression pattern in the root to determine whether it exhibited a pattern similar to that of IRX1 and IRX3. A GT8 family gene (At3g18660) and a gene encoding a protein of unknown function (At3g12955) are shown as an example. At3g18660 exhibits a similar expression pattern (Figure 3C), whereas At3g12955 exhibits an entirely different cell-specific expression pattern (Figure 3D). Only those genes judged to have a similar cell-specific expression pattern to IRX3 were selected for further study.
Figure 3.

Graphical Representation of Root Microarray Data (Birnbaum et al., 2003).
Stage 1 closest to the root tip; stage 3 furthest from the root tip. Cell layers in the root indicated are lateral root cap (lrc), epidermis (epi), endodermis and cortex (endo and cor), endodermis (end), and stele. (A) At4g18780 (IRX1); (B) At5g17420 (IRX3); (C) At3g18660; (D) At3g12955.
To further verify the coregulation of these genes with IRX3, the top 200 genes were used in a pairwise comparison using the two-gene scatterplot at the Nottingham Arabidopsis Stock Centre (NASC; http://nasc.nott.ac.uk/) and the Gene Correlator at the Genevestigator Web site (https://www.genevestigator.ethz.ch/) (Zimmermann et al., 2004). Both of these programs plot the expression of two selected genes from a large number of publicly available data sets. In addition, Gene Correlator generates a linear correlation coefficient (Table 1).
In general, any genes that did not exhibit a correlation coefficient of >0.6 were not selected for further study. However, there were exceptions, for example, a putative GT47 member (At2g28110) was retained despite a poor correlation (r2 = 0.01) because a member of the same family (At1g27440) did exhibit a good match with IRX3. Similarly, Xylem Insertion Mutant 12 (XIM12), an insertion in At5g16600, was selected for further analysis because it is a member of the MYB gene family that has been demonstrated to be involved in the regulation of secondary cell wall polymers (Tamagnone et al., 1998; Patzlaff et al., 2003). Some lines that did exhibit a good correlation were not selected for further study if there was no suitable insertion line available from the Salk or Flag collections (Alonso et al., 2003; Samson et al., 2004). In total, 16 lines were selected for further analysis.
To independently confirm the microarray data independently, 12 genes of the gene studied were analyzed using real-time RT-PCR. Consistent with the microarray data, all genes selected exhibited the highest level of expression in either the hypocotyls or at the base of the stem, low levels of expression in leaves, and a decline in expression levels going from the base to the tip of the stem (Figure 4).
Figure 4.

Real-Time RT-PCR Analysis of Selected Genes in Different Tissues.
The expression of genes was measured as the percentage of actin expression. For convenience, the results have been expressed as a proportion of the maximum expression level for that gene and are an average of three biological replicates.
Reverse Genetics
For those genes identified for further analysis, insertion lines were selected using the SIGnaL database (http://signal.salk.edu/). Where multiple insertions were available in the same genes, lines were selected on the basis of the position of the insertion and the highest likelihood of it disrupting gene function. In most cases, insertions were obtained within the exon of a gene or within the 5′ noncoding region (Table 1). Primers flanking the insertion site were used in conjunction with a primer from the left border of the T-DNA to identify lines homozygous for the T-DNA insertion (see Methods for details). Where problems with poor seed set or sterility occurred, the seeds from plants heterozygous for the insertion were collected.
To verify the effect of the insertion on mRNA expression, RT-PCR was performed using gene-specific primers designed to be close to the 5′ and 3′ ends of the gene. Line XIM43 exhibited no difference in the expression of the target gene compared with the wild-type control. This line contains an insertion in the promoter region, and this presumably does not significantly alter the expression of the target gene. Similarly, after 50 cycles of PCR, XIM29 and XIM30 generated a band of the predicted size, although at a greatly reduced level. Interestingly, both of these lines generated a phenotype (see below). These lines contain an insertion in the 5′ untranslated region and an intron of the target, respectively (Table 1). All of the other primer pairs generated a PCR fragment of the predicted size in the wild type, but that was undetectable in the corresponding insertion line (data not shown).
Phenotypic Analysis
Stem sections were cut from each line from the base of the mature inflorescence stem to determine the xylem morphology. The wild type is characterized by open xylem elements with a relatively round shape (Figure 5A). By contrast, the vessels from irx3-4 plants are irregular and frequently collapsed inwards such that some appear almost completely occluded (Figure 5B). Of the 16 insertion lines selected, nine could not be distinguished from the wild type (Table 1). Seven lines, however, exhibited a clear irx phenotype (Figure 5, Table 1). Because these lines did not correspond to any previously described irx mutants, they were designated irx6 to irx12 (Table 1). Three lines were classified as having a severe irx phenotype on the basis that collapsed vessels were obvious in all vascular bundles and plants examined and always resulted in severely irregular vessels. The mutants irx7, irx8, and irx9 were caused by insertions in a putative GT47 (At2g28110), a putative GT8 (At5g54690), and a putative GT43 (At2g37090) (Figure 5, Table 1). The remaining mutants exhibited varying degrees of severity. For example, irx6, irx10, and irx11, caused by insertions in a member of the COBRA gene family (COBL4), a GT47 family member (At1g27440), and a putative HD1 family transcription factor (At1g62990), respectively, all exhibit a clear irx phenotype (Figures 5C, 5G, and 5H). In these cases, however, the phenotype is less severe than that observed for irx3. Similarly, irx12 contains an insertion in a putative laccase (At2g38080) and exhibits a weak irx phenotype that appeared to vary in severity between plants and even between vascular bundles within the same plant (Figure 5I). The irx4 mutant exhibits alteration in lignin biosynthesis and is caused by a mutation in the cinnamoyl CoA reductase genes (Jones et al., 2001). The walls of these plants are characterized by much thicker diffuse appearance that stain poorly with toluidine blue (Jones et al., 2001). None of lines described in this study exhibited these characteristics.
Figure 5.

Cross Sections of Stem Vascular Bundles in Wild-Type and irx Mutant Plants.
Transverse stem sections stained with toluidine blue. A single representative vascular bundle is shown from each mutant. The phloem (ph) and xylem vessels (arrows) are indicated. (A) Columbia wild type; (B) irx3-4; (C) irx6; (D) irx7; (E) irx8; (F) irx9; (G) irx10; (H) irx11; (I) irx12. Bars = 50 μm.
In comparison with the wild type, irx3-4 plants are small, grow more slowly, are darker green, have narrower leaves, and were almost completely sterile (Figure 6). Similar phenotypes were obtained from insertions in the IRX1 and IRX5 genes (data not shown). This characteristic plant morphology was obtained for three other mutants: irx7, irx8, and irx9 (Figure 6). These lines correspond to the plants that exhibit a severe irx phenotype described above. Because of poor fertility, these lines were maintained by selecting heterozygotes and selecting homozygous insertion lines from their progeny. Removal of these plants from the growth chamber often resulted in severe wilting of the end of the inflorescence stems that was irreversible. Maintaining plants at very high humidity by permanently covering them with a plastic dome did help to generate plants that grew more vigorously, but all the phenotypes described above were still evident and mutants were very clearly distinguishable from the wild type.
Figure 6.

Whole-Plant Morphology of Wild-Type and irx Mutants.
All plants shown are 5 weeks old.
For all the other lines examined, the plants appeared morphologically wild-type (Figure 6); however, two lines did exhibit other phenotypes. irx6 plants containing an insertion in the COBL4 gene exhibited a normal growth habit (Figure 6) but resulted in a plant with dramatically reduced stem strength that caused the inflorescence stem to be easily broken. To a lesser extent, this was also true of XIM9 containing an insertion in a GT8 family gene (At3g18660). A weak stem is a characteristic of known secondary cell wall mutants (Turner and Somerville, 1997; Jones et al., 2001).
Cell Wall Analysis
Because the marker genes used for this analysis were all members of the cellulose synthase gene family, it was considered that some of these mutants may be deficient in cellulose synthesis. Consequently, the cellulose content from the stems of all 16 lines was measured. Consistent with previous analysis (Turner and Somerville, 1997), mutation in the genes for IRX1 and IRX3 caused dramatic reductions in cellulose content (Figure 7). Of the other lines, only irx6 caused by insertion in COBL4 exhibited a very large decrease in cellulose content, although the decrease is not as apparent as that observed for mutations in IRX3 (Figure 7). A strict comparison between these lines is, however, complicated by the fact that the COBL4 insertion line is derived from the Wassilewskija (Ws) background. irx7, irx8, and irx9 exhibited smaller but significant decreases in cellulose content (Figure 7). It is important to note, however, that these lines correspond to the plants with grossly altered morphology (Figure 6).
Figure 7.

Cellulose Content of Stems from Wild-Type and Insertion Mutant Lines.
Cellulose content is expressed as a proportion of the ethanol insoluble cell wall material collected at the late stage of stem development. Numbers correspond to insertion line number (see Table 1). Closed bars indicate measurement from irx1-5 and irx3-4 plants. Hatched bars highlight novel irx mutants. Shaded bars highlight Ws background. Asterisks show a significant reduction in cellulose. Standard error bars are shown (n = 4).
To overcome the potential problem with measuring cellulose content of plants with altered morphology, it is important to ensure that comparisons are made at the same developmental stage. Those lines that exhibited a reduction in cellulose content were examined at three stages of stem development. Wild-type plants exhibit an increase in cellulose content in the stem during development that reflects the increase in secondary cell wall deposition (Figure 8). Consistent with previous studies, plants with an insertion in IRX3 do not show this increase, and the cellulose content remains constant during stem development. Similarly, irx6 plants also exhibit no increase in cellulose during stem development (Figure 8). By contrast, irx7, irx8, and irx9 show a clear increase in cellulose content during development. The pattern of accumulation is similar to the wild type, but at each stage the cellulose content is less than that of corresponding wild-type plants (Figure 8).
Figure 8.

Cellulose Content of Developing Stems from Wild-Type and irx Mutant Plants.
Cellulose content is expressed as a proportion of the ethanol insoluble cell wall material. Standard error bars are shown (n = 4).
To facilitate characterization and classification, mutant lines were examined using the metabolic fingerprinting method of Fourier transform infrared (FTIR) spectroscopy (Goodacre et al., 2004). A PCA scores plot derived from FTIR spectra data clearly separated the mutant lines into three distinct clusters on the basis of secondary cell wall defects (Figure 9A). irx1, irx3, and irx5 (represented as cluster 2) and irx7, irx8, and irx9 (represented as cluster 3) are separated from other mutant lines, including the wild type, in principal component 1 (PC1; Figure 9A). The loadings plot for PC1 in the polysaccharide fingerprint region (1250 cm−1 to 800 cm−1) (Figure 9B) showed characteristic IR bands that correlated well with purified cellulose, with peaks at 1161 cm−1, 1109 cm−1, 1059 cm−1, 1034 cm−1, and 1059 cm−1 (Liang and Marchessault, 1959; Sugiyama et al., 1991; Kacurakova et al., 2002). The spectrum from cotton (Gossypium hirsutum) lintels is shown for reference. The scores of clusters 2 and 3 are negative on PC1 relative to the mean (Figure 9A), suggesting a decrease in cellulose in secondary cell walls. Separation between clusters 2 and 3 suggests that discrimination on PC2 is based on noncellulosic differences between the groups. The PC2 loading plot (Figure 9C) is characterized by positive peaks that can be designated to xylan-type polysaccharides at 1240 cm−1, 1128 cm−1, 1082 cm−1, 1045 cm−1, and 978 cm−1 as described by Kacurakova and colleagues (Kacurakova et al., 1998, 1999, 2000). A spectrum of purified birch (Betula spp) xylan (Figure 9C) is shown for reference, showing that many these peaks match those found in the PC2 loading plot. This suggests that the cell walls of irx7, irx8, and irx9 are deficient in xylan. Further analysis of the FTIR spectra using discriminate function analysis (Manley, 1994) to reduce within group variance and maximize between group variance demonstrated that irx12, a putative laccase, also formed a separate cluster (data not shown). Because no other irx mutants grouped with irx12, it is likely that this is the only line with a defect in phenylpropanoid metabolism.
Figure 9.

Cell Wall Analysis of Insertion Mutant Lines.
(A) PCA of FTIR spectra from wild-type and mutant stem material. PC1 accounted for 62.5% of the total explained variance, whereas PC2 accounted for 20.2%. Cluster 2 contains irx1, irx3, and irx5. Cluster 3 contains irx7, irx8, and irx9. Cluster 1 contains wild-type and all other insertion lines. Numbers refer to XIM line numbers. Circles are drawn as a guide and have no statistical significance.
(B) Loading plot of PC1 in the polysaccharide fingerprint region. A spectrum derived from cotton linters cellulose is also shown for reference. Peaks characteristic of cellulose (1161 cm−1, 1109 cm−1, 1059 cm−1, 1034 cm−1, and 1059 cm−1) are indicated.
(C) Loading plot of principal component 2 in the polysaccharide fingerprint region. A spectrum derived from birchwood xylan is also shown for reference. Peaks characteristic of xylan (1240 cm−1, 1082 cm−1, 1045 cm−1, and 978 cm−1) are indicated.
Figure 10 shows the noncellulosic carbohydrate composition of a crude cell wall fraction from stems. The proportion of sugars found in wild-type stems is in general agreement with previous studies (Turner and Somerville, 1997). The high xylose content, which is characteristic of secondary cell walls, reflects the typical composition of many angiosperms (Turner and Somerville, 1997). The cell wall sugar composition of cellulose-deficient mutants irx3 and irx6 plants were in approximate agreement with earlier studies (Turner and Somerville, 1997; Li et al., 2003).
Figure 10.

The Noncellulosic Carbohydrate Composition of Cell Wall Material from Stems of Wild-Type and Insertion Mutant Lines.
Individual sugars are expressed as a percentage of the total cell wall sugar. Material used was from the late stage of development. Standard error bars are shown (n = 5).
The largest changes are evident in irx7, irx8, and irx9, which all exhibited a large reduction in xylose. The irx7 noncellulosic sugar fraction contained only 28% of wild-type xylose, a characteristic repeated to a lesser extent in both irx8 (35% of the wild type) and irx9 (45% of the wild type). In addition to a decrease in xylose, significant increases in the remaining cell wall sugars are observed in all three lines. This increase in proportion may be partly explained by the loss of xylose. irx10 stems had similar, but less dramatic, alterations in sugar composition with a decrease in xylose (80% of the wild type) and a small increase in the percentage of all other sugars.
DISCUSSION
The secondary cell wall of Arabidopsis is composed predominantly of cellulose, lignin, and xylan, making it an attractive model for the study of these pathways. However, several other components are likely to be important in the secondary cell wall. This is exemplified by work on a laccase gene in poplar (Populus spp). Downregulation of this gene results in fragmented secondary cell walls, distorted xylem vessels, and the accumulation of phenolic compounds that would presumably normally be cross-linked into the wall to give its proper structural properties (Ranocha et al., 2002). Before this study, the role of these compounds in wall formation had received little attention. It is likely given the current paucity of knowledge with respect to secondary metabolism in plants that other such pathways contribute essential components to the secondary cell but are as yet poorly characterized. Genetic screens, based upon the irx phenotype, have led to the isolation of several important genes required for secondary cell wall biosynthesis, including those involved in cellulose and lignin biosynthesis (Turner and Somerville, 1997; Jones et al., 2001; Taylor et al., 2003). To date, however, only five complementation groups have been identified.
Estimates based upon the frequency of known mutations and the numbers of unique genes suggest that perhaps mutations in only one in 10 genes would give a clear phenotype (Meinke et al., 2003). It is clear that, superficially, the phenotype of some of the lines studied were indistinguishable from the wild type (Figure 6). Importantly, however, seven of the 16 genes selected in this study gave a clear irx phenotype. The comparative success of this study may be attributed in part at least to the combination of reverse genetics with expression data. As a consequence, it is possible to use a screen that targets specific processes in a single cell type at a particular stage of development. This kind of analysis entirely depends on availability of specific marker genes, such as IRX1 and IRX3, which are specific to the secondary cell wall and facilitated by the availability of tissue such as the stem, in which, at certain stages of development, the synthesis of the secondary cell wall is the major metabolic activity. This study offers empirical support for using the slope profile as a means of identify genes that covary and are likely to be involved in the same set of processes. In contrast with many of the clustering programs available, the slope profile does not require any assumptions and is mathematically simple.
The advantage of targeting specific processes with expression data followed by reverse genetic analysis of the genes is highlighted in this study. This is illustrated by insertions in the IRX3 gene and several other lines described in this study. These plants have greatly altered morphology that is presumably caused by the collapse of the xylem (Figure 5). Because most mutant analysis does not include cross sections of the xylem, the irx phenotype would not be obvious, and the altered morphology may be attributed to some other factor. Similarly, some mutants, such as irx6, appear morphologically normal (Figure 6) but exhibit a clear collapsed xylem phenotype that would be impossible to detect without making cross sections of the xylem. Why a collapse xylem causes alteration in plant morphology is unclear, but there is a correlation between the severity of the irx phenotype and the severity of the whole-plant phenotype. Those plants classified as very severe irx exhibit the most severe whole-plant morphology, whereas those plants with a less severe irx phenotype appear similar to the wild type (Figures 5 and 6).
Of the genes that do not give a phenotype, their correlation with the expression patterns of IRX1 and IRX3 would support a role for them in secondary cell wall formation. Of the nine lines that exhibited no phenotype, RT-PCR analysis suggests that one line contained normal levels of mRNA for the target gene; however, for the other eight lines, the corresponding mRNA was undetectable. It is not possible to preclude that these plants still make a truncated or altered mRNA that would lead to the production of protein that retains at least some activity. A more likely explanation of why some lines identified do not exhibit a clear phenotype is probably a combination of gene redundancy and a screening method that is specific for only one cell wall function. The former point can be illustrated by XIM54; this line is annotated as a gene of unknown function but contains a specific motif recognized by the program pfam (http://www.sanger.ac.uk/Software/Pfam/) known as DUF579. The Arabidopsis genome possesses 10 genes that contain this domain. Four of these genes, however, can be found within the list of 60 genes whose expression patterns most closely match that of IRX3. The extent to which redundancy contributes to the absence of an irx phenotype can only be assessed once lines containing two or more gene knockouts have been generated by crossing. The alternative explanation is the limitation of screening using the irx phenotype. This phenotype selects for plants whose secondary cell wall lacks the strength to withstand the forces generated within the xylem (Turner and Somerville, 1997). The irx phenotype, however, may not identify all possible secondary cell wall mutants. For example, a T-DNA insertion in At5g54160 that encodes caffeic acid O-methyltransferase 1 gene (AtOMT1) has been described previously by Goujon et al. (2003). No irx phenotype was observed; however, a reduction in syringyl lignin was detected in the vasculature of the plant (Goujon et al., 2003). The irx phenotype may also represent an inadequate method of screening for resistance of the cell wall to alteration in externally applied forces or to other secondary cell wall properties, such as cell–cell adhesion or hydrophobicity. Furthermore, these plants were grown in growth chambers under favorable conditions for plant growth. Exposing the plant to more adverse conditions, such as drought stress, may exacerbate any phenotype.
It is striking that only mutations in COBL4 gave a severe cellulose-deficient phenotype (Figure 7) in a manner similar to its rice homolog (Li et al., 2003). The data would be consistent with the COBRA gene family playing an essential, but as yet undetermined, role in cellulose deposition. Although insertions in this gene result in a dramatic reduction in cellulose synthesis, it is not as severe as irx3-4. This work highlights the differences that may occur when the same mutant is examined in different backgrounds. The original irx3-1 allele was isolated in a Landsberg erecta background, and although the plants were slightly smaller and darker green, they were fertile and grew normally. irx5-1 and irx1-1 plants exhibited a similar phenotype (Taylor et al., 2003). By contrast, irx1-5 and irx3-4 lines are T-DNA insertion mutants in the Columbia background. These plants exhibited a far more severe phenotype; they were much smaller, had narrow dark-green leaves, and were infertile. Both irx3-1 and irx3-4 are likely to cause a complete loss of IRX3 functions; consequently, the basis of the dramatic difference between the phenotype presumably results from differences between the ecotypes used. In this study, the only insertion available likely to give a knockout in COBL4 is in the Ws background. It is possible, therefore, that the less severe defects in irx6 compared with irx3 may be a result of differences in ecotype. There are, however, some parallels between the irx6 and the irx2 mutation (Turner and Somerville, 1997): they both have a very large decrease in stem strength, appear to form a secondary cell wall in the corners of the cell (Figure 5C; data not shown), and exhibit a less pronounced decrease in cellulose content compared with irx3. The data presented here support a role for the COBRA gene family in cellulose synthesis, but their exact function is yet to be defined.
irx7, irx8, and irx9 exhibit much smaller but significant changes in cellulose content of the stem. These plants, however, exhibit gross alteration in plant growth and morphology (Figure 6), and it is likely that this contributes to an overall reduction in secondary cell wall deposition in the stem. This would be analogous to the irx4 mutant caused by a defect in the gene encoding cinnamoyl CoA reductase. irx4 plants have large reduction in cell wall phenolics but also exhibit a decrease in cellulose that is presumably a result of alteration in plant morphology (Jones et al., 2001). This idea is supported by data that show cellulose accumulation during stem development. These lines all exhibit an increase in cellulose content during stem development in a manner that resembles the wild type (Figure 8). Furthermore, both the irx and whole-plant phenotypes of these three lines are very severe (Table 1, Figure 6). These phenotypes are considerably more pronounced than those exhibited by mutants, such as irx2-2, that have a larger decrease in cellulose content (Turner and Somerville, 1997). This would further support the idea that the irx phenotype is caused by a defect in a novel pathway and that the reduced cellulose is caused by the subsequent alteration in plant morphology.
Apart from COBL4, this study suggests that most genes that are specific for cellulose synthesis in secondary cell wall synthesis have been identified by forward genetics. It is clear, however, that not all genes involved in cellulose synthesis during secondary cell wall formation will be identified in our analysis. For example, KOR plays a role in cellulose synthesis during both primary and secondary cell wall formation (Lane et al., 2001; Sato et al., 2001; Szyjanowicz et al., 2004). As a consequence, the expression patterns of KOR and IRX3 will be different enough to stop them from grouping during the analysis. Similarly, genes involved in synthesizing the substrate for cellulose synthesis may not closely match the expression of IRX3.
More recent work has included analysis of a large number of publicly available data sets along with our own data. Using several clustering methods, as well as the slope profile, it has been shown that eight genes exhibit very similar expression patterns and cluster together very closely. This group includes IRX1, IRX3, and IRX5 together with COBL4, a chitinase-like gene, a laccase, a GT8 family member, and a GT43 family member. It is unlikely, however, that all these genes are involved in cellulose biosynthesis. An insertion in the laccase gene results in a weak irx phenotype (Figure 5). There is, however, no associated reduction in cellulose content (Figure 7). This gene is the homolog of a poplar gene, lac3, which has been demonstrated to be essential for secondary cell wall integrity (Ranocha et al., 2002). Although some studies have implicated laccases as playing a potential role in lignification, the downregulation of lac3 results in the accumulation of soluble phenolics, and it is the absence of these phenolics being cross-linked into the wall that causes the cell wall defect. The very close coregulation of IRX3 with a laccase suggests that the cross-linking of phenolics and the deposition of cellulose must be coordinately regulated to generate a functional secondary cell wall. Interestingly, four other laccase genes appear to exhibit some coregulation with IRX3. Whether these genes have an alternative function awaits further analysis.
In addition to the laccase, five other genes have an irx phenotype but have small or no alteration in cellulose content. These genes represent entirely novel aspects of secondary cell wall formation. One of these genes, At1g62990, exhibits similarity to the HD1 class of transcription factors and presumably regulates the expression of other genes that are likely to be required during the later stages of cell wall formation. At least four novel genes with homology to GTs have been identified as being essential for secondary cell wall formation. This includes two members of the GT47 family, one member of the GT43 family, and a member of the GT8 gene family.
The development of a method for using metabolic fingerprinting data, generated by FTIR, to study the secondary cell wall has greatly facilitated the study of these mutants. Using FTIR to study secondary cell walls creates particular problems with generating homogenous material that is thin enough to allow the infrared light to pass though during sampling. The validity of the method developed is clearly shown by the loadings plot of PC1. All cellulose-deficient mutants are separated from the wild type by PC1, and the loading plot exhibits a remarkably good match to that of purified cellulose (Figure 9B). The FTIR data (Figure 9A) also suggest that irx7, irx8, and irx9 group together and may form part of the same metabolic pathway. These mutants are separated from the known cellulose-deficient mutant by PC2. The loading plot of PC2 exhibits many of the characteristics of purified xylan (Figure 9C). This idea is confirmed by analysis of the cell wall sugar compositions (Figure 10); all three mutants exhibit a decrease in xylose. These mutants also exhibit complex changes in other cell wall sugars, in particular irx7 and irx9 exhibit an increase in the proportion of all other cell wall sugars. The alterations in morphology of these mutants mean that this data should be interpreted with caution because the results will also reflect changes in plant morphology. It is important to note, however, that the cellulose-deficient mutant irx3-4 exhibits a similar alteration in morphology (Figure 6) but does not exhibit any decrease in xylose (Figure 10). Exactly when and where the genes identified in this study function during secondary cell wall formation await further analysis.
The combination of expression analysis and reverse genetics has led to the identification of many genes that play a role in secondary cell wall synthesis. This targeted approach has led to the identification of seven novel secondary cell wall mutants. At least five of the genes identified define novel steps in secondary cell wall formation. The fact that these genes have not been identified by either biochemical studies or via forward genetic analysis emphasizes the efficiency of the approach taken in this study.
Since completing this work, it has come to our attention that Chris Somerville and colleagues have analyzed publicly available data to identify genes that are coexpressed with secondary cell wall CesA genes (Persson et al., 2005). Using a different method of analysis, they have identified genes that are coexpressed with CesA genes required for either primary or secondary cell wall formation. Of the top 25 genes identified as being coregulated with IRX3 (Table 1; Persson et al., 2005), 13 were common to both studies.
METHODS
Plant Material
All plants were germinated and grown on plates containing 0.8 to 1.0% agar (w/v) and MS media with B5 vitamins for 1 to 2 weeks before transferring nine plants to a 4-inch pot containing compost with vermiculite and perlite (10:1:1). They were then grown in continuous light at 22°C in controlled environment cabinets (Percival, Perry, IA) at a light intensity of 120 to 150 μE m−2. Material for the study of stem development was grown until the inflorescence stem contained two to three expanded siliques. The siliques and flowers were removed before the stems being divided into four sections of equal length. Hypocotyls and leaves were harvested at the same developmental stage as stems. All material used was from the Columbia background other than FLAG_428B03, which was in the Ws background.
Screening of Homozygote Plants with T-DNA Insertions
For those genes selected for further study, the SIGnal database (http://signal.salk.edu) was used to select lines containing T-DNA insertions most likely to cause a loss of gene function. For convenience, each insertion line selected was assigned a unique XIM number. Lines from the Salk collection were obtained from NASC (Nottingham University, UK) and ABRC (Ohio State University, Columbus, OH) (Alonso et al., 2003). One line (FLAG_428B03) containing an insertion in At5g15630 was obtained from the FLAG collection (Samson et al., 2004).
Plant DNA was extracted from one to two young leaves using a previously described miniprep procedure (Guidet et al., 1991). Mutant lines were confirmed for T-DNA insertions using the flanking primers (LP and RP) generated by the SIGnal T-DNA verification primer design Web site (http://signal.salk.edu/tdnaprimers.html) and primers from the T-DNA left border LBa1 (5′-GCGTGGACCGCTTGCTGCAACT-3′) and LBb1 (5′-TCAAACAGGATTTTCGCCTGCT-3′). For each line, four primer combinations were used: LP/RP/LBa1, LP/RP/LBb1, LP/RP, and LBa1/RP.
In the case of the insertion from the FLAG collection, only a single left border primer was used FLAG-LB1 (5′-CGGCTATTGGTAATAGGACACTGG-3′). The genotype of selected plants was verified in the next generation using a single reaction containing the flanking primers and one of the left border primers. The amplification conditions used for all screening were as follows: 95°C for 5 min; 35 cycles of 95°C for 15 s, 55°C for 30 s, and 72°C for 1 min; 4°C hold.
RNA Extraction and Expression Analysis
Material for RNA analysis was ground in liquid nitrogen, and RNA was isolated using the Qiagen RNAeasy kit (Crawley, UK) according to the manufacturer's instructions.
Real-time quantitative PCR was performed using an ABI Prism 7000 machine (Applied Biosystems, Warrington, UK). Primers and probes, for both Taqman and SYBR green assays, were designed using Primer Express (version 1.0) (Applied Biosystems). Taqman probes were labeled with 6-carboxyfluorescein at the 5′ end and tetramethylrhodamine at the 3′ end. Total RNA was treated with DNAase I (Invitrogen, Paisley, UK). First strand synthesis was performed in a volume of 20 μL, containing 1 μg of total RNA with 500 ng of poly-(dT) primer and 100 units of reverse transcriptase (Promega, Southampton, UK) at 42°C for 60 min. PCR conditions for both assays were as follows: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 60 s. For SYBR green assays only, a melting curve was produced at the end of every experiment to ensure that only single products were formed. The reliability of SYBR green primers was also examined by running the products on an agarose gel to ensure that only a single band was present. PCR reactions were performed in a volume of 25 μL, containing 12.5 μL of 2× quantitative PCR Mastermix (Eurogentec, Seraing, Belgium), 25 pmol each primer and 5 pmol probe (Taqman assays only). Data analysis was performed using the Sequence Detector (version 1.7) program (Applied Biosystems). Actin2 (At3g18780) expression was used to normalize the transcript level in each sample.
Array Measurements
For each biological replicate, material from nine plants was pooled to make a single sample for RNA purification. Three biological replicates were used for each developmental stage. Affymetrix GeneChip oligonucleotide arrays were used to analyze the gene expression of each developmental stage. Briefly, biotinylated cDNA samples from three biological replicates of each stage were synthesized and hybridized to Arabidopsis ATH1 genome oligonucleotide arrays (Affymetrix). Background correction, quantile normalization, and gene expression analysis were performed using RMAExpress (Bolstad et al., 2003). Further analyses, which included profile filtering, were performed using MaxdView (available from http://bioinf.man.ac.uk/microarray/maxd/). The microarray data were submitted in MIAME-compliant (minimum information about a microarray experiment) format to the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/) and have been assigned the accession number E-MEXP-265.
The volcano plot shown in Figure 2B was generated using the fold change that for each probe set was calculated as (mean reading at base of stem/mean reading at the tip of the stem). This was plotted against the P value generated from the standard Student's t test for each probe set. To identify genes with expression patterns similar to the IRX3 gene (At5g17420), profile filtering with the slope mathematical algorithm (MAXDVIEW) was performed using IRX3 as the target profile. To calculate the slope metric, the profile of the mean expression level (log2) for each plant was used. This slope metric calculates the sum of the differences of the first derivatives of the expression profiles (i.e., the slopes of lines joining expression values). The profile with the smallest sum is regarded as the most similar to the target profile (IRX3).
For a gene (X), the slope profile was calculated using the following general formula:
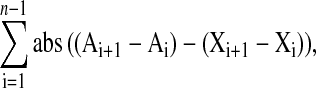 |
where abs is the absolute function abs (−Y) = Y, and Ai and Xi are the ith reading for reference gene A and the gene for comparison, X. In this case, the reference gene is IRX3 and n = 6 that corresponds to four stages of stem development, leaf, and hypocotyl. Those probes that most closely match IRX3 will have low slope profile.
Mutant Analysis
Stem sections, ∼200 μm thick, were hand cut using a razorblade and stained with toluidine blue O (Sigma-Aldrich, Poole, UK) as previously described (Turner and Somerville, 1997). Between 5 and 10 plants were examined for each mutant line. They were viewed on a Leica DMR light microscope (Leica Microsystems, Milton Keynes, UK) and photographed using a Spot RT digital camera (Diagnostic Instruments, Wigan, UK).
Metabolite Analyses
Cellulose measurements were performed with material from individual plants from the base of the primary inflorescence stem as previously described (Turner and Somerville, 1997). Three developmental stages were examined: early (plants with one to three expanded siliques), middle (plants with five to seven expanded siliques), and late (plants with 11 to 13 expanded siliques). For middle and late stages, a 10-cm segment from the base of the stem was used; however, for early staged plants, the entire inflorescence stem was analyzed.
For FTIR spectroscopy and gas chromatography analysis, a pool of five to seven stems was freeze-dried for 2 d and milled by rapid shaking with a ball bearing for 30 min using a tissue lyser (Qiagen) before analysis. For FTIR analysis, sample density was optimized such that good spectra with high signal-to-noise ratio were obtained. It was found that most optimal spectra were obtained at a dry weight of 50 mg/mL. Samples (5-μL aliquots) were evenly applied onto a silicon microplate containing 96 wells and oven dried at 50°C for 30 min (or until visibly dry). All samples were analyzed in quadruplicate.
A Bruker Equinox 55 FTIR spectrometer (Coventry, UK), fitted with a HTS-XT high-throughput microplate sampling accessory (Harrigan et al., 2004), was used to collect spectra over the wave number range of 4000 to 600 cm−1 as previously described (Timmins et al., 1998). Spectra were acquired at a rate of 20 s−1 and a resolution of 4 cm−1. To improve signal-to-noise ratio, 256 spectra were co-added and then averaged.
To reduce problems arising from baseline shifts, Matlab (The Math Works, Natick, MA) was used to correct for CO2 vibrations by removing the peaks at 2403 to 2272 cm−1 and filling with a trend. To account for any differences in sample thickness, normalization to the total area of spectra was performed. To reduce the dimensionality of the FTIR data, PCA was performed according to the NIPALS algorithm (Wold, 1996).
Analysis of monosaccharides in the noncellulose fraction of the cell wall fraction was performed using gas chromatography of alditol acetates as previously described (Reiter et al., 1993) using l-rhamnose, l-fuctose, l-arabinose, d-xylose, d-mannose, and d-galactose as standards.
Microarray data from this article have been deposited with the ArrayExpress data library under accession number E-MXP-265.
Acknowledgments
We are grateful to Andy Hayes and Leanne Wardleworth at the University of Manchester Microarray Facility for performing the microarray analysis. We would also like to thank Neil Taylor, Raymond Wightman, and Jon Pittman for their comments on the manuscript and Raymond Wightman for his assistance in analyzing the root microarray data (Birnbaum et al., 2003). Work performed by Leo Zeef and Leanne Wardleworth was funded by the Welcome Trust. David Brown was supported by a studentship from the Biotechnology and Biological Science Research Council. R.G. and J.E. are also very grateful to the Biotechnology and Biological Science Research Council for financial support.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantcell.org) is: Simon R. Turner (simon.turner@manchester.ac.uk).
Article, publication date, and citation information can be found at www.plantcell.org/cgi/doi/10.1105/tpc.105.031542.
References
- Alonso, J.M., et al. (2003). Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301 653–657. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408 796–815. [DOI] [PubMed] [Google Scholar]
- Birnbaum, K., Shasha, D.E., Wang, J.Y., Jung, J.W., Lambert, G.M., Galbraith, D.W., and Benfey, P.N. (2003). A gene expression map of the Arabidopsis root. Science 302 1956–1960. [DOI] [PubMed] [Google Scholar]
- Bolstad, B.M., Irizarry, R.A., Astrand, M., and Speed, T.P. (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19 185–193. [DOI] [PubMed] [Google Scholar]
- Bouton, S., Leboeuf, E., Mouille, G., Leydecker, M.T., Talbotec, J., Granier, F., Lahaye, M., Hofte, H., and Truong, H.N. (2002). Quasimodo1 encodes a putative membrane-bound glycosyltransferase required for normal pectin synthesis and cell adhesion in Arabidopsis. Plant Cell 14 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braam, J. (1999). If walls could talk. Curr. Opin. Plant Biol. 2 521–524. [DOI] [PubMed] [Google Scholar]
- Carpita, N., Tierney, M., and Campbell, M. (2001). Molecular biology of the plant cell wall: Searching for the genes that define structure, architecture and dynamics. Plant Mol. Biol. 47 1–5. [PubMed] [Google Scholar]
- Coutinho, P.M., Starn, M., Blanc, E., and Henrissat, B. (2003). Why are there so many carbohydrate-active enzyme-related genes in plants? Trends Plant Sci. 8 563–565. [DOI] [PubMed] [Google Scholar]
- Gardiner, J.C., Taylor, N.G., and Turner, S.R. (2003). Control of cellulose synthase complex localization in developing xylem. Plant Cell 15 1740–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodacre, R., Vaidyanathan, S., Dunn, W.B., Harrigan, G.G., and Kell, D.B. (2004). Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 22 245–252. [DOI] [PubMed] [Google Scholar]
- Goujon, T., Sibout, R., Pollet, B., Maba, B., Nussaume, L., Bechtold, N., Lu, F.C., Ralph, J., Mila, I., Barriere, Y., Lapierre, C., and Jouanin, L. (2003). A new Arabidopsis thaliana mutant deficient in the expression of O-methyltransferase impacts lignins and sinapoyl esters. Plant Mol. Biol. 51 973–989. [DOI] [PubMed] [Google Scholar]
- Guidet, F., Rogowsky, P., Taylor, C., Song, W., and Langridge, P. (1991). Cloning and characterization of a new rye-specific repeated sequence. Genome 34 81–87. [Google Scholar]
- Ha, M.A., MacKinnon, I.M., Sturcova, A., Apperley, D.C., McCann, M.C., Turner, S.R., and Jarvis, M.C. (2002). Structure of cellulose-deficient secondary cell walls from the irx3 mutant of Arabidopsis thaliana. Phytochemistry 61 7–14. [DOI] [PubMed] [Google Scholar]
- Harrigan, G.G., LaPlante, R.H., Cosma, G.N., Cockerell, G., Goodacre, R., Maddox, J.F., Luyendyk, J.P., Ganey, P.E., and Roth, R.A. (2004). Application of high-throughput Fourier-transform infrared spectroscopy in toxicology studies: Contribution to a study on the development of an animal model for idiosyncratic toxicity. Toxicol. Lett. 146 197–205. [DOI] [PubMed] [Google Scholar]
- Holland, N., Holland, D., Helentjaris, T., Dhugga, K.S., Xoconostle-Cazares, B., and Delmer, D.P. (2000). A comparative analysis of the plant cellulose synthase (CesA) gene family. Plant Physiol. 123 1313–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai, H., Masaoka, N., Ishii, T., and Satoh, S. (2002). A pectin glucuronyltransferase gene is essential for intercellular attachment in the plant meristem. Proc. Natl. Acad. Sci. USA 99 16319–16324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.A., and Takemoto, D. (2004). Plant innate immunity: Direct and indirect recognition of general and specific pathogen-associated molecules. Curr. Opin. Immunol. 16 48–62. [DOI] [PubMed] [Google Scholar]
- Jones, L., Ennos, A.R., and Turner, S.R. (2001). Cloning and characterization of irregular xylem4 (irx4): a severely lignin-deficient mutant of Arabidopsis. Plant J. 26 205–216. [DOI] [PubMed] [Google Scholar]
- Kacurakova, M., Belton, P.S., Wilson, R.H., Hirsch, J., and Ebringerova, A. (1998). Hydration properties of xylan-type structures: An FTIR study of xylooligosaccharides. J. Sci. Food Agric. 77 38–44. [Google Scholar]
- Kacurakova, M., Capek, P., Sasinkova, V., Wellner, N., and Ebringerova, A. (2000). FT-IR study of plant cell wall model compounds: Pectic polysaccharides and hemicelluloses. Carbohyd. Polym. 43 195–203. [Google Scholar]
- Kacurakova, M., Smith, A.C., Gidley, M.J., and Wilson, R.H. (2002). Molecular interactions in bacterial cellulose composites studied by 1D FT-IR and dynamic 2D FT-IR spectroscopy. Carbohydr. Res. 337 1145–1153. [DOI] [PubMed] [Google Scholar]
- Kacurakova, M., Wellner, N., Ebringerova, A., Hromadkova, Z., Wilson, R.H., and Belton, P.S. (1999). Characterisation of xylan-type polysaccharides and associated cell wall components by FT-IR and FT-Raman spectroscopies. Food Hydrocolloid 13 35–41. [Google Scholar]
- Lane, D.R., et al. (2001). Temperature-sensitive alleles of RSW2 link the KORRIGAN endo-1,4-beta-glucanase to cellulose synthesis and cytokinesis in Arabidopsis. Plant Physiol. 126 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y.H., Qian, O., Zhou, Y.H., Yan, M.X., Sun, L., Zhang, M., Fu, Z.M., Wang, Y.H., Han, B., Pang, X.M., Chen, M.S., and Li, J.Y. (2003). BRITTLE CULM1, which encodes a COBRA-like protein, affects the mechanical properties of rice plants. Plant Cell 15 2020–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, C.Y., and Marchessault, R.H. (1959). Infrared spectra of crystalline polysaccharides. 2. Native celluloses in the region from 640 to 1700 cm-1. J. Polym. Sci. 39 269–278. [Google Scholar]
- Madson, M., Dunand, C., Li, X.M., Verma, R., Vanzin, G.F., Calplan, J., Shoue, D.A., Carpita, N.C., and Reiter, W.D. (2003). The MUR3 gene of Arabidopsis encodes a xyloglucan galactosyltransferase that is evolutionarily related to animal exostosins. Plant Cell 15 1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley, B.F.J. (1994). Multivariate Statistical Methods: A Primer. (London: Chapman and Hall).
- Meinke, D.W., Meinke, L.K., Showalter, T.C., Schissel, A.M., Mueller, L.A., and Tzafrir, I. (2003). A sequence-based map of Arabidopsis genes with mutant phenotypes. Plant Physiol. 131 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouille, G., Robin, S., Lecomte, M., Pagant, S., and Hofte, H. (2003). Classification and identification of Arabidopsis cell wall mutants using Fourier-transform infrared (FT-IR) microspectroscopy. Plant J. 35 393–404. [DOI] [PubMed] [Google Scholar]
- Nicol, F., His, I., Jauneau, A., Vernhettes, S., Canut, H., and Hofte, H. (1998). A plasma membrane-bound putative endo-1,4-beta-D-glucanase is required for normal wall assembly and cell elongation in Arabidopsis. EMBO J. 17 5563–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen, K.M., Kauppinen, L., and Helariutta, Y. (2004). A weed for wood? Arabidopsis as a genetic model for xylem development. Plant Physiol. 135 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzlaff, A., McInnis, S., Courtenay, A., Surman, C., Newman, L.J., Smith, C., Bevan, M.W., Mansfield, S., Whetten, R.W., Sederoff, R.R., and Campbell, M.M. (2003). Characterisation of a pine MYB that regulates lignification. Plant J. 36 743–754. [DOI] [PubMed] [Google Scholar]
- Persson, S., Wei, H., Milne, J., Page, G.P., and Somerville, C.R. (2005). Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets. Proc. Natl. Acad. Sci. USA 102 8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquemal, J., Lapierre, C., Myton, K., O'Connell, A., Schuch, W., Grima-Pettenati, J., and Boudet, A.M. (1998). Down-regulation of cinnamoyl-CoA reductase induces significant changes of lignin profiles in transgenic tobacco plants. Plant J. 13 71–83. [Google Scholar]
- Ranocha, P., Chabannes, M., Chamayou, S., Danoun, S., Jauneau, A., Boudet, A.M., and Goffner, D. (2002). Laccase down-regulation causes alterations in phenolic metabolism and cell wall structure in poplar. Plant Physiol. 129 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter, W.D., Chapple, C.C.S., and Somerville, C.R. (1993). Altered growth and cell-walls in a fucose-deficient mutant of Arabidopsis. Science 261 1032–1035. [DOI] [PubMed] [Google Scholar]
- Roudier, F., Schindelman, G., DeSalle, R., and Benfey, P.N. (2002). The COBRA family of putative GPI-anchored proteins in Arabidopsis. A new fellowship in expansion. Plant Physiol. 130 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson, F., Brunaud, V., Duchene, S., De Oliveira, Y., Caboche, M., Lecharny, A., and Aubourg, S. (2004). FLAGdb(++): A database for the functional analysis of the Arabidopsis genome. Nucleic Acids Res. 32 D347–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, S., Kato, T., Kakegawa, K., Ishii, T., Liu, Y.G., Awano, T., Takabe, K., Nishiyama, Y., Kuga, S., Nakamura, Y., Tabata, S., and Shibata, D. (2001). Role of the putative membrane-bound endo-1,4-beta-glucanase KORRIGAN in cell elongation and cellulose synthesis in Arabidopsis thaliana. Plant Cell Physiol. 42 251–263. [DOI] [PubMed] [Google Scholar]
- Scheible, W.R., and Pauly, M. (2004). Glycosyltransferases and cell wall biosynthesis: Novel players and insights. Curr. Opin. Plant Biol. 7 285–295. [DOI] [PubMed] [Google Scholar]
- Schindelman, G., Morikami, A., Jung, J., Baskin, T.I., Carpita, N.C., Derbyshire, P., McCann, M.C., and Benfey, P.N. (2001). COBRA encodes a putative GPI-anchored protein, which is polarly localized and necessary for oriented cell expansion in Arabidopsis. Genes Dev. 15 1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama, J., Persson, J., and Chanzy, H. (1991). Combined infrared and electron diffraction study of the polymorphism of native celluloses. Macromolecules 24 2461–2466. [Google Scholar]
- Szyjanowicz, P.M.J., McKinnon, I., Taylor, N.G., Gardiner, J., Jarvis, M.C., and Turner, S.R. (2004). The irregular xylem 2 mutant is an allele of korrigan that affects the secondary cell wall of Arabidopsis thaliana. Plant J. 37 730–740. [DOI] [PubMed] [Google Scholar]
- Tamagnone, L., Merida, A., Parr, A., Mackay, S., Culianez-Macia, F.A., Roberts, K., and Martin, C. (1998). The AmMYB308 and AmMYB330 transcription factors from Antirrhinum regulate phenylpropanoid and lignin biosynthesis in transgenic tobacco. Plant Cell 10 135–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, N.G., Howells, R.M., Huttly, A.K., Vickers, K., and Turner, S.R. (2003). Interactions among three distinct CesA proteins essential for cellulose synthesis. Proc. Natl. Acad. Sci. USA 100 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, N.G., Laurie, S., and Turner, S.R. (2000). Multiple cellulose synthase catalytic subunits are required for cellulose synthesis in Arabidopsis. Plant Cell 12 2529–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins, E.M., Howell, S.A., Alsberg, B.K., Noble, W.C., and Goodacre, R. (1998). Rapid differentiation of closely related Candida species and strains by pyrolysis mass spectrometry and Fourier transform- infrared spectroscopy. J. Clin. Microbiol. 36 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, S.R., and Somerville, C.R. (1997). Collapsed xylem phenotype of Arabidopsis identifies mutants deficient in cellulose deposition in the secondary cell wall. Plant Cell 9 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorwerk, S., Somerville, S., and Somerville, C. (2004). The role of plant cell wall polysaccharide composition in disease resistance. Trends Plant Sci. 9 203–209. [DOI] [PubMed] [Google Scholar]
- Wold, H. (1996). Estimination of principal components and related models by iterative least squares. In Multivariate Analysis, K. Kirshnaiah, ed (New York: Academic Press), pp. 391–420.
- Zimmermann, P., Hirsch-Hoffmann, M., Hennig, L., and Gruissem, W. (2004). GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 136 2621–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]