

Abstract
Ebola virus causes severe hemorrhagic fever with high mortality rates in humans and nonhuman primates. Vascular instability and dysregulation are disease-decisive symptoms during severe infection. While the transmembrane glycoprotein GP1,2 has been shown to cause endothelial cell destruction, the role of the soluble glycoproteins in pathogenesis is largely unknown; however, they are hypothesized to be of biological relevance in terms of target cell activation and/or increase of endothelial permeability. Here we show that virus-like particles (VLPs) consisting of the Ebola virus matrix protein VP40 and GP1,2 were able to activate endothelial cells and induce a decrease in barrier function as determined by impedance spectroscopy and hydraulic conductivity measurements. In contrast, the soluble glycoproteins sGP and Δ-peptide did not activate endothelial cells or change the endothelial barrier function. The VLP-induced decrease in barrier function was further enhanced by the cytokine tumor necrosis factor alpha (TNF-α), which is known to induce a long-lasting decrease in endothelial cell barrier function and is hypothesized to play a key role in Ebola virus pathogenesis. Surprisingly, sGP, but not Δ-peptide, induced a recovery of endothelial barrier function following treatment with TNF-α. Our results demonstrate that Ebola virus GP1,2 in its particle-associated form mediates endothelial cell activation and a decrease in endothelial cell barrier function. Furthermore, sGP, the major soluble glycoprotein of Ebola virus, seems to possess an anti-inflammatory role by protecting the endothelial cell barrier function.
Ebola virus (EBOV) causes a severe hemorrhagic disease in humans and nonhuman primates, with mortality rates as high as 89% (14, 33). The hemorrhagic disease caused by EBOV is characterized by generalized fluid distribution problems, hypotension, coagulation disorders, and a tendency to bleed, finally resulting in fulminant shock. Vascular instability and dysregulation are hallmarks of the pathogenesis in EBOV hemorrhagic fever (HF). Endothelial disturbances can be caused indirectly, by proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α) released from EBOV-infected monocytes/macrophages, and directly, following virus infection of endothelial cells. Additionally, the EBOV transmembrane and soluble glycoproteins are regarded as major viral pathogenic determinants and are also thought to contribute to vascular dysregulation (8, 14, 16, 38, 47, 52, 63).
All glycoprotein forms are encoded by gene 4 of the EBOV genome. The primary product of this gene (80% of the transcripts) is a precursor of the nonstructural soluble glycoprotein (pre-sGP) that is posttranslationally cleaved by furin or a furin-like endoprotease into mature sGP and Δ-peptide, both of which are secreted products (54, 55). In addition to being observed in vitro, sGP has been detected in the blood of infected patients (34). RNA editing is necessary to express the precursor of the structural type I transmembrane glycoprotein (GP1,2) (34, 50). The precursor is posttranslationally cleaved by furin or a furin-like endoprotease into the disulfide-linked fragments GP1 and GP2 (35, 51, 53). The homotrimeric GP1,2 forms the spikes on the virus particle and is indispensable for receptor binding and fusion with the host cell membrane (18, 48, 59). In vitro studies have demonstrated that the full-length GP1,2 is cytotoxic to endothelial cells and thus may contribute to endothelial damage during EBOV HF (8, 52, 63). More recently it was demonstrated that a mutant EBOV lacking the editing site showed increased cytotoxicity, suggesting that editing might be a mechanism to regulate EBOV GP1,2 cytotoxicity (52). The active role of sGP has not been sufficiently elucidated. Previously it was suggested that sGP interacts with and inactivates neutrophils through binding to CD16b (23, 62), a concept that has been challenged by others (27). Due to the relatively high expression level of sGP during infection, its role in effectively binding antibodies that might otherwise be protective and/or in functioning as a mediator in the activation of target cells has been discussed (16, 34, 47). However, the functional role of the soluble glycoproteins in vascular dysregulation such as increased endothelial permeability, hemorrhage, and shock remains largely unknown (14, 38).
The vascular endothelium has multiple functions in maintaining homeostasis. It forms a unique selective barrier between the blood and tissue, controlling exchange of solutes and water. Balance of bodily solutes between blood and tissue and fluid homeostasis are regulated in two ways. Transport of macromolecules, such as albumin, through the endothelium follows a transcellular pathway (29). At the same time, water and small-solute exchange occurs by a paracellular route through endothelial junctions preferred in capillaries and postcapillary venules (2, 28). Under physiological conditions the intercellular junctions are impermeable to macromolecules. In inflamed tissue, the endothelial barrier function is decreased due to reduced intercellular adhesion, leading to enhanced extravasation of small solutes and water that causes edema formation (2, 28, 40). In severe cases intercellular gap formation, allowing extravasation of larger macromolecules and water into the interstitial space and further contributing to the severity of the pathological condition, can be observed (28).
The functional integrity of the endothelium is provided by endothelial adherens, tight, and gap junctions (5, 9, 38, 49). The composition of endothelial junctions is heterogeneous within different organs and within the microvascular bed (45). The common distribution of adherens junctions throughout the vascular bed makes them crucial structures for regulating the paracellular barrier function, particularly in small vessels. The postcapillary venules, displaying mainly adherens-type junctions, are the target site for increased paracellular permeability (2, 6, 28) and leukocyte extravasation during inflammation (10, 26, 49) and EBOV infection (13, 14, 36). The backbone of adherens junctions is formed by transmembrane vascular endothelial cadherin (VE-cadherin, also known as cadherin-5). VE-cadherin interacts with β- and γ-catenins, and this complex is connected via α-catenin to the actin filament cytoskeleton, which is crucially involved in the regulation of the endothelial barrier function (5, 9, 37, 57). An earlier study has shown that stimulation with tissue culture supernatants derived from Marburg virus-infected monocytes/macrophages caused a reorganization of the VE-cadherin-catenin complex and was associated with intercellular gap formation (13). This phenomenon is comparable to changes observed under stimulation of endothelial cells with inflammatory mediators such as TNF-α (58). While the molecular mechanisms for the breakdown of endothelial barrier function are not completely understood, it is clear that the VE-cadherin-catenin complex and actin filaments are crucially involved. Increases in endothelial permeability are associated with changes in both VE-cadherin organization and formation of actin filament stress fibers that are able to contract, thereby providing the driving force for intercellular gap formation (5, 37, 57). The vascular endothelium also directs immune responses through the induction of cytokines, chemokines, and cellular receptors that recruit or activate immune cells. Proinflammatory cytokines (e.g., TNF-α, IFN-γ) activate the endothelium, causing expression of cell adhesion molecules (CAMs) (e.g., E-selectin, ICAM-1, VCAM-1). These molecules mediate the initial steps in leukocyte transmigration and mediate rolling and tight adhesion of leukocytes to the endothelium (46, 49, 60).
In the present paper we address the question of whether the soluble EBOV glycoproteins, sGP and Δ-peptide, influence endothelial activation and endothelial barrier function. We found that neither sGP nor Δ-peptide was able to activate endothelial cells or cause significant changes in barrier function. In contrast, virus-like particles (VLPs) expressing the transmembrane glycoprotein GP1,2 at the surface and thus mimicking the morphology of virus particles were able to activate endothelial cells and induce a moderate but sustained decrease in barrier function. Most interestingly, sGP displayed an unexpected protective role in endothelial layer integrity when endothelial cells were treated with TNF-α, suggesting a possible anti-inflammatory role of sGP during EBOV HF.
(This work was performed by V. M. Wahl-Jensen in partial fulfillment of the thesis requirement for a Ph.D. from the University of Manitoba, Winnipeg, Manitoba, Canada, 2004.)
MATERIALS AND METHODS
Cell culture and virus.
Endothelial cells were isolated from human umbilical veins and cultured as previously described (40). Cells were passaged once and grown to confluence in endothelial growth medium (Promocell, Heidelberg, Germany). All culture dishes were coated with cross-linked gelatin as described elsewhere (39). The human embryonic kidney cell line 293T and Vero E6 cells (ATCC 1568) were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, l-glutamine (2 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml). All cells were incubated at 37°C in a humidified 5% CO2 environment. Zaire ebolavirus (ZEBOV) strain Mayinga was kindly provided by the Special Pathogens Branch, Centers for Disease Control and Prevention, Atlanta, Ga. Virus stocks were freshly prepared in Vero E6 cells using Dulbecco's modified Eagle medium with 2% fetal calf serum. Harvesting was performed at a time when no obvious cytopathic effect was seen, in order to limit contamination with released cellular proteins. Mock-infected Vero E6 cells were treated the same way in order to prepare a control (mock stock). Virus titration was performed as previously described (47).
Infection of endothelial cells.
Confluent monolayers of human endothelial cells on coverslips were either mock infected or infected with ZEBOV at a multiplicity of infection of 10 (diluted in endothelial basal medium). After an adsorption period of 1 h, the inoculum was replaced with new medium (endothelial basal medium containing 5% human serum) and the cultures were incubated for various times at 37°C in a CO2 incubator.
Production and purification of recombinant proteins and VLPs.
Influenza virus hemagglutinin epitope-tagged (HA-tagged) recombinant ZEBOV sGP and Δ-peptide were generated through transient transfection and purified as described previously (56). Briefly, soluble glycoproteins were expressed in 293T cells and immunoaffinity purified using the HA tag. The dimeric and monomeric conformations of sGP and Δ-peptide, respectively, were conserved and were similar to those of wild-type proteins produced during virus infection (56). Purified proteins were quantitated using the DC protein assay (Bio-Rad, Mississauga, Ontario, Canada). ZEBOV VLPVP40/GP and VLPVP40 were generated as previously described by transient transfection of 293T cells with a plasmid(s) encoding ZEBOV VP40 and/or EBOV GP1,2 and quantitated by electron microscopy particle counts and a DC protein assay (56). Prior to use, all recombinant glycoproteins, VLPs, and media were analyzed for endotoxin presence using the Limulus amebocyte lysate test (BioWhittaker, Walkersville, Md.) according to the manufacturer's instructions. The relative molar concentrations of glycoproteins were calculated as follows: sGP, 6 × 1012 molecules per μg; Δ-peptide, 4.3 × 1013 molecules per μg; GP1,2 in VLPVP40/GP, 9 × 105 molecules per particle.
Treatment of endothelial cells with proteins and VLPs.
Recombinant proteins or VLPs were added to the culture medium of confluent endothelial cells at 10-μg/ml or 50-μg/ml quantities for proteins or at a 1:1 ratio of VLPs to cells. Cells were incubated at 37°C for 6, 12, or 24 h posttreatment in a 95% humidified, 5% CO2 environment. Recombinant human TNF-α (100 ng/ml) (R&D Systems, Minneapolis, Minn.) served as a positive control, while HA-peptide (Roche, Laval, Quebec, Canada), used to elute the HA-tagged soluble glycoproteins from the affinity column, and purified supernatants of control vector-transfected cells served as negative controls.
Immunofluorescence assays.
For immunofluorescence analysis, polyclonal goat antibodies to ICAM-1 and VCAM-1, mouse monoclonal antibodies to E-selectin and PECAM-1 (all from R&D Systems, Minneapolis, Minn.), and a monoclonal antibody to VE-cadherin (BD Biosciences, Heidelberg, Germany) were used. After the appropriate treatment, endothelial cells were fixed with 2% formaldehyde followed by permeabilization with 0.1% Triton X-100 for 10 min at room temperature. Cells were incubated with primary antibodies at a dilution of 1:100 overnight at 4°C. A corresponding secondary antibody conjugated to Alexa 488 (Molecular Probes, Eugene, Oreg.) or Cy3 (Jackson ImmunoResearch Laboratories, Bar Harbor, Maine) was applied for 1 h at a 1:400 dilution. Actin was visualized by incubating the cells with phalloidin conjugated with tetramethyl rhodamine isocyanate (TRITC; Sigma, Deisenhofen, Germany) for 30 min at room temperature. Stained cells were postfixed with 2% paraformaldehyde and analyzed using a Zeiss microscope.
RT-PCR.
Endothelial cells were treated with VLPs or mock supernatants as described above. At 0, 6, 12, and 24 h, cells were disrupted using the guanidinium isothiocyanate-based RLT buffer of an RNeasy Mini kit (QIAGEN, Hilden, Germany). RNA was isolated according to the protocol for animal cells provided by the manufacturer. cDNA was synthesized using a First Strand cDNA synthesis kit for reverse transcription-PCR (RT-PCR) (Roche, Mannheim, Germany) according to the manufacturer's instructions followed by conventional PCR with the following primers: ICAM-1 fwd, 5′-CAGTGACTGTCACTCGAGATCT-3′; ICAM-1 rev, 5′-CCTCTTGGCTTAGTCATGTGAC-3′; VCAM-1 fwd, 5′-CTGGAG GATGCAGACAGGAAG-3′; VCAM-1 rev, 5′-CCAATCTGAGCAGCAATCCGG-3′; E-sel fwd, 5′-AGT GGCCACGGTGAATGTGTA-3′; E-sel rev, 5′-CCCAGATGAGG TACACTGAAG-3′; GAPDH fwd, 5′-GGTTTTTCTAGACGGCAGGTCA-3′; GAPDH rev, 5′-TGGCAAA TTCCATGGCACCGTCA-3′. The PCR included 94°C for 30 s, 65°C for 30 s, and 72°C for 1 min for 19 to 22 cycles, with a final elongation of 7 min at 72°C.
Impedance spectroscopy.
Chambered slides for impedance spectroscopy were prepared by applying photosensitive lacquer (CRC Industries, Iffenzheim, Germany) on indium tin oxide (ITO)-coated polyester film (75 mm by 25 mm by 2 mm) (Delta Technologies Ltd., Stillwater, Minn.) Electrode areas were generated by exposing masked ITO slides to UV light for 3 min. Electrode areas were cleared of lacquer by treatment with a sodium hydroxide solution (0.7%, wt/vol). Chambers removed from LabTek slides (Nunc International, Naperville, Ill.) were fixed to the ITO with silicon, coated with cross-linked gelatin as described above, and seeded with endothelial cells. The transendothelial electrical resistance (TER) was determined as previously described (42). Briefly, an alternating voltage was applied, and the impedance magnitude was measured at frequencies between 10 Hz and 1 MHz between the electrode area of the ITO slide and a counterelectrode. The TER was calculated from the resultant spectra (42). Prior to addition of secreted glycoproteins or VLPs, the cells were allowed to equilibrate for approximately 1 h or until a constant TER spectrum was observed in order to establish a baseline resistance. Only the endothelial layers displaying more then 10 Ω cm2 were used for experiments. At the termination of all experiments, a 3 mM solution of EGTA, which induces a decrease in TER, was added to all wells as a control. All electrical resistance data are presented as normalized to baseline resistance values (TER/TER0). TER data are shown as means ± standard errors. Data were compared by analysis of variance in conjunction with Bonferroni's adjustment (SigmaStat; SPSS, Erfurt, Germany). Values were considered to be statistically significant when P was <0.05.
Transwell filter tracer assay and hydraulic conductivity.
The transwell filter tracer assays were performed on Costar filters (diameter, 6.5 mm; pore size, 0.4 μm) (Costar, Corning, N.Y.) by using fluorescein isothiocyanate (FITC)-labeled dextran (Sigma, Deisenhofen, Germany) as the tracer substance as described elsewhere (13). Briefly, endothelial cells were treated with VLPs, and 0.5 μg/ml of FITC-dextran (4 kDa, neutral charge) was added to the upper compartment. Following an incubation time of 0, 3, 10, or 20 h, the FITC-dextran was measured in the lower compartment of the transwell filter system by fluorescence photometry. All steps were performed at 37°C.
Hydraulic conductivities of VLP-treated endothelial cell monolayers were measured and calculated as described elsewhere (40). Briefly, cells were cultured on commercially available Costar (Corning, N.Y.) polycarbonate filters (pore size, 0.4 μm; diameter, 6.5 mm). Endothelial cells were treated with VLPVP40/GP or VLPVP40, and after 20 h a hydrostatic pressure of 10 cm of water column was applied to the upper side of the transwell filter system using a glass tube inserted into the filter. Water flux (Jv,) was measured for the first 30 min, and hydraulic conductivity (Lp) was calculated according to Starling's equation, Lp = Jv · A−1 · ΔP−1, where A indicates the filter area (in cm2) and ΔP the applied hydrostatic pressure (cm of water column). Constant hydrostatic pressure was maintained by medium adjustment to the upper compartment of the filter. Hydraulic conductivity was expressed as cm · s−1 · cm H2O−1. Measurements were performed in four independent experiments.
RESULTS
Activation of endothelial cells by Ebola virus glycoproteins.
It was recently reported that supernatants from ZEBOV-infected Vero E6 cells, but not mock-infected supernatants, were capable of activating primary human endothelial monolayers (20). This activation was demonstrated by transcriptional upregulation of the CAMs ICAM-1 and VCAM-1, although expression of these and other CAMs on the surfaces of cells was not addressed. Whether this activation could be the result of an interaction between cellular receptors and ZEBOV soluble glycoproteins or particle-associated GP1,2, independent of soluble host factors, remained uninvestigated. Therefore, we first investigated the upregulation of specific cell markers following EBOV infection of endothelial cells. Endothelial cells were infected with ZEBOV at a multiplicity of infection of 10. Expression of ICAM-1, VCAM-1, and E-selectin was demonstrated on the transcriptional level by RT-PCR (data not shown) and on the protein level by indirect immunofluorescence staining 12 h (Fig. 1) and 24 h postinfection. These results confirmed previously published data (20) but additionally demonstrated the expression of CAMs on the surfaces of infected endothelial cells.
FIG. 1.

EBOV-induced expression of CAMs in primary endothelial cells. Mock-infected (controls) and ZEBOV-infected endothelial cells were fixed at 12 h and stained with monoclonal antibodies to E-selectin, ICAM-1, and VCAM-1. Original magnification, ×400.
To specifically determine the ability of ZEBOV secreted glycoproteins to activate primary endothelial cells, we expressed, characterized, and purified the major secreted glycoproteins, sGP and Δ-peptide. Both proteins were determined to be authentic proteins produced during natural virus infection (56). To assess the ability of particle-associated GP1,2 to induce activation, we simultaneously transfected VP40 and/or GP1,2 plasmids into 293T cells using a previously established protocol to generate ZEBOV VLPVP40/GP and VLPVP40, the latter of which served as a control (Fig. 2C) (56). Additionally, VLPs were characterized by electron microscopy (data not shown). All preparations were tested for the presence of endotoxin, and levels were found to universally occur below 0.21 endotoxin unit/ml, values equivalent to or less than that of media used in all experiments.
FIG. 2.
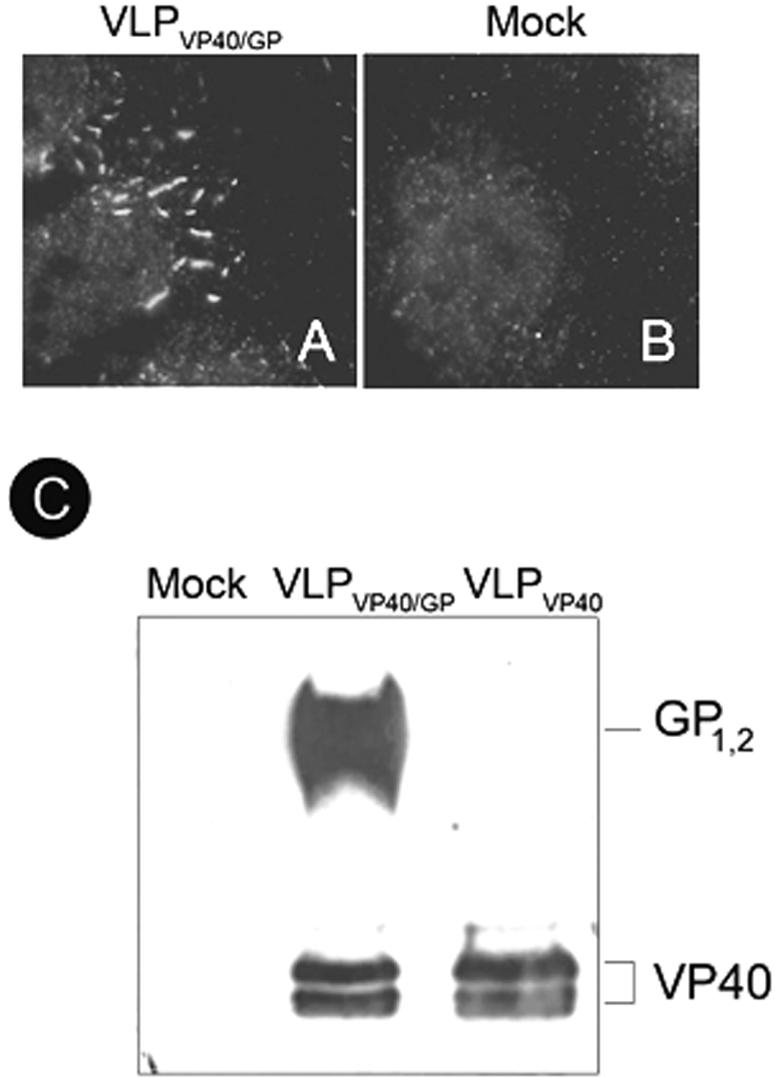
Adhesion of virus-like particles to endothelial cells. Endothelial cells were treated with purified VLPVP40/GP for 1.5 h, washed extensively, and stained with a monoclonal antibody directed against the transmembrane glycoprotein GP1,2 (ZGP12/1.1). Binding of VLPVP40/GP to endothelial cells (A) is shown in comparison to a control (Mock) (B). Original magnification, ×1,000. The different VLP preparations were characterized by Western blotting using anti-GP1,2 and anti-VP40 antibodies (C), indicating the absence of GP1,2 in the VLPVP40.
In order to further dissect whether soluble glycoproteins play a role in the upregulation of CAMs and whether replication is needed for the activation, we treated endothelial cells with either VLPs (1:1 ratio with cells) or secreted glycoproteins (10 μg/ml or 50 μg/ml) for 6, 12, or 24 h; 100 ng/ml of TNF-α was used as a positive control to demonstrate endothelial activation. Like live virus, VLPVP40/GP bound to endothelial cells, as demonstrated by immunofluorescence analysis (Fig. 2A and B), and strongly activated endothelial cells, as measured by upregulation of ICAM-1, VCAM-1, and E-selectin (Fig. 3A and B). Neither sGP nor Δ-peptide caused upregulation of ICAM-1, VCAM-1, or E-selectin over a time period of 24 h at any concentration tested (50 μg/ml and 10 μg/ml) (Fig. 3A, shown for 12 h after treatment with 50 μg/ml of glycoproteins). The time kinetics of endothelial activation were studied on the transcriptional level by RT-PCR using RNA isolated from cells either treated with the VLPs, mock treated, or treated with the soluble glycoproteins. In VLPVP40/GP-treated cells, transcripts for ICAM-1, VCAM-1, and E-selectin appeared early, with maximal levels of expression at 6 h, and declined thereafter (Fig. 3B). In contrast, sGP and Δ-peptide (data not shown) as well as VLPVP40 (Fig. 3C, shown for E-selectin expression) did not upregulate the expression of CAMs, confirming the protein expression data shown in Fig. 3A. These results demonstrated that replication is not necessary to induce activation and that activation is induced by VLPs containing GP1,2.
FIG. 3.

Endothelial cell activation through soluble glycoproteins and virus-like particles. (A) Endothelial cells were treated with soluble glycoproteins (sGP or Δ-peptide) at a concentration of 50 μg/ml or with VLPVP40/GP (1 particle/cell). Twelve hours posttreatment, cells were fixed and permeabilized, and activation was measured by immunofluorescence analysis using monoclonal antibodies directed against E-selectin, VCAM-1, and ICAM-1. TNF-α was used as a positive control (100 ng/ml). PECAM-1 staining was used to confirm the integrity of the endothelial monolayer. Only TNF-α and VLPVP40/GP induced expression of CAMs. Magnification, ×400. (B and C) The kinetics of VLP-induced endothelial cell activation was further assessed on the transcriptional level by RT-PCR using primers described in Materials and Methods. Cells were either treated with VLPVP40/GP or VLPVP40 (1 particle/cell) or mock treated and harvested for RNA isolation at the indicated time points (hours). Transcription of CAMs was clearly upregulated, with maximal levels at 6 h, after VLPVP40/GP (B) but not mock (B) or VLPVP40 (C) treatment.
Change of barrier function by Ebola virus glycoproteins.
In order to directly assess the abilities of ZEBOV sGP and Δ-peptide as well as VLPs to affect the endothelial cell barrier function, we used impedance spectroscopy to detect changes in TER. Endothelial cells were cultured on electrodes, secreted glycoproteins and VLPs were added to the confluent monolayers, and the TER was recorded over a period of 24 h. Even at high concentrations (50 μg/ml), sGP and Δ-peptide were unable to cause long-lasting changes in endothelial barrier function (Fig. 4A). The rapid transient drop in TER immediately after administration of sGP was concentration independent and therefore seems to be nonspecific. This was followed by a slightly prolonged TER decrease that was not statistically significant. The application of VLPVP40/GP decreased the barrier function within the first 1 to 2 h (Fig. 4B) and was maintained at a 17% decrease for the duration of the experiment. This decrease in TER was found to be statistically significant (P < 0.05), and the extent of the decrease was comparable to that for endothelial cell treatment with TNF-α at concentrations ranging from 0.1 to 1 ng/ml (Fig. 4C).
FIG. 4.

Effects of soluble glycoproteins, virus-like particles, and TNF-α on endothelial barrier function. Confluent endothelial cell monolayers were treated with soluble glycoproteins (A), VLPs (1 particle/cell) (B), or different doses of TNF-α (C), and the TER was measured using impedance spectroscopy. Treatment of endothelial cells with sGP administered at 10 or 50 μg/ml or with Δ-peptide administered at 50 μg/ml showed no significant long-lasting changes in TER (A). Purified HA-peptide served as a negative control and was added at an equivalent concentration. In contrast to VLPVP40- and mock-treated endothelial cells, treatment with VLPVP40/GP resulted in a decrease in the endothelial barrier function (B). Change in barrier function was significant (P < 0.05) from the time points marked by asterisks. In addition, confluent endothelial monolayers were treated with increasing concentrations of TNF-α, and a dose-dependent decrease in TER was observed (C).
To further characterize the permeability-decreasing effect of VLPVP40/GP, we measured endothelial permeability by two additional methods using a transwell filter system. Firstly, we used a 4-kDa FITC-dextran tracer to investigate macromolecular permeability through an endothelial monolayer grown on a filter. We did not observe a significant increase in FITC-dextran permeability following treatment with VLPs (data not shown). However, determination of the hydraulic conductivity clearly indicated a nearly threefold increase in water permeability following treatment of endothelial cells with VLPVP40/GP but not VLPVP40 (Fig. 5). The results indicate that VLPVP40/GP is able to increase endothelial permeability for water but not macromolecules and are in accordance with the moderate TER increase demonstrated in Fig. 4.
FIG. 5.
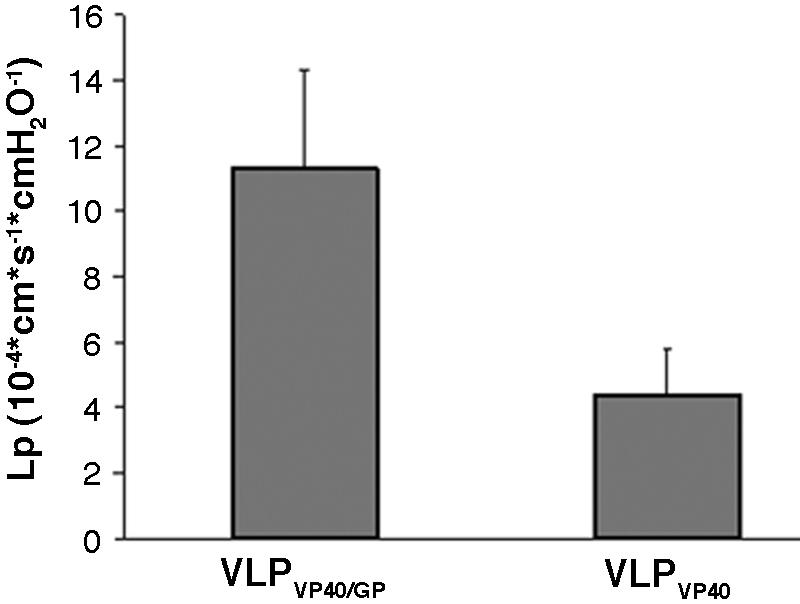
Effect of virus-like particles on hydraulic conductivity (Lp) of endothelial cells. Confluent endothelial cells cultured on polycarbonate filter membranes were treated with VLPVP40/GP or VLPVP40. In contrast to VLPVP40, VLPVP40/GP increased the hydraulic conductivity approximately threefold.
In order to investigate if the change in barrier function was associated with a redistribution of the adherens-type junction proteins, VE-cadherin, PECAM-1, and the actin filament system were visualized after treatment of endothelial monolayers with VLPVP40/GP (1:1 ratio to cells) or different concentrations (10 μg/ml [data not shown] or 50 μg/ml) of the soluble glycoprotein sGP or Δ-peptide for 24 h. Subsequently, cells were fixed and stained for VE-cadherin and actin filaments by using a VE-cadherin monoclonal antibody and TRITC-labeled phalloidin specific for actin filaments, respectively. Neither of the two secreted glycoproteins caused changes in VE-cadherin arrangement or actin reorganization (Fig. 6), effects that are typically seen after treatment of endothelial cells with harsh proinflammatory mediators such as thrombin or high concentrations of TNF-α. VLPVP40/GP treatment did not result in a marked VE-cadherin rearrangement but caused a moderate increase in actin stress fiber formation (Fig. 6). This probably contributes to the moderate changes in barrier function observed by impedance spectroscopy and hydraulic conductivity (Fig. 4 and 5).
FIG. 6.

Effects of soluble glycoproteins and virus-like particles on VE-cadherin and actin distribution in endothelial cells. Confluent endothelial monolayers were treated with either 50 μg/ml sGP or Δ-peptide, VLPVP40/GP (1 particle/cell), or TNF-α (1 ng/ml or 100 ng/ml), which served as a positive control. Only TNF-α at 100 ng/ml was able to cause morphological rearrangement of VE-cadherin and actin stress fibers (arrows). VLPVP40/GP and TNF-α at lower concentrations (1 ng/ml) caused a moderate increase in actin filament stress fiber formation. In control cells the junction-associated actin filaments were seen (arrowheads). Magnification, ×400.
Combined effects of viral glycoproteins and TNF-α on endothelial barrier function.
During an EBOV infection, the endothelium of the host is targeted by cytokines produced by activated monocytes/macrophages (primary target cells) (14) and soluble viral glycoproteins that are released into the bloodstream (34). Thus, it seems likely that the endothelial barrier function is affected as a result of a combined function of all these different cellular and viral mediators. Therefore, we tested the effects of sGP and Δ-peptide on endothelial cell integrity in the presence of TNF-α using impedance spectroscopy. We first determined the amount of TNF-α that produces a moderate but reliable decrease in endothelial cell barrier function (Fig. 4C). For the studies with the soluble glycoproteins, we used 1 ng/ml TNF-α, a dose that reproducibly decreased the barrier function by approximately 30%.
When administered simultaneously, VLPVP40/GP and TNF-α had an additive effect on the decrease in endothelial barrier function that was not observed with VLPVP40 (Fig. 7A). This indicates the importance of particle-associated GP1,2 for this process. As demonstrated above (Fig. 4A), sGP did not affect the integrity of the endothelial cell monolayer within 24 h of incubation. However, when administered simultaneously with TNF-α, sGP reversed the barrier-decreasing effect of TNF-α by about 20% and thus allowed for recovery of the endothelial monolayer (Fig. 7B). This effect was statistically significant after 17 h (P < 0.05). Furthermore, this effect was sGP specific, since neither VLPVP40/GP (Fig. 7A) nor Δ-peptide (data not shown) had a protective effect on endothelial cell barrier function if applied simultaneously with TNF-α.
FIG. 7.

Effect of simultaneous application of TNF-α and VLPs or sGP on endothelial cells. (A) Endothelial cells were treated with TNF-α (1 ng/ml) and VLPs (1 particle/cell) or sGP (10 μg/ml). Simultaneous application of TNF-α and VLPVP40/GP led to further decreases in TER (approximately 15%), indicating their additive effect on the barrier function. (B) Simultaneous treatment with TNF-α and sGP led to a 15% to 20% recovery of the TNF-α-induced changes in barrier function. Changes in TER were significant (P < 0.05) from the time point marked by asterisks.
DISCUSSION
Of all the viral hemorrhagic fevers, those caused by EBOV are the most severe (14, 33). The dramatic clinical presentation in humans and nonhuman primates as well as extensive laboratory data generated in the past has led to the idea that filovirus-induced disease is as much an immune syndrome as a vascular disease (14, 38). While the clinical picture has become clearer through in vivo experimental studies, the molecular mechanisms, particularly with respect to vascular dysregulation, remain elusive. Although vascular instability and dysregulation are thought to be disease-decisive symptoms, endothelial cells are largely considered secondary target cells during EBOV infection. Primary replication occurs within monocytes/macrophages and dendritic cells (20, 22, 47), which become activated upon infection and produce active mediator molecules such as proinflammatory cytokines and chemokines but also release soluble viral glycoproteins, particularly sGP, which has been detected in the plasma of human patients (34). The pathogenic role of secreted cytokines in EBOV pathogenesis has been investigated in the past (3, 20, 22, 24, 47), but little is known regarding the role of the secreted glycoproteins in endothelial dysfunction. Nevertheless, it has long been hypothesized that the secreted glycoproteins, particularly sGP and Δ-peptide, may play an important role during EBOV pathogenesis and can function as mediators of endothelial and immune dysregulation (13, 15, 19).
In the past several studies have morphologically described the involvement of the endothelium in experimentally infected nonhuman primates and rodent models as well as in postmortem human material (4, 19, 31, 32, 64). Destruction of endothelial cells could be found only in postmortem human material (64) and some nonhuman primate models (30-32), but all investigators concluded that endothelial cell function was impaired during infection. To investigate the influence of the soluble and transmembrane glycoproteins on endothelial cell activation and barrier function, we used immunofluorescence analyses and RT-PCR as well as impedance spectroscopy and transwell filter systems, respectively. Impedance spectroscopy is a highly sensitive biophysical assay that provides a unique possibility to study the endothelial barrier function under resting (25) and shear stress conditions with high time resolution (11, 42). This technique determines the TER of a cultured endothelial cell monolayer and predominantly reflects the changes in paracellular permeability (42). Endothelial cell barrier function is frequently studied in transwell filter systems by analyzing the passage of tracer substances (10, 12, 13). In contrast to impedance spectroscopy, which reliably detects changes in barrier function of about 2% (42), tracer systems are limited in time resolution and sensitivity and thus allow for the detection of strong effects only.
In the present work we clarified the long-standing question as to whether the EBOV secreted glycoproteins sGP and Δ-peptide are able to influence endothelial cell functions. We determined that, in addition to its ability to induce transcriptional activation of ICAM-1 and VCAM-1 (20), ZEBOV infection of endothelial cells resulted in surface expression of those molecules as well as of E-selectin (Fig. 1). We found that, in contrast to sGP and Δ-peptide, VLPVP40/GP, produced by transfection of VP40 and GP1,2, and nearly identical in morphology to infectious virus particles, activated endothelial cells in culture and decreased the endothelial cell barrier function (Fig. 3, 4, and 5). These effects were not observed using VLPs lacking GP1,2 (VLPVP40), indicating the importance of the transmembrane glycoprotein GP1,2 in the context of a virion particle for endothelial cell activation (Fig. 3, 4, and 5). The permeability-increasing effect of VLPVP40/GP might allow for extravasation of small solutes and water and thus might contribute to the development of edema and shock observed during EBOV HF. The moderate permeability increase was confirmed by a moderate change in endothelial cell morphology following VLPVP40/GP treatment, as indicated by largely intact VE-cadherin staining along the cell junctions but formation of actin stress fibers (Fig. 6). A similar morphological change associated with permeability increase was also observed after treatment of endothelial cells with low concentrations of TNF-α (1 ng/ml) (Fig. 4 and 6). The data are in agreement with our earlier studies showing that Marburg virus infection did not enhance macromolecular permeability in a transwell filter system (13).
Geisbert et al. (20) recently demonstrated upregulation of mRNA transcripts of several genes associated with endothelial cell activation, including cyclooxygenase-2, inducible nitric oxide synthase, ICAM-1, and VCAM-1, following ZEBOV infection of endothelial cells. At the same time, gamma-irradiated EBOV almost failed to activate endothelial cells (only cyclooxygenase-2 mRNA transcripts were upregulated), and it was therefore proposed that endothelial cell activation was dependent on virus replication. This raises the question as to whether or not VLPs and virus subjected to gamma irradiation, a method commonly used to inactivate infectious high-containment agents, can be used interchangeably to represent replication-deficient virus particles. It is known that gamma irradiation changes structural features of proteins dramatically, particularly at high doses (17, 21). For example, alpha-crystallin activity was reduced by 40% after a dose of 4,000 Gy, and a dose of 60,000 Gy was used to inactivate EBOV (20). Thus, it seems reasonable to assume that the structure of glycoproteins on gamma-irradiated EBOV may be altered, resulting in insufficient binding of virus particles to endothelial cells and, in turn, insufficient activation, an explanation that still needs to be addressed experimentally.
During an EBOV infection, large amounts of proinflammatory cytokines are secreted from infected primary target cells (20, 22, 47), with the level of TNF-α in the blood of infected patients reaching 5 to 7 ng/ml. Therefore, it is reasonable to assume that the endothelial barrier function during EBOV infection may be affected as a result of a combined action of different cellular and viral mediators. To verify this hypothesis, we studied the effect of the primary soluble glycoprotein sGP on endothelial barrier function in the presence of TNF-α (Fig. 7B). In order to detect potential cumulative effects on endothelial cell barrier function, we used a TNF-α concentration of 1 ng/ml, which induced an approximately 30% decrease in barrier function (Fig. 4C). The moderate effect of VLPVP40/GP on endothelial barrier function was further enhanced in the presence of TNF-α, thus contributing to the severity of endothelial damage even in the absence of direct endothelial cell infection. Surprisingly, when sGP was administered simultaneously with TNF-α, it caused a recovery of endothelial cell barrier function starting after approximately 10 h (Fig. 7B). This finding is intriguing and suggests that sGP may have an anti-inflammatory role in the course of EBOV pathogenesis. During EBOV infection, areas of focal tissue destruction can be seen in multiple organs. Interestingly, these areas largely lack infiltration of leukocytes, although neutrophil aggregation within the vascular system is observed in infected nonhuman primates (31, 32). This might indicate that the activation of the endothelium with the recruitment of neutrophils occurs but that the transmigration process is blocked or impaired, which could be related to the observed anti-inflammatory effect of sGP. TNF-α plays a pivotal role in establishing and orchestrating inflammatory and immune responses to infection (1, 43). As a consequence, many viruses have evolved countermeasures, such as some of the large DNA viruses (i.e., poxviruses) that encode immunomodulatory proteins which directly inhibit or modify antiviral activities of proinflammatory cytokines (7, 41). Future studies will have to address the mechanism behind the potential anti-inflammatory action of sGP. In particular, it must be clarified whether the effect is specific for TNF-α and, if so, whether sGP directly interferes with TNF-α or components of the TNF signaling pathway. In this study we used a TNF-α concentration (1 ng/ml) lower than that observed in vivo during infection, but the effect of TNF-α on endothelial cells in vivo can be less pronounced. It was shown that under shear stress, which resembles in vivo physiological conditions, endothelial cells are less responsive to TNF-α (44, 61). Thus, it would also be of interest to determine the effect of sGP in vivo.
In conclusion, we demonstrated that neither of the two major EBOV secreted glycoproteins, sGP and Δ-peptide, was responsible for endothelial cell activation and/or reduced endothelial cell barrier function. In contrast, VLPs containing the EBOV transmembrane glycoprotein GP1,2 were potent activators of endothelial cells and also induced changes in endothelial cell barrier function, indicating that the transmembrane glycoprotein GP1,2 in the context of a virus particle is an important pathogenic determinant. Interestingly, sGP was found to modulate the endothelial cell inflammatory response, a phenomenon that might play a critical role in EBOV pathogenesis.
Acknowledgments
The excellent technical help of Sylvia Grossklaus and Christine Mund is gratefully acknowledged. We thank Ayato Takada for kindly providing the monoclonal antibody ZGP12/1.1 and Rainer Koch for assistance in statistical evaluations.
This study was supported by grants from the Canadian Institutes of Health Research (CIHR) (MOP-43921, awarded to H.F.), the Priority program of the SPP 1130 “Infection of the endothelium” (DFG-grant SCHN/430 3-1, awarded to H.-J.S.), and the “Med-drive-program” of the Medical Faculty Carl-Gustav Carus, University of Dresden (awarded to J.S.). V.M.W.-J. was supported by fellowships from the Manitoba Health Research Council (MHRC) and the Department of Medical Microbiology, University of Manitoba.
REFERENCES
- 1.Alcami, A., and U. H. Koszinowski. 2000. Viral mechanisms of immune evasion. Immunol. Today 21:447-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander, J. S., and J. W. Elrod. 2002. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J. Anat. 200:561-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baize, S., E. M. Leroy, A. J. Georges, M. C. Georges-Courbot, M. Capron, I. Bedjabaga, J. Lansoud-Soukate, and E. Mavoungou. 2002. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 128:163-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baskerville, A., S. P. Fisher-Hoch, G. H. Neild, and A. B. Dowsett. 1985. Ultrastructural pathology of experimental Ebola haemorrhagic fever virus infection. J. Pathol. 147:199-209. [DOI] [PubMed] [Google Scholar]
- 5.Bazzoni, G., and E. Dejana. 2004. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84:869-901. [DOI] [PubMed] [Google Scholar]
- 6.Bogatcheva, N. V., J. G. Garcia, and A. D. Verin. 2002. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry (Moscow) 67:75-84. [DOI] [PubMed] [Google Scholar]
- 7.Brunetti, C. R., M. Paulose-Murphy, R. Singh, J. Qin, J. W. Barrett, A. Tardivel, P. Schneider, K. Essani, and G. McFadden. 2003. A secreted high-affinity inhibitor of human TNF from Tanapox virus. Proc. Natl. Acad. Sci. USA 100:4831-4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan, S. Y., M. C. Ma, and M. A. Goldsmith. 2000. Differential induction of cellular detachment by envelope glycoproteins of Marburg and Ebola (Zaire) viruses. J. Gen. Virol. 81:2155-2159. [DOI] [PubMed] [Google Scholar]
- 9.Dejana, E. 2004. Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5:261-270. [DOI] [PubMed] [Google Scholar]
- 10.Dejana, E., R. Spagnuolo, and G. Bazzoni. 2001. Interendothelial junctions and their role in the control of angiogenesis, vascular permeability and leukocyte transmigration. Thromb. Haemost. 86:308-315. [PubMed] [Google Scholar]
- 11.DePaola, N., J. E. Phelps, L. Florez, C. R. Keese, F. L. Minnear, I. Giaever, and P. Vincent. 2001. Electrical impedance of cultured endothelium under fluid flow. Ann. Biomed. Eng. 29:648-656. [DOI] [PubMed] [Google Scholar]
- 12.Esser, S., K. Wolburg, H. Wolburg, G. Breier, T. Kurzchalia, and W. Risau. 1998. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J. Cell Biol. 140:947-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldmann, H., H. Bugany, F. Mahner, H. D. Klenk, D. Drenckhahn, and H. J. Schnittler. 1996. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 70:2208-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldmann, H., S. Jones, H. D. Klenk, and H. J. Schnittler. 2003. Ebola virus: from discovery to vaccine. Nat. Rev. Immunol. 3:677-685. [DOI] [PubMed] [Google Scholar]
- 15.Feldmann, H., V. E. Volchkov, V. A. Volchkova, and H. D. Klenk. 1999. The glycoproteins of Marburg and Ebola virus and their potential roles in pathogenesis. Arch. Virol. Suppl. 15:159-169. [DOI] [PubMed] [Google Scholar]
- 16.Feldmann, H., V. E. Volchkov, V. A. Volchkova, U. Ströher, and H. D. Klenk. 2001. Biosynthesis and role of filoviral glycoproteins. J. Gen. Virol. 82:2839-2848. [DOI] [PubMed] [Google Scholar]
- 17.Fujii, N., K. Hiroki, S. Matsumoto, K. Masuda, M. Inoue, Y. Tanaka, M. Awakura, and M. Akaboshi. 2001. Correlation between the loss of the chaperone-like activity and the oxidation, isomerization and racemization of gamma-irradiated alpha-crystallin. Photochem. Photobiol. 74:477-482. [DOI] [PubMed] [Google Scholar]
- 18.Garbutt, M., R. Liebscher, V. Wahl-Jensen, S. Jones, P. Moller, R. Wagner, V. Volchkov, H. D. Klenk, H. Feldmann, and U. Ströher. 2004. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J. Virol. 78:5458-5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geisbert, T. W., L. E. Hensley, T. Larsen, H. A. Young, D. S. Reed, J. B. Geisbert, D. P. Scott, E. Kagan, P. B. Jahrling, and K. J. Davis. 2003. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 163:2347-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geisbert, T. W., H. A. Young, P. B. Jahrling, K. J. Davis, T. Larsen, E. Kagan, and L. E. Hensley. 2003. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 163:2371-2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grieb, T., R. Y. Forng, R. Brown, T. Owolabi, E. Maddox, A. McBain, W. N. Drohan, D. M. Mann, and W. H. Burgess. 2002. Effective use of gamma irradiation for pathogen inactivation of monoclonal antibody preparations. Biologicals 30:207-216. [DOI] [PubMed] [Google Scholar]
- 22.Hensley, L. E., H. A. Young, P. B. Jahrling, and T. W. Geisbert. 2002. Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol. Lett. 80:169-179. [DOI] [PubMed] [Google Scholar]
- 23.Kindzelskii, A. L., Z. Yang, G. J. Nabel, R. F. Todd III, and H. R. Petty. 2000. Ebola virus secretory glycoprotein (sGP) diminishes Fc gamma RIIIB-to-CR3 proximity on neutrophils. J. Immunol. 164:953-958. [DOI] [PubMed] [Google Scholar]
- 24.Leroy, E. M., S. Baize, V. E. Volchkov, S. P. Fisher-Hoch, M. C. Georges-Courbot, J. Lansoud-Soukate, M. Capron, P. Debre, J. B. McCormick, and A. J. Georges. 2000. Human asymptomatic Ebola infection and strong inflammatory response. Lancet 355:2210-2215. [DOI] [PubMed] [Google Scholar]
- 25.Lo, C. M., C. R. Keese, and I. Giaever. 1995. Impedance analysis of MDCK cells measured by electric cell-substrate impedance sensing. Biophys. J. 69:2800-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luscinskas, F. W., S. Ma, A. Nusrat, C. A. Parkos, and S. K. Shaw. 2002. The role of endothelial cell lateral junctions during leukocyte trafficking. Immunol. Rev. 186:57-67. [DOI] [PubMed] [Google Scholar]
- 27.Maruyama, T., M. J. Buchmeier, P. W. H. I. Parren, and D. R. Burton. 1998. Ebola virus, neutrophils and antibody specificity. Science 282:845a. [Google Scholar]
- 28.Michel, C. C., and F. E. Curry. 1999. Microvascular permeability. Physiol. Rev. 79:703-761. [DOI] [PubMed] [Google Scholar]
- 29.Minshall, R. D., C. Tiruppathi, S. M. Vogel, and A. B. Malik. 2002. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem. Cell Biol. 117:105-112. [DOI] [PubMed] [Google Scholar]
- 30.Ryabchikova, E. I., L. V. Kolesnikova, and N. Rassadkin Iu. 1998. Microscopic study of species specific features of hemostatic impairment in Ebola virus infected monkeys. Vestn. Ross. Akad. Med. Nauk. 3:51-55. [PubMed] [Google Scholar]
- 31.Ryabchikova, E., L. V. Kolesnikova, and S. V. Luchko. 1999. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 179:S199-S202. [DOI] [PubMed] [Google Scholar]
- 32.Ryabchikova, E., L. V. Kolesnikova, and S. V. Netesov. 1999. Animal pathology of filoviral infections. Curr. Top. Microbiol. Immunol. 235:145-173. [DOI] [PubMed] [Google Scholar]
- 33.Sanchez, A., A. S. Khan, S. R. Zaki, G. J. Nabel, T. G. Ksiazek, and C. J. Peters. 2001. Filoviridae: Marburg and Ebola viruses, p. 1279-1304. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 34.Sanchez, A., S. G. Trappier, B. W. Mahy, C. J. Peters, and S. T. Nichol. 1996. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. USA 93:3602-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez, A., Z. Y. Yang, L. Xu, G. J. Nabel, T. Crews, and C. J. Peters. 1998. Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J. Virol. 72:6442-6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schnittler, H., U. Stroeher, T. Afanasieva, and H. Feldmann. 2004. The role of endothelial cells in filovirus hemorrhagic fever, p. 279-304. In H. Feldmann (ed.), Ebola and Marburg viruses: molecular and cellular biology. Horizon Biosciences, Norfolk, United Kingdom.
- 37.Schnittler, H. J. 1998. Structural and functional aspects of intercellular junctions in vascular endothelium. Basic Res. Cardiol. 93(Suppl. 3):30-39. [DOI] [PubMed] [Google Scholar]
- 38.Schnittler, H. J., and H. Feldmann. 2003. Viral hemorrhagic fever—a vascular disease? Thromb. Haemost. 89:967-972. [PubMed] [Google Scholar]
- 39.Schnittler, H. J., R. P. Franke, U. Akbay, C. Mrowietz, and D. Drenckhahn. 1993. Improved in vitro rheological system for studying the effect of fluid shear stress on cultured cells. Am. J. Physiol. 265:C289-C298. [DOI] [PubMed] [Google Scholar]
- 40.Schnittler, H. J., A. Wilke, T. Gress, N. Suttorp, and D. Drenckhahn. 1990. Role of actin and myosin in the control of paracellular permeability in pig, rat and human vascular endothelium. J. Physiol. 431:379-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schreiber, M., K. Rajarathnam, and G. McFadden. 1996. Myxoma virus T2 protein, a tumor necrosis factor (TNF) receptor homolog, is secreted as a monomer and dimer that each bind rabbit TNFα, but the dimer is a more potent TNF inhibitor. J. Biol. Chem. 271:13333-13341. [DOI] [PubMed] [Google Scholar]
- 42.Seebach, J., P. Dieterich, F. Luo, H. Schillers, D. Vestweber, H. Oberleithner, H. J. Galla, and H. J. Schnittler. 2000. Endothelial barrier function under laminar fluid shear stress. Lab. Investig. 80:1819-1831. [DOI] [PubMed] [Google Scholar]
- 43.Seet, B. T., J. B. Johnston, C. R. Brunetti, J. W. Barrett, H. Everett, C. Cameron, J. Sypula, S. H. Nazarian, A. Lucas, and G. McFadden. 2003. Poxviruses and immune evasion. Annu. Rev. Immunol. 21:377-423. [DOI] [PubMed] [Google Scholar]
- 44.Sheikh, S., Z. Gale, G. E. Rainger, and G. B. Nash. 2004. Methods for exposing multiple cultures of endothelial cells to different fluid shear stresses and to cytokines, for subsequent analysis of inflammatory function. J. Immunol. Methods 288:35-46. [DOI] [PubMed] [Google Scholar]
- 45.Simionescu, M. 1980. Structural and functional differentiation of microvascular endothelium. Ciba Found. Symp. 71:39-60. [DOI] [PubMed] [Google Scholar]
- 46.Springer, T. A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76:301-314. [DOI] [PubMed] [Google Scholar]
- 47.Ströher, U., E. West, H. Bugany, H. D. Klenk, H. J. Schnittler, and H. Feldmann. 2001. Infection and activation of monocytes by Marburg and Ebola viruses. J. Virol. 75:11025-11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takada, A., C. Robison, H. Goto, A. Sanchez, K. G. Murti, M. A. Whitt, and Y. Kawaoka. 1997. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 94:14764-14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vestweber, D. 2002. Regulation of endothelial cell contacts during leukocyte extravasation. Curr. Opin. Cell Biol. 14:587-593. [DOI] [PubMed] [Google Scholar]
- 50.Volchkov, V. E., S. Becker, V. A. Volchkova, V. A. Ternovoj, A. N. Kotov, S. V. Netesov, and H. D. Klenk. 1995. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 214:421-430. [DOI] [PubMed] [Google Scholar]
- 51.Volchkov, V. E., H. Feldmann, V. A. Volchkova, and H. D. Klenk. 1998. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 95:5762-5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Volchkov, V. E., V. A. Volchkova, E. Muhlberger, L. V. Kolesnikova, M. Weik, O. Dolnik, and H. D. Klenk. 2001. Recovery of infectious Ebola virus from complementary DNA: RNA editing of the GP gene and viral cytotoxicity. Science 291:1965-1969. [DOI] [PubMed] [Google Scholar]
- 53.Volchkov, V. E., V. A. Volchkova, U. Ströher, S. Becker, O. Dolnik, M. Cieplik, W. Garten, H. D. Klenk, and H. Feldmann. 2000. Proteolytic processing of Marburg virus glycoprotein. Virology 268:1-6. [DOI] [PubMed] [Google Scholar]
- 54.Volchkova, V. A., H. Feldmann, H. D. Klenk, and V. E. Volchkov. 1998. The nonstructural small glycoprotein sGP of Ebola virus is secreted as an antiparallel-orientated homodimer. Virology 250:408-414. [DOI] [PubMed] [Google Scholar]
- 55.Volchkova, V. A., H. D. Klenk, and V. E. Volchkov. 1999. Delta-peptide is the carboxy-terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology 265:164-171. [DOI] [PubMed] [Google Scholar]
- 56.Wahl-Jensen, V., S. K. Kurz, P. R. Hazelton, H. J. Schnittler, U. Ströher, D. R. Burton, and H. Feldmann. 2005. Role of Ebola virus secreted glycoproteins and virus-like particles in activation of human macrophages. J. Virol. 79:2413-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wojciak-Stothard, B., and A. J. Ridley. 2002. Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 39:187-199. [DOI] [PubMed] [Google Scholar]
- 58.Wong, R. K., A. L. Baldwin, and R. L. Heimark. 1999. Cadherin-5 redistribution at sites of TNF-α and IFN-γ-induced permeability in mesenteric venules. Am. J. Physiol. 276:H736-H748. [DOI] [PubMed] [Google Scholar]
- 59.Wool-Lewis, R. J., and P. Bates. 1998. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J. Virol. 72:3155-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Worthylake, R. A., and K. Burridge. 2001. Leukocyte transendothelial migration: orchestrating the underlying molecular machinery. Curr. Opin. Cell Biol. 13:569-577. [DOI] [PubMed] [Google Scholar]
- 61.Yamawaki, H., S. Lehoux, and B. C. Berk. 2003. Chronic physiological shear stress inhibits tumor necrosis factor-induced proinflammatory responses in rabbit aorta perfused ex vivo. Circulation 108:1619-1625. [DOI] [PubMed] [Google Scholar]
- 62.Yang, Z., R. Delgado, L. Xu, R. F. Todd, E. G. Nabel, A. Sanchez, and G. J. Nabel. 1998. Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science 279:1034-1037. [DOI] [PubMed] [Google Scholar]
- 63.Yang, Z. Y., H. J. Duckers, N. J. Sullivan, A. Sanchez, E. G. Nabel, and G. J. Nabel. 2000. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 6:886-889. [DOI] [PubMed] [Google Scholar]
- 64.Zaki, S. R., and C. S. Goldsmith. 1999. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 235:97-116. [DOI] [PubMed] [Google Scholar]