

Abstract
Homologs of peroxin 16 genes (PEX16) have been identified only in Yarrowia lipolytica, humans (Homo sapiens), and Arabidopsis (Arabidopsis thaliana). The Arabidopsis gene (AtPEX16), previously reported as the SSE1 gene, codes for a predicted 42-kD membrane peroxin protein (AtPex16p). Lin et al. (Y. Lin, J.E. Cluette-Brown, H.M. Goodman [2004] Plant Physiol 135: 814–827) reported that SSE1/AtPEX16 was essential for endoplasmic reticulum (ER)-dependent oil and protein body biogenesis in peroxisome-deficient maturing seeds and likely also was involved in peroxisomal biogenesis based on localization of stably expressed green fluorescent protein::AtPex16p in peroxisomes of Arabidopsis plants. In this study with Arabidopsis suspension-cultured cells, combined in vivo and in vitro experiments revealed a novel dual organelle localization and corresponding membrane association/topology of endogenous AtPex16p. Immunofluorescence microscopy with antigen affinity-purified IgGs showed an unambiguous, steady-state coexistence of AtPex16p in suspension cell peroxisomes and ER. AtPex16p also was observed in peroxisomes and ER of root and leaf cells. Cell fractionation experiments surprisingly revealed two immunorelated polypeptides, 42 kD (expected) and 52 kD (unexpected), in homogenates and microsome membrane pellets derived from roots, inflorescence, and suspension cells. Suc-gradient purifications confirmed the presence of both 42-kD and 52-kD polypeptides in isolated peroxisomes (isopycnic separation) and in rough ER vesicles (Mg2+ shifted). They were found peripherally associated with peroxisome and ER membranes but not as covalently bound subunits of AtPex16p. Both were mostly on the matrix side of peroxisomal membranes and unexpectedly mostly on the cytosolic side of ER membranes. In summary, AtPex16p is the only authentic plant peroxin homolog known to coexist at steady state within peroxisomes and ER; these data provide new insights in support of its ER-related, multifunctional roles in organelle biogenesis.
Peroxisomes are structurally simple organelles with their single membrane surrounding a rather nondescript proteinaceous matrix, which does not possess any DNA or allied protein-synthesizing machinery. Nevertheless, they constitute an important, metabolically plastic, and diverse complement to other common organelles within virtually all eukaryotic cell types (Subramani, 1998; Veenhuis et al., 2000; Parsons et al., 2001; Baker and Graham, 2002). A unique feature of this organelle's functional plasticity, which is more prevalent for plant (Kamada et al., 2003; Reumann, 2004) than other (Emanuelsson et al., 2003) peroxisomes, is that constitutive and induced changes in metabolic capabilities are accomplished exclusively via regulated posttranslational acquisitions of nuclear-encoded matrix and membrane proteins (Mullen, 2002; Sparkes and Baker, 2002; Trelease, 2002; Lazarow, 2003; Veenhuis et al., 2003). Hence, targeting, intracellular sorting, and import of these proteins are critically important features for biogenesis/differentiation of peroxisomes in all organisms.
Pioneering advances in elucidating molecular and structural mechanisms related to peroxisomal biogenesis and differentiation were made through genetic studies with five different yeasts, namely Saccharomyces cerevisiae, Pichia pastoris, Hansenula polymorpha, Yarrowia lipolytica, and Candida boidinii (Elgersma and Tabak, 1996; Subramani et al., 2000; Titorenko and Rachubinski, 2001a, 2001b; Wang et al., 2004). Analyses of peroxisomal mutant phenotypes led to the identification of peroxin (PEX) genes coding for peroxins (Pex). Consecutive numbers have been assigned to each PEX gene (Distel et al., 1996). To date, at least 32, 17, and 23 predicted or demonstrated PEX genes have been identified from studies or database searches of yeasts (Vizeacoumar et al., 2004), mammals (Lazarow, 2003), and plants (Mullen et al., 2001a; Charlton and Lopez-Huertas, 2002; http://lsweb.la.asu.edu/rtrelease), respectively.
About one-half of the 23 predicted plant PEX genes have been shown via subcellular localizations and/or reverse genetics experiments to code for actual peroxin homologs of yeast or mammalian peroxins. For example, AtPex2p, AtPex3p, AtPex10p, AtPex14p, and AtPex16p were localized in peroxisomes via (immuno)fluorescence microscopy (Hayashi et al., 2000; Hu et al., 2002; Sparkes et al., 2003; Hunt and Trelease, 2004; Lin et al., 2004). In genetic studies, the Atpex5 null mutant exhibited defects in glyoxysomal function and in the intracellular transport of matrix proteins bearing the type 1 peroxisomal targeting signals (Zolman et al., 2000). The Atpex6 mutant displayed a peroxisome-defective phenotype and a reduced peroxisomal matrix protein import (Zolman and Bartel, 2004). When analyzed for import of peroxisomal proteins, the Atpex14 mutant (ped2) showed a reduced import of matrix proteins into the types of plant peroxisomes (Hayashi et al., 2000). Embryo lethality was a common feature of T-DNA transposon knockout mutants Atpex2/ted3 (Hu et al., 2002), Atpex10 (Schumann et al., 2003; Sparkes et al., 2003), and Atpex16/shrunken seed 1 (sse1; Lin et al., 1999). Assignment of gene products as functional peroxin homologs was limited in the latter studies.
Nevertheless, Lin et al. (1999) discovered that the SSE1 gene coded for a predicted AtPex16p homolog, which they suggested functioned in the biogenesis of protein and oil bodies. This is of particular interest because both organelles are derived from endoplasmic reticulum (ER; Huang, 1992; Sarmiento et al., 1997). A role in peroxisomal biogenesis was not advanced because a distinctive phenotype was not observed in the developing seeds prior to their demise. More recently, Lin et al. (2004) found that AtPex16/sse1 mutant embryos indeed lacked normal peroxisomes and that a green fluorescent protein (GFP)::AtPex16p fusion protein complemented the AtPex16/sse1 phenotype. The complemented transgenic plants exhibited GFP::AtPex16p autofluorescence in root hair and embryo peroxisomes. These observations supported their contention that AtPex16p/sse1 was a peroxin homolog.
Pex16p has been examined thus far only in two other organisms, namely humans (Homo sapiens) and Y. lipolytica. HsPex16p functioned in peroxisome membrane assembly of immature preperoxisomes in CHO-K1 culture cells (Honsho et al., 2002), and loss of HsPex16p blocked the synthesis of peroxisomes and peroxisomal membrane import in cells from a Zellweger syndrome patient (South and Gould, 1999). YlPex16p acted as a negative regulator of early intermediates in peroxisomal division, and Ylpex16 mutants did not assemble functional peroxisomes, likely due to their inability to import a full complement of matrix proteins (Eitzen et al., 1997; Guo et al., 2003). Interestingly, AtPEX16 partially complemented this Ylpex16 mutant, suggesting some similarity in function of the Yarrowia and Arabidopsis (Arabidopsis thaliana) Pex16p (Lin et al., 1999). These studies implicate Pex16 homologs in the early stages of peroxisomal formation (assembly) in all three species.
A unique feature of YlPex16p is its association with ER. Radiolabeled YlPex16p (and YlPex2p) was detected in ER and then in ER-derived vesicles that moved under the control of protein secretory genes to Y. lipolytica peroxisomes (Titorenko and Rachubinski, 1998a). Within the ER, YlPex16p was N-linked core glycosylated with a single oligosaccharide chain. These profound findings contributed to the reemergence of models enlisting the participation of ER in peroxisomal biogenesis (Kunau and Erdmann, 1998; Titorenko and Rachubinski, 1998b; Baerends et al., 2000; Mullen et al., 2001a; Tabak et al., 2003). Evidence for involvement of plant ER in peroxisomal biogenesis came from studies on the localizations of endogenous peroxisomal ascorbate peroxidase (APX) and AtPex10p within subdomains of plant ER and on the indirect sorting of APX to peroxisomes through ER subdomain(s) (Mullen et al., 1999, 2001b; Nito et al., 2001; Lisenbee et al., 2003a, 2003b; Flynn et al., 2005). In these and other studies, peroxins overwhelmingly were localized at steady state to either peroxisomes or ER. In this study, data from microscopy and cell fractionation/biochemical experiments revealed a coexistence of AtPex16p in peroxisomes and ER at steady state. Interestingly, 42-kD AtPex16p polypeptides and immunorelated 52-kD polypeptides were associated with the membranes of each organelle. The definitive existence of endogenous AtPex16p within peroxisomes of roots, leaves, and suspension cells fortifies its role as a peroxin homolog, functioning as such within ER and/or peroxisome membranes. The distribution of AtPex16p throughout most of the ER, rather than within restricted peroxisomal ER subdomain(s) reinforces its suggested role in oil body biogenesis. Together, the results support at least a bifunctional role for AtPex16p in organelle biogenesis.
RESULTS
Amino Acid Sequence Comparisons of Pex16p Homologs
Figure 1 shows an amino acid sequence alignment of five Pex16p homologs. At present, only humans are known to possess more than one isoform. Surprisingly, the PEX16 gene has been identified in only one (Y. lipolytica) of the five yeast species, which has been examined extensively for peroxisomal biogenesis. Analyses of the alignment reveal that AtPex16p has only an 18% to 26% amino acid identity with the other homologs, although their deduced polypeptide molecular masses are quite similar, ranging from 38 to 44 kD. Two membrane helices (underlined, Fig. 1) are predicted within MmPex16p and AtPex16p (HMMTOP 2.0 program; Tusnady and Simon, 1998, 2001). Three membrane helices are predicted in HsPex16p (isoforms 1 and 2). From biochemical evidence, South and Gould (1999) concluded that HsPex16p (isoform 1) was an integral membrane protein. Eitzen et al. (1997) reported that YlPex16p possessed four membrane helices with only one of them considered as a membrane-spanning domain. They concluded from biochemical results, however, that YlPex16p was a peripheral membrane protein.
Figure 1.

Alignment (ClustalW) of deduced amino acid sequences for Pex16p homologs in humans (Hs; two isoforms), Mus musculus (Mm), Y. lipolytica (Yl), and Arabidopsis (At). Gaps (marked with dashes) were introduced to maximize the sequence alignment. Predicted membrane helices are underlined. Clusters of basic amino acid residues are bolded. Predicted N-linked glycosylation sites are boxed.
The cluster of basic amino acid residues (bolded, Fig. 1) along with the N-terminal-most (first) transmembrane domain in HsPex16p (isoform 1) was identified as a necessary membrane-targeting signal (Honsho et al., 2002). A similar positional relationship (basic cluster and transmembrane domain) is conserved only among the three mammalian isoforms. In AtPex16p, three clusters of basic residues (bolded) are apparent, one upstream of the first predicted membrane helix, another between the two predicted membrane helices, and the third downstream of the second predicted membrane helix. A cluster of basic residues does not exist in YlPex16p.
YlPex16p was reported to be an N-linked glycoprotein with a single oligosaccharide chain (Titorenko and Rachubinski, 1998a). Accordingly, an N-glycosylation site exits in YlPex16p (boxed residues, Fig. 1). A putative glycosylation site also occurs in AtPex16p but not in any of the other homologs.
Characterization of AtPex16p Antiserum and Affinity-Purified IgGs, and Identification of Immunorelated 42-kD and 52-kD Polypeptides in Plant and Suspension Cells
Figure 2 (top) presents representative immunoblot results used to assess the specificity and fidelity of anti-AtPex16p antiserum (lane 1) and of affinity-purified IgGs prepared from this antiserum (lanes 2 and 3). Clarified homogenates of Arabidopsis suspension cells were used as the source of antigens on the blots. Lane 1 shows that the AtPex16p antiserum reacts most strongly with a 42-kD polypeptide band, consistent with the predicted molecular mass of AtPex16p. Several other bands of lower and higher molecular mass also are apparent in this lane. Preimmune serum applied to similar blots in place of antiserum did not produce any chemiluminescence signals.
Figure 2.

Rabbit anti-AtPex16p IgGs, affinity purified against Immobilon-bound 42-kD polypeptides (anti-AtPex16p-42 IgGs), are specific for 42- and 52-kD polypeptides in cell fractions of Arabidopsis suspension cells, roots, and leaves. Top, Arabidopsis suspension cell (7 d) homogenates (1,500g, 15 min supernatants from 5,000 psi pressure cell-ruptured cells) were subjected to SDS-PAGE and polypeptides were electroblotted to Immobilon and immunodetected via chemiluminescence with rabbit anti-AtPex16p antiserum (AtPex16p antiserum; 1:1,000, lane 1), PA-purified IgGs (PA anti-AtPex16p IgGs; 1:1,000, lane 2), and 42-kD polypeptide affinity-purified IgGs (anti-AtPex16p-42 IgGs; 1:100, lane 3). Bottom, Roots and flowers excised from 4-week-old Arabidopsis plants were powdered in liquid nitrogen, and then homogenized with a mortar and pestle in a buffered medium. Membrane-free supernatants (S, 200,000g, 1 h; lanes 1 and 3) and corresponding microsomal membrane pellets (MP; lanes 2 and 4), prepared from clarified homogenates, were subjected to SDS-PAGE immunoblot analyses. Samples were detergent (DOC) solubilized, TCA precipitated, and 150 μg protein was applied per well. Chemiluminescence signals were produced from polypeptide-bound anti-AtPex16p-42 IgGs (1:100, overnight incubation).
In lane 2 (top), Protein A (PA)-purified IgGs (rabbit PA anti-AtPex16p IgGs) recognize two distinguishable bands on the blots, namely a 42-kD and a 52-kD polypeptide band. Some diffuse signals in a lower molecular range also are observed. IgGs in this PA anti-AtPex16p IgG antibody preparation were further purified. Bands with the 42-kD polypeptides were excised from Immobilon blots, incubated with PA anti-AtPex16p IgGs, and bound IgGs were eluted from the 42-kD polypeptides. These antibodies are referred to as anti-AtPex16p-42 IgGs throughout this article.
Lane 3 (top) exhibits surprising and interesting results with the anti-AtPex16p-42 IgGs. They recognize not only 42-kD polypeptides as expected, but also 52-kD polypeptides (slightly weaker) in a well-separated band. No other bands are observed, indicative of a collection of highly purified and specific IgGs in the antibody probe. Evidence that these IgGs were affinity purified against only 42-kD polypeptides was that the same results were obtained with replicate preparations of these IgGs using different Immobilon blots with well-separated 42- and 52-kD bands. Hence, the polypeptides in the 52-kD band are immunorelated to the 42-kD AtPex16p polypeptides.
The question arose as to whether immunorelated 42- and 52-kD polypeptides also existed in Bright Yellow (BY)-2 cells and/or in one or more parts of Arabidopsis plants. Anti-AtPex16p-42 IgGs recognized multiple bands on blots of BY-2 cell homogenates (data not shown); hence, it was not possible to determine whether immunorelated NtPex16p polypeptides existed in BY-2 cells, nor could we reliably use these antibodies for immunological studies with BY-2 cells. The bottom section of Figure 2 presents immunoblot results with cell fractions from Arabidopsis plant parts. Both 42- and 52-kD polypeptide bands were detected in microsomal membrane fractions recovered from flowers and roots (lanes 2 and 4). Only 42-kD polypeptides were found in the supernatant fractions of these plant parts (lanes 1 and 3). However, 42- or 52-kD polypeptide bands were not detected on blots of clarified homogenates made from roots, flowers, leaves, or siliques, or in 200,000g (1 h) supernatants or microsomal pellets derived from leaves or siliques (data not shown). These negative results likely were due to the common technical difficulty of not being able to apply sufficient amounts of 42- and 52-kD polypeptides (in protein-rich samples) per well of SDS gels. Nevertheless, the presence of the 42- and 52-kD polypeptides in at least some plant parts clearly indicates that these immunorelated polypeptides are not uniquely expressed in Arabidopsis suspension cells.
Association of AtPex16p-42 and 52-kD Polypeptides with Microsome Membranes
Discovering two AtPex16p immunorelated polypeptides, one of which (42 kD) is a predicted membrane protein (Fig. 1), prompted a comparison of the membrane associations of these two polypeptides. Both polypeptides are present in clarified homogenates (Fig. 3, lane 1); the 42-kD polypeptide band is more prominent than the 52-kD band (see also Fig. 2, top, lane 3). Similar to our surprising results with flowers and roots (Fig. 2, bottom), 42-kD polypeptides (but not 52-kD polypeptides) were solubilized in the homogenizing medium (Fig. 3, lane 2). Both were in microsome pellets as expected (Fig. 3, lane 3). This result also was similar to those obtained with roots and flowers (Fig. 2, bottom). Calnexin, an ER marker, was prevalent in the microsome pellet but not found in the supernatant (data not shown), indicating that the presence of 42-kD polypeptides in the supernatant was not due to an inefficient sedimentation of microsomes. The 42-kD band consistently was found in homogenizing medium supernatants and microsomal membrane pellets whether suspension cells (frozen in liquid nitrogen) were disrupted with a mortar and pestle or disrupted at varying pressures in a French pressure cell (data not shown).
Figure 3.

AtPex16p-42 and 52-kD polypeptides are variously associated with Arabidopsis suspension cell microsomal membranes. Clarified homogenates (CH) prepared in HEPES-buffered homogenizing medium (HM; 1,500g, 15 min supernatant of 5,000 psi pressure cell-ruptured cells; lane 1) were centrifuged (200,000g, 1 h) to produce a membrane-free supernatant (Su; lane 2) and microsomal membrane pellet (MP; lane 3). Equal portions of separate water-insoluble microsomal pellets (4 mg protein per 2 mL) were incubated with 0.2 m KCl, 0.1 m Na2CO3 (pH 11.5), or 0.05% (w/v) DOC for 1 h. Supernatant (S) and pellet (P) fractions (200,000g, 30 min) were recovered and analyzed from each treatment (lanes 4–9). These fractions and those from HM were subjected to SDS-PAGE immunoblot analyses (as for Fig. 2) using anti-AtPex16p-42 IgGs (1:100, overnight) as probes for all samples. Protein samples (150 μg) were added per well of SDS gels.
Incubation of microsomal membrane pellets in 0.2 m KCl resulted in release of a portion of membrane-bound 42-kD polypeptides into the KCl-soluble supernatant without any accompanying 52-kD polypeptides (Fig. 3, lane 4). All 52-kD polypeptides along with another portion of 42-kD polypeptides remained in the KCl-insoluble membrane pellet (lane 5). Incubations of other, separate microsome pellets (each with the same protein content) in twice the volume of 0.2 m, 0.4 m, or 1.0 m KCl yielded the same results as shown in lanes 4 and 5 (data not shown). Incubation of another separate microsomal membrane pellet in alkaline sodium carbonate solubilized all of the 42- and 52-kD polypeptides (compare lanes 6 and 7) that were in the water-insoluble membranes (lane 3). Incubation of another microsomal membrane pellet in 0.05% (w/v) deoxycholate (DOC) solubilized both polypeptides (lane 8), with a portion of both remaining in the detergent-insoluble membranes (lane 9). Increasing the concentrations of DOC from 0.05% to 0.5% (w/v) resulted in a complete release of all 42- and 52-kD polypeptides (data not shown).
In summary, both 42- and 52-kD polypeptides are associated with membranes, albeit variously, suggesting that they are not covalently bound subunits of a potentially heteromeric AtPex16p. The varied solubility (in water, KCl, alkaline carbonate, and DOC) of the AtPex16p-42 polypeptides is surprising and unusual for membrane-associated polypeptides. Interpretations as to whether these polypeptides are peripherally or integrally associated with membranes are discussed.
Evaluation of Affinity-Purified (PA and 42-kD Antigen) IgGs for Immunofluorescence Microscopy
Figure 4 displays representative confocal optical sections of Arabidopsis suspension cells transiently expressing mycAtPex16p. Figure 4A shows one representative transformed cell. Overexpressed mycAtPex16p was visualized (with anti-myc antibodies) in two compartments, a reticular and a punctate compartment. However, Figure 4B shows that no anti-AtPex16p-42 immunofluorescence was detected in the same dual-incubated transformed cell using our standard tube procedure. Low IgG concentrations are characteristic of the affinity-purified anti-Pex16p-42 IgG preparations. Therefore, a portion of the same batch of transiently transformed cells was dual labeled with PA anti-AtPex16p IgGs (higher IgG concentration) plus anti-myc antibodies. Figure 4C reveals two myc-labeled compartments (reticular and punctate) similar to those shown in Figure 4A. However, in the same transformed cell (Fig. 4D), there is a virtual perfect colocalization of the reticular and punctate immunofluorescence patterns. These results demonstrate that these two antibodies are labeling the same overexpressed mycAtPex16p antigen. Control experiments included applications of preimmune antiserum in place of the primary IgGs, omitting primary IgGs or secondary IgGs, or adding irrelevant IgGs. In no cases was background fluorescence observed (data not shown). In summary, the colocalization of anti-myc antibodies and PA anti-AtPex16p IgGs (Fig. 4, C and D) is compelling direct evidence for the monotypic fidelity of the PA affinity-purified IgGs for AtPex16p.
Figure 4.

PA anti-AtPex16p IgGs specifically recognize myc-epitope-tagged AtPex16p transiently expressed in Arabidopsis suspension cells processed for immunofluorescence microscopy via our standard tube procedure. Arabidopsis suspension cells, biolistically bombarded with pRTL2/mycAtPEX16 and held for 2.5 h expression, were formaldehyde fixed, cell walls permeabilized and digested with pectolyase and cellulase, membranes permeabilized with Triton X-100, and organelles immunolabeled with primary and secondary antibodies in a tube prior to spreading these cells on microscope slides for examination of the transformed immunofluoescent cells (about 0.2% of the cells on the slides). A to D, All images are representative confocal laser optical sections. A and B, Results of dual labeling a representative cell transiently expressing mycAtPex16p. A, Cell illustrating a reticular (ER) and punctate (peroxisome) Cy2 immunofluorescence image labeled with anti-mouse myc epitope antibodies (1: 500, 1 h) and B, the same cell examined for Cy5 bound to anti-AtPex16p-42 IgGs (1:10, overnight). No Cy5 fluorescence attributable to endogenous AtPex16p was observed in the transformed or neighboring nontransformed cells. C and D, Fluorescence images of a cell from the same population of bombarded cells dual labeled with C, anti-mouse myc epitope antibodies (1:500, 1 h), and D, rabbit PA anti-AtPex16p IgGs (1:1,000, 2 h). Colocalization is apparent between the myc (Cy2) and AtPex16p (Cy5) labeled antigens in both reticulate and punctate patterns observed throughout the cytoplasm surrounding the unlabeled nucleus (n). Bar = 5 μm.
Localizations of Endogenous AtPex16p in Arabidopsis Suspension and Root Cells via Immunofluorescence Microscopy
As indicated above, endogenous AtPex16p was not detected in the nontransformed cells with either of the affinity-purified antibodies when employing our tube procedure for labeling cells for immunofluorescence microscopy. These observations plus the need to apply at least 150 μg protein per gel well to detect AtPex16p on immunoblots (Figs. 2 and 3) indicate that AtPex16p occurs in relatively low abundance. A modified, on-slide procedure was employed for other low abundance membrane proteins (Lisenbee et al., 2003a; Flynn et al., 2005).
Figure 5, A and B, are representative images of a cluster of eight to nine cells dual immunolabeled with anti-AtPex16p-42 IgGs (green) and anti-catalase IgGs (red; peroxisomes). AtPex16p was observed in both the reticular (arrowheads) and punctate peroxisomal compartments (arrows) in all cells (Fig. 5A). The merged image (Fig. 5C) convincingly shows that a portion of the endogenous AtPex16p is located in the catalase-containing peroxisomes (yellow). Virtually all cells exhibited peroxisomal labeling after a 1-h incubation in catalase antibodies, whereas only approximately 15% to 25% of the cells were immunolabeled after an overnight incubation in relatively high concentration (1:10) of anti-AtPex16p-42 IgGs. These undesirable parameters prompted us to apply PA anti-AtPex16p IgGs (specific and higher concentration, Fig. 4, C and D) to a different portion of the same batch of cells. Figure 5D shows a representative image of clustered cells immunolabeled with PA anti-AtPex16p IgGs at a comparatively low concentration (1:1,000) for only 2 h. Under these conditions, the PA anti-AtPex16p IgGs recognized similar punctate (arrows) and reticular (arrowheads) patterns as did the anti-AtPex16p-42 IgGs (Fig. 5A), and the patterns were observed in virtually all of the cells on the microscope slides. Therefore, PA anti-AtPex16p IgGs were used routinely for immunofluorescence localizations of endogenous AtPex16p (e.g. Fig. 5, D, G, and J). Figure 5E shows the punctate pattern characteristic of catalase-containing peroxisomes (arrows), and the merged image (Fig. 5F) shows colocalization of a portion of the endogenous AtPex16p with catalase in the peroxisomes (yellow).
Figure 5.

Immunofluorescence images of dual-labeled cells reveal the coexistence of endogenous AtPex16p in peroxisomes and ER. A to O, Representative confocal optical sections of nontransformed Arabidopsis cells processed for microscopy via the on-slide procedure whereby formaldehyde-fixed and pectolyase-cellulase-digested cells were spread on microscope slides, membranes were permeabilized with Triton X-100, and then primary and secondary antibodies and/or Concanavalin A-Alexa 594 were added to the cells (rather than to fixed/permeabilized cells in microfuge tubes as was done for cells shown in Fig. 4). A to C: A, Green Cy2 punctate (arrows) and reticular patterns (arrowheads; rabbit anti-AtPex16p-42 IgGs; 1:10, overnight). B, The same cells exhibit a Cy5 punctate peroxisomal pattern (mouse anti-catalase monoclonal IgG; 1:500, 1 h). C, Merged image shows colocalized (yellow) catalase and AtPex16p in peroxisomes. D to F: D, Different portion of the same population of cells revealed similar Cy2 punctate (arrows) and reticular patterns (arrowheads; rabbit PA anti-AtPex16p IgGs; 1:500, 1 h). E, Same cells with Cy5 peroxisomes (mouse anti-catalase IgGs; 1:500, 1 h). F, Merged image shows colocalized catalase and Atpex16p in peroxisomes. G to I: G, Different portion of the same population of cells (rabbit PA anti-AtPex16p IgGs; 1:500, 1 h). H, Same cells (mouse anti-BiP monoclonal antibodies; 1:500, 1 h). I, Merged image produced a yellow reticulate pattern indicative of ER localization of AtPex16p. J to L: J, Reticulate pattern (PA anti-AtPex16p IgGs; 1:500, 1 h). K, Reticulate pattern (Concanavalin A-Alexa 594; 1:500, 1 h). L, Merged image (yellow) indicative of the ER localization of AtPex16p. M to O. M, Reticulate pattern (mouse anti-BiP monoclonal antibodies; 1:500, 1 h). N, Reticulate pattern (Concanavalin A-Alexa 594; 1:500, 1 h). O, Merged image produced a yellow reticulate pattern demonstrating that both Concanavalin A and BiP are colocalized in the same ER compartment. Bar = 5 μm.
Due to the resemblance of the reticular compartment to ER, cells were dual labeled with ER markers. Figure 5G shows AtPex16p in reticular compartment and punctate peroxisomes as before. Figure 5H shows the same cells labeled with mouse anti-binding protein (BiP) monoclonal antibodies. In the merged image (Fig. 5I), the two antigens are colocalized in the reticular compartment (yellow-orange). However, not the entire BiP-localized ER compartment possesses AtPex16p as evidenced by the red BiP fluorescence (Fig. 5I). Another portion of these cells also was dual labeled for endogenous AtPex16p and ConcanavalinA (another ER marker) conjugated to Alexa 594. Figure 5J shows the punctate and reticular fluorescence pattern displayed by PA anti-AtPex16p IgGs. Figure 5K shows a reticular pattern labeled with ConcanavalinA-Alexa594. In the merged image (Fig. 5L), AtPex16p is colocalized with ConcanavalinA to the same extent as it is with ER BiP (compare Fig. 5, I and L). To test whether ConcanavalinA labeled the same ER (sub)domains as anti-ER BiP antibodies, cells were dual labeled with anti-BiP and ConcanavalinA-Alexa-594. Immunofluorescence displayed in Figure 5M is attributable to anti-ER BiP antibodies, whereas the fluorescence signal in Figure 5N is attributable to ConcanavalinA-Alexa 594. In the merged image (Fig. 5O), a near perfect colocalization (yellow) is observed for both ER markers in the same reticular compartment. Control experiments verified that background fluorescence was not being observed when primary or secondary antibodies were omitted, or when irrelevant primary antibodies (e.g. rabbit anti-pea reversibly glycosylated protein antiserum, mouse anti-maize ATPase monoclonal antibodies) were added with secondary antibodies (data not shown). In summary, endogenous AtPex16p resides within most of the cellular ER compartment and all peroxisomes of each cell (Fig. 5, C and F) indicating a steady-state coexistence within these two organelles.
It was of interest to learn whether there was a similar coexistence in Arabidopsis plants. We examined root cells of nontransformed plants because AtPex16p-42 and 52-kD polypeptides were identified in subfractions of roots (Fig. 2, bottom). Figure 6, A and B, reveal that endogenous AtPex16p colocalizes with peroxisomal catalase within root cells. Endogenous AtPex16p and peroxisomal catalase also were colocalized in green leaf peroxisomes (data not shown). However, AtPex16p was not observed also in a reticular cytoplasmic compartment reminiscent of ER in root or leaf cells. Attempts to visualize ER in these cells with Concanavalin A-Alexa594 or anti-castor calnexin antiserum also were unsuccessful, indicating that these hand-sectioned, fixed tissues were not amenable to ER fluorescence microscopy imaging. The peroxisomal localizations provide direct evidence for the in vivo localization of endogenous AtPex16p in peroxisomes of Arabidopsis plant cells.
Figure 6.

AtPex16p exists in Arabidopsis root cell peroxisomes. A and B, Representative confocal optical sections of Arabidopsis root cells dual labeled with rabbit PA anti-AtPex16p IgGs (1:100, overnight) and mouse monoclonal anti-catalase IgGs (1:2, overnight). Hand-sectioned roots from 4-week-old plants were fixed in formaldehyde, digested partially in a mixture of pectinase and cellulase, and cell membranes were permeabilized in Triton X-100. A, Punctate immunofluorescence (Cy2) pattern of endogenous AtPex16p (arrows). B, Superimposable punctate peroxisomal catalase immunofluorescence (Cy5) pattern in the same cells (arrows). Bar = 5 μm.
In Vitro Localizations of Endogenous AtPex16p: Suc Gradient-Isolated Organelles
Cell fractionation experiments were carried out with suspension cells to compare in vitro localizations with our immunofluorescence localizations (Fig. 5). Clarified homogenates containing organelles (except nuclei and plastids) were applied to Suc density gradients. Figure 3 in Lisenbee et al. (2003a) shows a representative gradient profile illustrating the positions with marker proteins of peroxisomes, mitochondria, and ER. Figure 7 in this article presents a representative immunoblot of pooled fractions collected from a similar Suc gradient. Lane 1 reveals that the clarified homogenate (applied sample) possesses AtPex16p-42 and 52-kD polypeptides, peroxisomal membrane APX, and the ER marker calnexin. Lane 2 shows that AtPex16p-42 and 52-kD polypeptides are recovered in pooled isolated peroxisomes with APX but not with calnexin (ER). Lane 3 shows that these two polypeptides also are in pooled fractions collected at the top of the gradient that likely possess membrane vesicles of varied origin including ER (calnexin is present). Lane 4 shows as anticipated that AtPex16p-42, 52-kD polypeptides, APX, nor calnexin exist in the pooled soluble fractions collected above the gradient.
Figure 7.

AtPex16p-42 and 52-kD polypeptides exist in Suc-gradient-isolated peroxisomes and vesicles. Arabidopsis suspension cell (7 d) clarified homogenates (applied sample; 1,500g, 15 min supernatant of 1,000 psi pressure cell ruptured cells; lane 1) were layered onto 30% to 59% (w/w) Suc gradients and centrifuged at 50,000g (90 min) in a vTi-50 rotor. Proteins in fractions with intact peroxisomes (catalase activities, 48%–54% w/w Suc; lane 2), vesicles (32%–44% w/w Suc; lane 3), and non-particulate, soluble fractions (10%–15% w/w Suc; lane 4) were DOC solubilized, TCA precipitated, separated in SDS gels, electroblotted onto Immobilon, and then probed with anti-AtPex16p-42 IgGs (1:100, overnight), anti-cucumber peroxisomal APX IgGs (1:1,000, 1 h), or anti-castor calnexin antiserum (1:5,000, 1 h). Polypeptides were visualized via chemiluminescence. A total of 150, 25, or 25 μg protein was added per well of SDS gels used for blots probed for AtPex16p, APX, or calnexin, respectively.
Mg2+-induced shift experiments were conducted to more specifically resolve and/or confirm the membrane site of AtPex16p found in microsomal pellets (Fig. 3, lane 3), membrane vesicle fractions (Fig. 7, lane 3), and reticular immunofluorescence images (Figs. 4 and 5). Microsomal pellets with or without Mg2+ were applied to Suc density gradients (with or without Mg2+); following centrifugation, the distribution of AtPex16 and calnexin polypeptides was compared in the two gradients (Fig. 8). Mg2+ maintains polysome binding to membrane vesicles derived from rough ER, whereas the absence of Mg2+ dislodges polysomes from rougher vesicles making them less dense than polysome-bound vesicles. In the (−) Mg2+ gradient (Fig. 8), both AtPex16-42 and 52-kD polypeptides, coincident with calnexin, were found in fractions ranging from about 15% to 41% (w/w) Suc. However, in the (+) Mg2+ gradient, both the AtPex16p-42 and 52-kD polypeptides, coincident with calnexin, were found in heavier density fractions and in the pellet (P) in response to a Mg2+- induced shift. The 42-kD polypeptide bands were difficult to see in the same fractions as the 52-kD polypeptides due to their relatively lower abundance. However, longer chemiluminescence film exposures, which blurred 52-kD bands, clearly revealed 42-kD bands in these fractions (data not shown). These results indicate that both AtPex16p polypeptides are present in rough ER-derived microsomal vesicles.
Figure 8.

Rough ER vesicles that undergo a Mg2+ -induced shift in Suc gradients possess both AtPex16p-42 and 52-kD polypeptides. Arabidopsis suspension cell (7 d) clarified homogenates (1,500g, 15 min supernatant of 2,000 psi pressure cell-ruptured cells) were prepared in homogenizing medium with 5 mm MgCl2 (+Mg2+) or 2 mm EDTA (−Mg2+). Microsomes (200,000g, 1 h) were layered onto 15% to 45% (w/w) Suc gradients with 5 mm MgCl2 or 2 mm EDTA and centrifuged at 125,000g (2.5 h) in a SW28.1 rotor. Proteins in the 1 mL fractions were DOC solubilized, TCA precipitated, applied to SDS gels (about 150 μg protein per well), electroblotted to Immobilon, and probed with anti-AtPex16p-42 IgGs (1:100, overnight) or anti-castor calnexin antiserum (1:5,000, 1 h) followed by chemiluminescence detection of polypeptides. A pellet (P) at the bottom of both gradient tubes was resuspended in homogenizing medium, proteins were DOC solubilized, TCA precipitated, and applied (150 μg protein) to the left-most well of SDS gels.
The Mg2+-gradient pellet, which possessed abundant AtPex16p-42 and 52-kD polypeptide was fixed and resin embedded for thin-section electron microscopy examinations (Lisenbee et al., 2003a). Figure 9 shows a representative electron micrograph of the pellet collected at the bottom of the gradient tube confirming that Mg2+ gradient pellets were composed entirely of polysome-studded membranes characteristic of rough ER vesicles.
Figure 9.
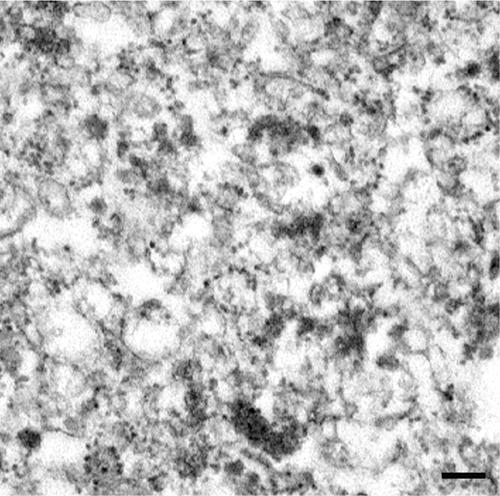
Representative electron micrograph of the pellet possessing prominent Mg2+-shifted AtPex16p-42 and 52-kD polypeptide bands (P, Fig. 8) indicating that both of these polypeptides exist within rough ER vesicles. These and other thin-sectional views were made in the mid region of the pellet, and all views revealed a preponderance of polysome-bearing membrane vesicles characteristic of rough ER vesicles throughout the sections (courtesy of C. Lisenbee). Bar = 0.2 μm.
In summary, these in vitro data demonstrate that endogenous AtPex16p-42 and 52-kD polypeptides are localized to peroxisomes and rough ER vesicles, thus confirming interpretations of images obtained from in vivo immunofluorescence experiments (Figs. 4 and 5).
Membrane Association and Topological Orientation of AtPex16p-Isolated Peroxisomes and Rough ER Vesicles
The membrane associations of AtPex16p-42 and 52-kD polypeptides with purified peroxisomal and rough ER vesicle membranes were determined using the same solution solubility approach that was conducted with crude microsome pellets (Fig. 3). A necessary but inconsequential difference was that the peroxisomal and ER vesicle fractions were incubated sequentially in the solutions rather than individually as with the microsomal fractions. Immunoblot results shown in Figure 10 illustrate the results of these experiments. Lane 1 shows that AtPex16p-42 polypeptides in peroxisomes and ER vesicles were almost completely extracted (solubilized) in 0.2 m KCl, whereas the 52-kD polypeptides were only partly extracted. Sequential treatment of the KCl-insoluble pellet with alkaline Na2CO3 led to the extraction of more, but not all, of the 52-kD polypeptide from the membranes in both peroxisomes and rough ER vesicles (lane 2). Incubation of the KCl/Na2CO3-insoluble pellet in DOC resulted in removal of essentially all of the remaining 52-kD polypeptides from both peroxisomes and rough ER (lane 3). The DOC-insoluble pellet did not possess 42- or 52-kD polypeptides (lane 4). Positive controls gave expected results. Neither peroxisomal APX nor ER calnexin were solubilized in KCl; instead, they were solubilized partially in alkaline sodium carbonate and then in DOC. Some APX and calnexin remained in the final membrane pellet. Catalase was included as a peroxisomal matrix protein control. Portions remaining after bursting peroxisomes in buffer were solubilized partially in KCl, and the remainder was extracted in alkaline carbonate (data not shown). These results were similar to those shown in Lisenbee et al. (2003a).
Figure 10.

Both the AtPex16p-42 and 52-kD polypeptides exhibit similar behavior (solubilities) in reagents used to assess their association with peroxisome and ER vesicle membranes. Pooled Suc gradient-isolated peroxisomes and Mg2+-shifted rough ER vesicles (pellet plus Suc fractions 43%–38% [w/v], Fig. 8) were treated sequentially with two volumes of solutions made to final concentrations of 0.2 m KCl (lane 1), 0.1 m sodium carbonate, pH 11.5 (lane 2), and 0.05% (w/v) DOC (lane 3), ending with a detergent-insoluble pellet (lane 4). Solubilized proteins were TCA precipitated, applied to SDS gels, electroblotted, and membranes probed with anti-AtPex16p-42 IgGs (1:100, overnight), anti-cucumber peroxisomal APX IgGs (1:1,000, 1 h), or anti-castor calnexin antiserum (1:5,000, 1 h) for chemiluminescence detections. A total of 150, 25, or 25 μg proteins were added per well of SDS gels used for detections of AtPex16p, APX, or calnexin, respectively.
Figure 11 presents immunoblot results of Proteinase K digestions with and without Triton X-100 to determine the topological orientation of AtPex16p-42 and 52-kD polypeptides in isolated, intact peroxisomes and rough ER vesicles. The procedure employed was similar to that described in detail by Lisenbee et al. (2003a). Data for a representative concentration of applied Proteinase K are shown. Both polypeptide bands were present in the untreated samples (lanes 1 and 4, top sections), with a relatively greater proportion of 42- to 52-kD polypeptides in peroxisomes, and the reverse proportionality in ER vesicles. These proportions consistently were observed in replicate experiments. AtPex16p-42 and 52-kD polypeptides in peroxisomes were not digested in Proteinase K in the absence of detergent (lane 2, top). However, the multiple, lower molecular mass bands repeatedly observed only in this lane (lane 2) may reflect a partial degradation of the 52-kD polypeptides. On the other hand, lane 5 (top) illustrates that both 42- and 52-kD polypeptides in ER vesicles were digested in the absence of Triton X-100, indicating that these polypeptides were on the outer surface of the ER vesicles. Other polypeptides protected from Proteinase K digestions were not observed in the lower part of these gels. Both polypeptides in peroxisomes and ER vesicles were digested in the presence of the detergent (lanes 3 and 6, top).
Figure 11.

Both AtPex16p-42 and 52-kD polypeptides are located mostly on the matrix side of the peroxisomal boundary membrane, whereas both polypeptides are located on the cytosolic side of rough ER vesicles. Pooled Suc gradient-isolated intact peroxisomes (54%–48% w/w Suc; lanes 1–3) and Mg2+-shifted rough ER vesicles (pellet plus Suc fractions 43%–38% w/w, Fig. 8; lanes 4–6) were subjected to Proteinase K digestion (4:1 [w/w] sample protein:Proteinase K) with (+) and without (−) pretreatment in 1% (v/v) Triton X-100. Proteins in all samples were solubilized in DOC, TCA precipitated, applied to SDS gels, electroblotted onto Immobilon, and probed with anti-AtPex16p-42 IgGs (1:100, overnight), anti-cucumber peroxisomal APX IgGs (1:1,000, 1 h), anti-castor calnexin antiserum (1:5,000, 1 h), anti-cottonseed catalase IgGs (1:1,000, 1 h), or anti-maize BiP monoclonal antibodies (1:1,000, 1 h) for chemiluminescence detections. Protein samples (150 μg) were added per well to SDS gels probed for AtPex16p, whereas 25 μg proteins per well was added to SDS gels used for blots probed with APX, catalase, calnexin, and BiP.
Positive controls for the peroxisomal digestion experiments were cytosolic-facing, membrane-bound peroxisomal APX and matrix catalase. Peroxisomal APX was digested with Proteinase K without Triton X-100 (lane 1, 2, and 3, middle) in accordance with Lisenbee et al. (2003a). Catalase was protease protected until the boundary membranes were solubilized in detergent as expected for a matrix protein (lanes 1, 2, and 3, bottom). Positive controls for the ER vesicle digestions were mostly lumenal-facing integral membrane-bound calnexin (Huang et al., 1993) and lumenal BiP. As expected, calnexin was digested partially without added detergents (top band, lane 5; middle section) and then a lower molecular mass band was digested upon addition of detergent as shown previously (Lisenbee et al., 2003a). The anti-calnexin antibody also recognizes lumenal calreticulin; thus the results observed in lane 5 likely are digestions of both calreticulin and lumenal-facing calnexin. ER BiP was protease protected until the addition of Triton X-100 (lanes 4, 5, and 6, bottom).
In summary, the collective results presented in Figures 10 and 11 indicate that both AtPex16p-42 and 52-kD polypeptides are associated similarly with peroxisomal and ER membranes, although they have different topological orientations in peroxisomes (mostly inside) and ER vesicles (mostly outside). Interpretations of whether they are integral or peripheral membrane polypeptides in these two organelles are discussed.
DISCUSSION
New insights are presented on the functional characteristics and subcellular localizations of endogenous AtPex16p in Arabidopsis suspension cells and plants. Lin et al. (1999, 2004) accumulated evidence for AtPex16p/sse1 being a plant peroxin homolog. Their conclusion was based on three lines of evidence: (1) absence of peroxisomes in sse1 mutant embryos, (2) complementation of both the shrunken seed and lethal phenotypes of sse1 mutants with GFP::AtPEX16 chimeras, and (3) the localization of overexpressed GFP::AtPex16p to aggregated peroxisomes in complemented transgenic plants. Localization to altered peroxisomes was not a mitigating factor in our study. That is, specific affinity-purified IgGs directly demonstrated the existence of endogenous AtPex16p within individual peroxisomes in leaf, root, and suspension cells in nontransformed plants and cultured cells. Our data corroborate and significantly extend the assertion of Lin et al. (2004) that the sse1 protein, AtPex16p, functions as a peroxin in Arabidopsis. Additionally, we found that endogenous AtPex16p-42 polypeptides are immunorelated to 52-kD polypeptides, both of which are membrane-associated polypeptides (proteins) that coexist at steady state within ER and peroxisomes.
Is There a Structural and/or Functional Relationship(s) between AtPex16p-42 and 52-kD Polypeptides?
BLAST searches of Arabidopsis databases reveal only one PEX16 gene that encodes a polypeptide of approximately 42 kD and not a similar 52-kD polypeptide. In addition, reverse transcription-PCR of total RNA extracted from Arabidopsis suspension cells revealed only one band, i.e. no other splice variant was observed (data not shown). Also, Lin et al. (2004) reported a single mRNA band for PEX16 in Arabidopsis flowers, siliques, roots, emerging leaves, and developing and germinated seeds. However, antibodies affinity purified to the AtPex16p-42 polypeptides specifically and consistently recognized two polypeptide bands on immunoblots, the expected 42-kD polypeptide band and the unexpected 52-kD polypeptide band in fractions derived from Arabidopsis plant parts and suspension cells (Figs. 2, 3, 7, and 8). Specificity of the PA and 42-kD-antigen affinity-purified anti-AtPex16p IgGs was demonstrated by their sole recognition in transformed cells of transiently expressed mycAtPex16p-42 (Fig. 4, C–E). Hence, several lines of evidence convincingly indicate that these 52-kD polypeptides are immunorelated to the authentic AtPex16p-42 polypeptides.
Antibodies specific to the endogenous human and Y. lipolytica Pex16p homologs recognized on blots only one authentic polypeptide band, a 38-kD HsPex16p (South and Gould, 1999) and a 44-kD YlPex16p band (Eitzen et al., 1997). Titorenko and Rachubinski (1998a) concluded from results of endo H digestions and tunicamycin treatments that YlPex16p in the ER lumen was N-linked core glycosylated with one chain before moving in vesicles to peroxisomes where YlPex16p resided as a glycoprotein. A single predicted glycosylation site is apparent in AtPex16p similar to the one observed in YlPex16p (Fig. 1). Deglycosylated YlPex16p was observed on immunoblots as 42-kD polypeptides compared to 44-kD glycosylated YlPex16p (Titorenko and Rachubinski, 1998a). Thus, the 52-kD polypeptides likely are not single-chain, N-glycosylated forms of AtPex16p-42 polypeptides. Database searches did not reveal predicted sites for prenylation or other lipid anchors such as glycosylphosphotidylinositol. Numerous attempts to ascertain a reliable amino acid sequence via proteomics procedures were unsuccessful. Hence, no clues are available on the identity of the immunorelated 52-kD polypeptides.
Within this context, the question arose as to whether the 42- and 52-kD polypeptides formed a heteromeric AtPex16p. Although their topological orientations were essentially the same in each membrane compartment, they exhibited significantly different solubility behaviors. For example, the 52-kD polypeptides always were recovered in membrane fractions derived from homogenates of plant parts or suspension cells (Figs. 2, bottom, and 3, lane 3), whereas AtPex16p-42 polypeptides consistently were recovered in both water-soluble and membrane fractions (e.g. Fig. 2, bottom section and Fig. 3, lanes 2 and 3).
Besides revealing a difference in solubility behavior, the solubility of the 42-kD polypeptides in water may be indicative of a cytosolic fraction of these polypeptides. Titorenko and Rachubinski (2000) reported that YlPex6p, a peripheral membrane protein located on the cytosolic surface of preperoxisomal P2 vesicles, was recovered in membrane and water-soluble fractions. Their explanation was that this peroxin shuttled during peroxisomal biogenesis between the surface of the P2 vesicles and the cytosol. This phenomenon probably does not apply to our plant cells, because no direct evidence has been presented in plants for the existence of differentiated populations of preperoxisomal vesicles or for shuttling to/from the cytosol.
Another possible explanation comes from the recent demonstrations that HsPex16p and other peroxisomal membrane proteins (PMPs) bind in vitro to Pex19p, which functions as a cytosolic chaperone/carrier to prevent precipitation of PMPs during intracellular translocations (Shibata et al., 2004). The Pex19p-PMP complexes were recovered in water-soluble fractions, whereas HsPex16p and other PMPs alone in membranes. Pex19p interactions with nascent PMPs were (including peroxins) have not been described in plants, but likely occur.
The differential solubility of 42- and 52-kD polypeptides also may be attributable to AtPex16p being a weakly bound peripheral membrane protein, of which a portion becomes dislodged from the membrane during cellular disruptions and ends up in the homogenization medium. The AtPex16p-42 polypeptides remaining in the microsomal membranes were almost completely solubilized in 0.2 m KCl (Fig. 3, lane 4). Such salt treatments extract peripheral membrane proteins bound to the membranes via electrostatic attractions (Fujiki et al., 1982; Overvoorde and Grimes, 1994; Takeda and Kasamo, 2002). In separate experiments, alkaline Na2CO3 solubilized all of the AtPex16p-42 kD in microsomal membranes (Fig. 3, lane 6). This solution denatures proteins without disrupting the organization of the bilayer and releases peripheral membrane proteins, ER lumen proteins, and peroxisomal matrix proteins (Fujiki et al., 1982; Eitzen et al., 1997; Mothes et al., 1997; Obrdlik et al., 2000; Lisenbee et al., 2003a). These combined data indicate that AtPex16p-42 polypeptides are weakly bound peripheral membrane polypeptides (proteins), not partially cytosolic, and may well explain their recoveries in homogenizing medium and microsomal membranes.
Properties of the 52-kD polypeptides are demonstrably different than AtPex16p-42 polypeptides. The 52-kD polypeptides were not solubilized in water (homogenizing medium) or 0.2 m KCl. Nevertheless, their solubility in alkaline Na2CO3 indicated they were peripherally associated with the microsomal, ER, and peroxisomal membranes, but more tightly bound than AtPex16p-42 polypeptides. The positive integral membrane controls, i.e. peroxisomal APX and ER calnexin that were not solubilized in sequential KCl and alkaline Na2CO3 treatments, support our conclusions for peripheral membrane associations. Combined, the data do not indicate a heteromeric combination of 52- and 42-kD polypeptides to form functional AtPex16p. We suspect that 52-kD polypeptides are posttranslationally modified AtPex16p polypeptides that complement the function of AtPex16p-42 in ER and peroxisomes.
Significance of the Coexistence of AtPex16p in Peroxisomes and ER
In all other studies, Pex16p homologs were observed at steady state only in peroxisomes. Our combined in vitro and in vivo results with specific anti-AtPex16p IgGs convincingly revealed a coexistence of AtPex16p in both ER and peroxisomes in Arabidopsis suspension cells. Lin et al. (2004) may not have observed a similar coexistence of overexpressed GFP::AtPex16p in their transgenic Arabidopsis plants because of the abnormal distribution of organelles in the cells (e.g. single enlarged peroxisomes). We observed endogenous AtPex16p within normal peroxisomes in wild-type Arabidopsis leaf and root cells, but not also within ER due to technical limitations. The 42- and 52-kD polypeptides were immunodetected with the same IgGs in plant and suspension cells. It is our contention, among these minor discrepancies, that the organellar coexistence of AtPex16p is a feature common to both Arabidopsis plant and suspension-cultured cells.
The steady-state coexistence of AtPex16p suggests that it serves more than one function in cells. An indicator that it has a different role in each compartment is the distinctly different membrane topological orientation of AtPex16p-42 (and 52-kD) polypeptides in ER (cytosolic side) and peroxisomes (matrix side). However, these orientations per se do not point to specific functions.
Insights into a main function for the peroxisomal AtPex16p come from the following: YlPex16p, which also is a peripheral membrane protein facing the matrix of peroxisomes, is required for assembly and proliferation of Y. lipolytica peroxisomes from preperoxisomal vesicles (Eitzen et al., 1997; Guo et al., 2003). Ylpex16, a mutant defective in formation of peroxisomes, was partially complemented by AtPEX16 (SSE1; Lin et al., 1999). Although HsPex16p is an integral membrane protein, it also participates in formation of peroxisomes from non-ER-derived preperoxisomal vesicles (South and Gould, 1999). These similarities point to a role of peroxisomal AtPex16p in plant peroxisome assembly.
A function of the ER-associated AtPex16p may be participation in ER-dependent oil and protein body formation in developing seeds, which initially was put forward by Lin et al. (1999). Since Arabidopsis suspension cells do not possess protein bodies, the ER-associated AtPex16p may participate only in production of prevalent oil bodies in these cells. Such an ER-related function also was surmised for YlPex16p in Y. lipolytica cells grown on oleic acid (Lin et al., 1999, 2004).
Although separate, single functions for AtPex16p (sse1) have been proposed above, the possibility exists for more than one function of AtPex16p in each compartment. For example, the same or a different resident ER AtPex16p may mediate sorting of selected PMPs (e.g. peroxisomal APX) and/or peroxins (e.g. peroxisomal AtPex16p as is YlPex16p) indirectly to peroxisomes from ER. Thus, AtPex16p may also function as an early peroxin within subdomains of ER. These considerations suggest that AtPex16p has an overall bifunctional role within cells or that it serves a bifunctional role within ER and yet another role within peroxisomes, making it multifunctional within cells. These possible scenarios currently are under investigation.
MATERIALS AND METHODS
Arabidopsis Plant Growth and Suspension Cell Cultures
Arabidopsis (Arabidopsis thaliana ecotype Columbia) seeds were surface sterilized in 2% NaOCl and grown on a 4:1:1 mixture of seed starter soil, perlite, and vermiculite, respectively. The seeds were vernalized for 3 d at 4°C and then grown under long-day conditions of 16-h-light (100 μE m−2 s−1)/8-h-dark cycle at 22°C for 4 weeks.
Suspension cell cultures of Nicotiana tabacum L. cv BY-2 and Arabidopsis var. Landsberg erecta were grown and maintained as described previously by Lee et al. (1997) and Lisenbee et al. (2003a), respectively.
Production of AtPex16p Antiserum, and Affinity Purifications of IgGs
Antiserum produced in rabbits, which were immunized with purified Arabidopsis Pex16/sse1 protein (AtPex16p) that was overexpressed in Escherichia coli, was a kind gift from Yun Lin and H.M. Goodman (Harvard Medical School, Boston). Briefly, overexpressed AtPex16p with a hexahistidine tag at the C terminus was purified from E. coli extracts on a Ni metal affinity column. Bands with AtPex16 42-kD polypeptides (predicted molecular mass) were excised from SDS gels and injected with adjuvant into two rabbits. Hyperimmune antiserum was collected from both animals; antiserum from one of the rabbits was used for affinity purifications of IgGs for this study.
PA anti-AtPex16p IgGs were prepared via Protein A-Sepharose column chromatography from the anti-AtPex16p antiserum. Kunce et al. (1988) describes details of the procedure.
Anti-AtPex16-42 IgGs were prepared from PA anti-AtPex16p IgGs to produce antigen affinity-purified (42-kD polypeptides) IgGs as follows: Clarified homogenates of Arabidopsis suspension cells were prepared from 7-d-old suspension cell cultures. Pelleted cells resuspended in 1.5 volumes of homogenizing medium (25 mm HEPES-KOH, pH 7.5, 3 mm dithiothreitol, and 0.5 mm phenylmethylsulfonyl fluoride) were ruptured in one passage through a French pressure cell (1-inch diameter piston) at 5,000 psi, and then centrifuged in a Sorvall SS-34 fixed angle rotor (Sorvall Products, Newtown, CT) at 1,500g for 15 min. To the supernatant, sodium DOC (Sigma-Aldrich, St. Louis) was added to 0.05% (w/v) final concentration, and proteins were precipitated by incubation for 30 min (4°C) in 10% w/v TCA (final concentration) and centrifugation at 10,000g for 15 min (4°C) in a Sorvall SS-34 rotor. Proteins were resuspended in appropriate amounts of SDS sample buffer (125 mm Tris-HCl, pH 6.8, 20% [v/v] glycerol, 4% [w/v] SDS, and 0.00125% [w/v] bromphenol blue), and suspensions were neutralized with solid Tris. Just before loading SDS gels, freshly prepared 500 mm dithiothreitol was added to 10 mm (final concentration), the reduced samples were boiled for 8 min and then added to wells of 12% precast Mini-Protean II polyacrylamide gels (Bio-Rad, Hercules, CA). Protein (150 μg), estimated using the Coomassie Blue dye-binding method (standard assay) with bovine plasma gamma globulin (Bio-Rad) as the standard, was added to each well. The SDS gels were run at 120 v for about 35 to 40 min until the bromphenol dye front reached the bottom of the gel. Separated proteins were then electroblotted with a semi-dry transfer apparatus (Bio-Rad) onto polyvinylidene difluoride membranes (Immobilon P; Millipore, Bedford, MA) as described by Lisenbee et al. (2003a). Time of electroblotting per four gels was 90 min. To visualize proteins, the entire membrane was incubated in Ponceau S (Roche, Mannheim, Germany) for 10 min. Membranes were destained in phosphate-buffered saline (PBS) until the 42-kD bands were visible and their position marked with ink. Ponceau S stain was completely removed with six PBS washes and then two Immobilon membrane strips about 3 mm wide, each including 42-kD polypeptides from six wells, were cut out of the membrane blot. The strips were incubated (blocked) overnight in PBS containing 5% (w/v) bovine serum albumin (BSA; Sigma-Aldrich), washed three times in PBS, and then incubated in PA anti-AtPex16p IgGs (1:20) in PBS containing 1% (w/v) BSA for 4 h at room temperature. Following three washes in PBS using gentle inversion rocking, IgGs bound to the 42-kD polypeptides on the strips were eluted in Gly elution buffer (0.2 m Gly [Sigma], 1 mm EGTA [Sigma], pH 2.7) for 10 min. The eluant was neutralized with an equal volume (approximately 0.2 mL) of neutralizing buffer (100 mm Tris-HCl, pH 8.0, containing 0.1% [w/v] BSA and 0.02% (w/v) Na azide).
In Vitro Cell Fractionation Analyses: Clarified Homogenate and Microsomal Membrane Preparations; SDS-PAGE and Immunoblot Analyses; Suc Density Gradient Organelle Isolations; and Mg2+-Induced Shift Subfractionations, Membrane Protein Associations, and Topology
Lisenbee et al. (2003a) describe in detail most of the methods and procedures employed with suspension cells in this section. Pertinent modifications/variations in methodology and new procedures with suspension cells and plant parts are given in the above section and below.
Preparations of clarified homogenates (1,500g, 15 min supernatants of pressure cell-disrupted cells) from 7-d-old Arabidopsis cell cultures are similar to the ones described in the section directly above. The psi applied to disrupt Arabidopsis cells in one passage through the pressure cells varied depending on the experiments, i.e. 5,000 psi (preparations of microsomal membrane pellets, Figs. 2 and 3), 1,000 psi (isolations of peroxisomes in Suc density gradients, Figs. 7, 10, and 11), or 2,000 psi (Mg2+-induced shift experiments Figs. 8, 9, 10, and 11). Microsomal membrane pellets were derived from 1,500g (20 min) supernatants by centrifugation in a Beckman 70.1 Ti rotor at 200,000g for 1 h (4°C).
Cell fraction of plant parts was as follows. Roots, leaves, flowers, and siliques were harvested from 4-week-old Arabidopsis plants. These plant parts were frozen in liquid nitrogen and ground to a powder in a mortar. A volume of each powdered plant parts (roots, 5 mL; leaves; 10 mL, flowers; 8 mL, siliques; 5 mL) was mixed with an equal volume of homogenizing medium (see above section) and was ground further in a mortar to produce homogenates. Each homogenate was then centrifuged at 2,000g (30 min, 4°C) in a Sorvall SS-34 rotor, and the resulting supernatant was centrifuged at 20,000g (45 min, 4°C) in a Beckman 70.1 Ti rotor. This 20,000g supernatant was centrifuged in the same rotor at 200,000g for 1 h at 4°C to yield microsomal pellets. Each microsomal pellet was resuspended in 500 μL of homogenizing medium.
For immunoblot analyses of polypeptides separated by SDS-PAGE and electroblotted to Immobilon membranes, samples from all experiments, unless noted otherwise in figure legends, were made to 0.05% (w/v) sodium DOC, proteins were precipitated with 10% (w/v) TCA, separated in 12% SDS gels, and electroblotted as described in more detail in the above section. In all experiments aimed at detecting AtPex16p on blots, 150 μg protein was applied to each SDS gel well, while 25 μg protein was applied per well for detection of APX, calnexin, catalase, and BiP on blots. For chemiluminescence detections of the various antibody probes, Immobilon membranes were blocked in 5% (w/v) nonfat dry milk in Tris-buffered saline plus Tween 20 (TBST) overnight at 4°C. Primary and secondary antibodies were applied separately in 1% (w/v) nonfat dry milk with three exchanges of TBST after each antibody application. Components in an Immunstar chemiluminescence kit (Bio-Rad) were used to visualize immuno-reactive proteins (recorded on Kodak X-OMAT autoradiography film [Eastman-Kodak, Rochester, NY]).
Rabbit primary and goat anti-rabbit secondary antibodies (and concentrations) were as follows: rabbit anti-AtPex16p antiserum (1:1,000), rabbit PA anti-AtPex16p IgGs (1:1,000), rabbit anti-AtPex16p-42 IgGs (1:100), rabbit anti-castor calnexin antiserum (1:5,000; provided by Sean Coughlan; Coughlan et al., 1997), rabbit PA anti-cottonseed catalase IgGs (1:1,000; Kunce et al., 1988), rabbit PA anti-cucumber peroxisomal APX IgGs (1:1,000; Corpas et al., 1994), rabbit anti-maize BiP monoclonal antibodies (1:1,000; provided by Eliot Herman), goat anti-rabbit alkaline phosphatase conjugate (1:10,000; Bio-Rad). All antibodies were applied in 1% (w/v) nonfat dry milk in TBST separately for 1 h at room temperature, except anti- AtPex16p-42 IgGs, which were applied overnight at room temperature.
Microsomal membrane pellets (200,000g, 1 h) were incubated individually, and in some cases sequentially, in different solutions to assess the association of AtPex16p and/or other polypeptides with these membranes. Pellets possessing approximately 4 mg protein were incubated in 0.2 m KCl, 0.1 m sodium carbonate (pH 11.5), and/or 0.05% (w/v) sodium DOC. To assess whether the concentration of KCl, or ratio of protein to KCl, variously affected the solubilization of AtPex16p, pellets (4 mg protein) were incubated in 2 mL of 0.2 m KCl, or 4 mL of 0.2 m, 0.4 m, or 1 m KCl in 25 mm HEPES-KOH, pH 7.5. All samples (in varying concentrations of KCl, 0.1 m Na2CO3 (pH 11.5), or 0.05 or 0.5% (w/v) DOC in water were incubated for 1 h at 4°C with brief vortexing every 10 min. Following incubations in each of these solutions, the samples were centrifuged at 200,000g (30 min) in a Beckman 70.1 Ti rotor, the supernatants and pellets (resuspended in 25 mm HEPES, pH 7.5) were processed for SDS-PAGE, and polypeptides were analyzed on immunoblots. Isolated peroxisomes (equilibrium Suc density gradients) and rough ER vesicles (Mg2+-induced shift Suc gradients) were incubated sequentially in the lower concentrations of these solutions (Fig. 10) as described above in this section. Protein concentrations were not measured in each case but are known to be significantly lower than in microsomal pellets; therefore, solubilizations in the lower concentrations were not influenced by the ratio of protein-to-solution concentration.
Procedures and methods for isolation of peroxisomes and membranes vesicles in Suc density gradients (Figs. 10 and 11), separations of ER vesicles in Suc gradients ± Mg2+ (Fig. 8), and Proteinase K digestions and Triton X-100 membrane solubilizations to assess membrane protein topologies (Fig. 11) were followed as described by Lisenbee et al. (2003a). However, in each case, the methods for SDS-PAGE, protein determination, and detergent solubilization of proteins were done as described in the above section.
Microprojectile Bombardment, and Processing of Suspension Cells and Plant Parts for Immunofluorescence Confocal Laser Scanning Microscopy
Arabidopsis cells were harvested 4 d post subculture, were fixed in 4% (w/v) formaldehyde (prepared from paraformaldehyde, Ted Pella, Redding, CA) for 1 h, and further processed for immunolabeling. Alternatively, harvested cells were transiently transformed via biolistic bombardment with pRTL2/mycAtPEX16, and then fixed in 4% (w/v) formaldehyde 2.5 h post bombardment. Details of biolistic bombardment and processing of wild-type and transiently transformed suspension cells for immunofluorescence labeling are described in Lisenbee et al. (2003a, 2003b) and Flynn et al. (2005). An important modification included in Flynn et al. (2005) that improved cell morphology was to incubate formaldehyde-fixed, nontransformed, and transiently transformed Arabidopsis cells in 0.1% (w/v) Pectolyase Y-23 (Seishin Pharmaceutical, Tokyo) and 0.05% (rather than 1.0%; w/v) Cellulase RS (Karlan Research Products, Santa Rosa, CA). The cells were then processed for immunofluorescence labeling using our standard tube procedure or a modified on-slide procedure. The latter procedure differed from our tube procedure in that a portion of the formaldehyde-fixed, pectolyase/cellulase digested cells (processed together in a tube) were removed and adhered to a poly-l-lysine-coated microscope slide, permeabilized in 0.5% Triton X-100, and then immunolabeled in primary and secondary antibodies. This on-slide procedure is now being used routinely in our laboratory for immunofluorescence detection of what apparently are low abundance (membrane) proteins, which are not labeled using the tube procedure (Figs. 4 and 5).
Roots and green leaves harvested from 4-week-old Arabidopsis plants were hand sectioned with a single-edge razor blade, placed in wells of a spot plate, and then fixed in 4% (w/v) formaldehyde for 30 min (room temperature). After three washings in PBS, cell walls were digested in 0.1% (w/v) Pectinase (cat. no. P2401, Sigma-Aldrich) and 1% (w/v) Cellulase Y-C (Seishin Pharmaceutical, Tokyo Japan, distributed by Karlan Research Products, Santa Rosa, CA) for 30 min at 30°C followed by three washes in PBS. Membranes were permeabilized in 0.5% Triton X-100 (Sigma-Aldrich) in PBS and then washed in three changes of PBS. Sections were incubated in TBST with 5% (w/v) protease-free BSA (no. A-3059, Sigma-Aldrich) for 30 min. Primary (overnight incubations) and secondary (1 h incubations) antibodies diluted in TBST-5% BSA were applied to the sections. The sections were removed from the wells and then mounted on slides and coverslipped in 90% (v/v) glycerol with n-propyl gallate to prevent photo bleaching.
Antibody and ConcanavalinA-Alexa594 concentrations and room temperature incubation times used for microscopy of the plant parts were as follows: Sources not given in the immunoblot section above are given below: rabbit PA anti-AtPex16p IgGs (1:100, overnight), mouse monoclonal anti-salicylic acid BiP (catalase) antibody (1:2, overnight; Chen et al., 1993), rabbit anti-castor calnexin antiserum (1:200, overnight), ConcanavalinA-Alexa594 (1:10, overnight; Molecular Probes, Eugene, OR).
Antibody and ConcanavalinA-Alexa594 concentrations and incubation times at room temperature used for suspension cells were as follows: rabbit anti-AtPex16p-42 IgGs (1:10, overnight); rabbit PA anti-AtPex16p IgGs (1:1,000, 2 h); mouse anti-c myc monoclonal antibodies (1:500, 1 h; purified 9E10, Covance Research Products, Berkley, CA), mouse monoclonal anti-salicylic acid BiP (catalase) antibody (1:5, 1 h), mouse anti-Hsc 70 monoclonal antibodies (BiP, 1:2,000, 1 h; StressGen, San Diego), Concanavalin A-Alexa594 (1:500, 1 h), goat anti-rabbit Cy2 (1:500, 1 h), goat anti-mouse Cy2 (1:500, 1 h), goat anti-rabbit Cy5 (1:500, 1 h), and goat anti-mouse Cy5 (1:500, 1 h). All of the cyanine-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (Westgrove, PA).
Wild-type suspension cells, plant parts, and transiently transformed suspension cells were imaged using a Leica DM RBE microscope equipped with Leica TCS NT scanning head (Leica, Heidelberg). The images were assembled and adjusted for brightness and contrast using Adobe Photoshop 7.0 (Adobe Systems, Mountain View, CA).
AtPEX16 Synthesis and Plasmid Construction
Total RNA was isolated from 4-d-old Arabidopsis suspension cells using RNeasy mini Kit (Qiagen, Valencia, CA) following the manufacturer's instructions with the following exception: Rather than starting with 100 mg of plant material, about 100 to 200 mg of Arabidopsis cells constituted the starting material. AtPEX16 cDNA was synthesized first from total RNA using RNeasyPlant Mini Kit (Qiagen) and then amplified with the Access reverse transcription-PCR system (Promega, Madison, WI) using the forward primers that replaced the start Met with an in-frame NheI site (5′-CATGCTAGCGAAGCTTATAAGCAATGGG-3′) and a reverse primer that introduced an in-frame XbaI site at the 3′ end of the stop codon (5′-CTGCTCTAGAACCTCACGATCCCGATATGTAAGTG-3′). These synthetic oligonucleotides were synthesized and purchased from Genetech Biosciences (Tempe, AZ). The resulting PCR product was cloned into the pCR2.1 TA shuttle vector (Invitrogen, San Diego) to yield pCR2.1/ NheI-AtPEX16-XbaI, which was sequenced (Arizona State University DNA laboratory) before insertion into the plant expression vector pRTL2/mycX prepared and donated by Dr. Robert Mullen (University of Guelph, Ontario, Canada). This plasmid has an in-frame XbaI site that follows DNA coding for a single copy of a myc epitope. pCR2.1/ NheI-AtPEX16-XbaI was then digested with Nhe1 and Xba1 and ligated into Xba1-digested pRTL2/mycX vector to yield pRTL2/mycAtPEX16 with 35S cauliflower mosaic virus promoter.
Distribution of Materials
Upon request, all novel materials described in this publication will be made available in a timely manner for noncommercial research purposes. No restrictions or conditions will be placed on the use of any materials described in this article that would limit their use in noncommercial research purposes.
Acknowledgments
We thank Dr. Yun Lin and Dr. H. M. Goodman for providing an ample supply of anti-AtPex16p antiserum. Michael Heinze and Cayle Lisenbee are thanked for their tutelage and insightful discussions, especially for the cell fraction experiments. Heather Gustafson's assistance with cell fractionation experiments and maintenance of cell cultures are greatly appreciated. Special thanks and sincere gratitude are given to Matthew Lingard for his thorough reading, insightful criticisms, and perceptive editing of the manuscript and for his gracious help and discussions in presentation of data in the figures.
This work was supported by the National Science Foundation (grant no. MCB–0091826 to R.N.T.) and in part by the William N. and Myriam Pennington Foundation. The Graduate Program in Molecular and Cellular Biology at Arizona State University also funded research assistantships for S.K.K.
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.105.061291.
References
- Baerends RJS, Faber KN, Kiel JAKW, Van der Klei IJ, Harder W, Veenhuis M (2000) Sorting and function of peroxisomal membrane proteins. FEMS Microbiol Rev 24: 291–301 [DOI] [PubMed] [Google Scholar]
- Baker A, Graham IA (2002) Plant Peroxisomes: Biochemistry, Cell Biology and Biotechnological Applications. Kluwer Academic Publishers, Dordrecht, The Netherlands
- Charlton W, Lopez-Huertas E (2002) PEX genes in plants and other organisms. In A Baker, I Graham, eds, Plant Peroxisomes. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 385–226
- Chen Z, Ricigliano JW, Klessig DF (1993) Purification and characterization of a soluble salicylic acid-binding protein from tobacco. Proc Natl Acad Sci USA 90: 9533–9537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpas FJ, Bunkelmann J, Trelease RN (1994) Identification and immunochemical characterization of a family of peroxisome membrane-proteins (Pmps) in oilseed glyoxysomes. Eur J Cell Biol 65: 280–290 [PubMed] [Google Scholar]
- Coughlan SJ, Hastings C, Winfrey R Jr (1997) Cloning and characterization of the calreticulin gene from Ricinus communis L. Plant Mol Biol 34: 897–911 [DOI] [PubMed] [Google Scholar]
- Distel B, Erdmann R, Gould SJ, Blobel G, Crane DI, Cregg JM, Dodt G, Fujiki Y, Goodman JM, Just WW, et al (1996) Unified nomenclature for peroxisome biogenesis factors. J Cell Biol 135: 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitzen GA, Szilard RK, Rachubinski RA (1997) Enlarged peroxisomes are present in oleic acid-grown Yarrowia lipolytica overexpressing the PEX16 gene encoding an intraperoxisomal peripheral membrane peroxin. J Cell Biol 137: 1265–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y, Tabak HF (1996) Proteins involved in peroxisome biogenesis and functioning. Biochim Biophys Acta 1286: 269–283 [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Elofsson A, von Heijne G, Cristóbal S (2003) In silico prediction of the peroxisomal proteome in fungi, plants and animals. J Mol Biol 330: 443–456 [DOI] [PubMed] [Google Scholar]
- Flynn CR, Heinze M, Schumann U, Gietl C, Trelease RN (2005) Compartmentalization of the plant peroxin, AtPex10p, within subdomain(s) of ER. Plant Sci 168: 635–652 [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol 93: 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Kit YY, Nicaud JM, Le Dall MT, Sears SK, Vali H, Chan H, Rachubinski RA, Titorenko VI (2003) Peroxisome division in the yeast Yarrowia lipolytica is regulated by a signal from inside the peroxisome. J Cell Biol 162: 1255–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Nito K, Toriyama-Kato K, Kondo M, Yamaya T, Nishimura M (2000) AtPex14p maintains peroxisomal functions by determining protein targeting to three kinds of plant peroxisomes. EMBO J 19: 5701–5710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honsho M, Hiroshige T, Fujiki Y (2002) The membrane biogenesis peroxin Pex16p: topogenesis and functional roles in peroxisomal membrane assembly. J Biol Chem 277: 44513–44524 [DOI] [PubMed] [Google Scholar]
- Hu JP, Aguirre M, Peto C, Alonso J, Ecker J, Chory J (2002) A role for peroxisomes in photomorphogenesis and development of Arabidopsis. Science 297: 405–409 [DOI] [PubMed] [Google Scholar]
- Huang AHC (1992) Oil bodies and oleosins in seeds. Annu Rev Plant Physiol 43: 177–200 [Google Scholar]
- Huang LQ, Franklin AE, Hoffman NE (1993) Primary structure and characterization of an Arabidopsis thaliana calnexin like protein. J Biol Chem 268: 6560–6566 [PubMed] [Google Scholar]
- Hunt JE, Trelease RN (2004) Sorting pathway and molecular targeting signals for the Arabidopsis peroxin 3. Biochem Biophys Res Commun 314: 586–596 [DOI] [PubMed] [Google Scholar]
- Kamada T, Nito K, Hayashi H, Mano S, Hayashi M, Nishimura M (2003) Functional differentiation of peroxisomes revealed by expression profiles of peroxisomal genes in Arabidopsis thaliana. Plant Cell Physiol 44: 1275–1289 [DOI] [PubMed] [Google Scholar]
- Kunau W-H, Erdman R (1998) Peroxisome biogenesis: Back to the endoplasmic reticulum? Curr Biol 8: R299–R302 [DOI] [PubMed] [Google Scholar]
- Kunce CM, Trelease RN, Turley RB (1988) Purification and biosynthesis of cottonseed (Gossypium hirsutum L.) catalase. Biochem J 251: 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarow PB (2003) Peroxisome biogenesis: advances and conundrums. Curr Opin Cell Biol 15: 489–497 [DOI] [PubMed] [Google Scholar]
- Lee MS, Mullen RT, Trelease RN (1997) Oilseed isocitrate lyases lacking their essential type I peroxisomal targeting signal are piggybacked to glyoxysomes. Plant Cell 9: 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Cluette-Brown JE, Goodman HM (2004) The peroxisome deficient Arabidopsis mutant sse1 exhibits impaired fatty acid synthesis. Plant Physiol 135: 814–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Sun L, Nguyen LV, Rachubinski RA, Goodman HM (1999) The pex16p homolog SSE1 and storage organelle formation in Arabidopsis seeds. Science 284: 328–330 [DOI] [PubMed] [Google Scholar]
- Lisenbee CS, Heinze M, Trelease RN (2003. a) Peroxisomal ascorbate peroxidase resides within a subdomain of rough endoplasmic reticulum in wild-type Arabidopsis cells. Plant Physiol 132: 870–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisenbee CS, Karnik SK, Trelease RN (2003. b) Overexpression and mislocation of a tail-anchored GFP redefines the identity of peroxisomal ER. Traffic 4: 491–501 [DOI] [PubMed] [Google Scholar]
- Mothes W, Heinrich SU, Graf R, von Heijne G, Brunner J, Rapport TA (1997) Molecular mechanism of membrane protein integration into the endoplasmic reticulum. Cell 89: 523–533 [DOI] [PubMed] [Google Scholar]
- Mullen RT (2002) Targeting and import of matrix proteins into peroxisomes. In A Baker, I Graham, eds, Plant Peroxisomes. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 339–384
- Mullen RT, Flynn CR, Trelease RN (2001. a) How are peroxisomes formed? The role of the endoplasmic reticulum and peroxins. Trends Plant Sci 6: 256–261 [DOI] [PubMed] [Google Scholar]
- Mullen RT, Lisenbee CS, Flynn CR, Trelease RN (2001. b) Stable and transient expression of chimeric peroxisomal membrane proteins induces an independent “zippering” of peroxisomes and an endoplasmic reticulum subdomain. Planta 213: 849–863 [DOI] [PubMed] [Google Scholar]
- Mullen RT, Lisenbee CS, Miernyk JA, Trelease RN (1999) Peroxisomal membrane ascorbate peroxidase is sorted to a membranous network that resembles a subdomain of the endoplasmic reticulum. Plant Cell 11: 2167–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrdlik P, Neuhaus G, Merkle T (2000) Plant heterotrimeric G protein beta subunit is associated with membranes via protein interactions involving coiled-coil formation. FEBS Lett 476: 208–212 [DOI] [PubMed] [Google Scholar]
- Overvoorde PJ, Grimes HD (1994) Topographical analysis of the plasma membrane-associated sucrose binding-protein from soybean. J Biol Chem 269: 15154–15161 [PubMed] [Google Scholar]
- Nito K, Yamaguchi K, Kondo M, Hayashi M, Nishimura M (2001) Pumpkin peroxisomal ascorbate peroxidase is localized on peroxisomal membranes and unknown membranous structures. Plant Cell Physiol 42: 20–27 [DOI] [PubMed] [Google Scholar]
- Parsons M, Furuya T, Pal S, Kessler P (2001) Biogenesis and function of peroxisomes and glycosomes. Mol Biochem Parasitol 115: 19–28 [DOI] [PubMed] [Google Scholar]
- Reumann S (2004) Specification of the peroxisome targeting signals type 1 and type 2 of plant peroxisomes by bioinformatics analyses. Plant Physiol 135: 783–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmiento C, Ross JHE, Murphy DJ (1997) Expression and subcellular targeting of a soybean oleosin in transgenic rapeseed: implications for the mechanism of oil-body formation in seeds. Plant J 11: 783–796 [DOI] [PubMed] [Google Scholar]
- Schumann U, Wanner G, Veenhuis M, Schmid M, Gietl C (2003) AthPEX10, a nuclear gene essential for peroxisome and storage organelle formation during Arabidopsis embryogenesis. Proc Natl Acad Sci USA 100: 9626–9631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, Kashiwayama Y, Imanaka T, Kato H (2004) Domain architecture and activity of human Pex19p, a chaperone-like protein for intracellular trafficking of peroxisomal membrane proteins. J Biol Chem 279: 38486–38494 [DOI] [PubMed] [Google Scholar]
- South ST, Gould SJ (1999) Peroxisome synthesis in the absence of preexisting peroxisomes. J Cell Biol 144: 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkes IA, Baker A (2002) Peroxisomes biogenesis and protein import in plants, animals and yeasts: enigma and variations? Mol Membr Biol 19: 171–185 [DOI] [PubMed] [Google Scholar]
- Sparkes IA, Brandizzi F, Slocombe SP, El-Shami M, Hawes C, Baker A (2003) An Arabidopsis pex10 null mutant is embryo lethal, implicating peroxisomes in an essential role during plant embryogenesis. Plant Physiol 133: 1809–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani S (1998) Components involved in peroxisome import, biogenesis, proliferation, turnover, and movement. Physiol Rev 78: 171–188 [DOI] [PubMed] [Google Scholar]
- Subramani S, Koller A, Snyder WB (2000) Import of peroxisomal matrix and membrane proteins. Annu Rev Biochem 69: 399–418 [DOI] [PubMed] [Google Scholar]
- Tabak HF, Murk JL, Braakman I, Geuze HJ (2003) Peroxisomes start their life in the endoplasmic reticulum. Traffic 4: 512–518 [DOI] [PubMed] [Google Scholar]
- Takeda Y, Kasamo K (2002) Transmembrane topography of plasma membrane constituents in mung bean (Vigna radiata L.) hypocotyl cells: the large scale asymmetry of surface peptides. Biochim Biophys Acta 1558: 14–25 [DOI] [PubMed] [Google Scholar]
- Titorenko VI, Rachubinski RA (1998. a) Mutants of the yeast Yarrowia lipolytica defective in protein exit from the endoplasmic reticulum are also defective in peroxisome biogenesis. Mol Cell Biol 18: 2789–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko VI, Rachubinski RA (1998. b) The endoplasmic reticulum plays an essential role in peroxisome biogenesis. Trends Biochem Sci 23: 231–233 [DOI] [PubMed] [Google Scholar]
- Titorenko VI, Rachubinski RA (2000) Peroxisomal membrane fusion requires two AAA family ATPases, Pex1p and Pex6p. J Cell Biol 150: 881–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko VI, Rachubinski RA (2001. a) Dynamics of peroxisome assembly and function. Trends Cell Biol 11: 22–29 [DOI] [PubMed] [Google Scholar]
- Titorenko VI, Rachubinski RA (2001. b) The life cycle of the peroxisome. Nat Rev Mol Cell Biol 2: 357–368 [DOI] [PubMed] [Google Scholar]
- Trelease RN (2002) Peroxisomal biogenesis and acquisition of membrane proteins. In A Baker, I Graham, eds, Plant Peroxisomes. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 305–337
- Tusnady GE, Simon I (1998) Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol 283: 489–506 [DOI] [PubMed] [Google Scholar]
- Tusnady GE, Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17: 849–850 [DOI] [PubMed] [Google Scholar]
- Veenhuis M, Kiel JAKW, Van der Klei IJ (2003) Peroxisome assembly in yeast. Microsc Res Tech 61: 139–150 [DOI] [PubMed] [Google Scholar]
- Veenhuis M, Salomons FA, Van der Klei IJ (2000) Peroxisome biogenesis and degradation in yeast: a structure/function analysis. Microsc Res Tech 51: 584–600 [DOI] [PubMed] [Google Scholar]
- Vizeacoumar FJ, Torres-Guzman JC, Aitchison JD, Rachubinski RA (2004) Pex30p, Pex31p, and Pex32p form a family of peroxisomal integral membrane proteins regulating peroxisome size and number in Saccharomyces cerevisiae. Mol Biol Cell 15: 665–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, McMahon MA, Shelton SN, Nampaisansuk M, Ballard JL, Goodman JM (2004) Multiple targeting modules on peroxisomal proteins are not redundant: discrete functions of targeting signals within Pmp47 and Pex8p. Mol Biol Cell 15: 1702–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolman BK, Bartel B (2004) An Arabidopsis indole-3-butyric acid-response mutant defective in PEROXIN6, an apparent ATPase implicated in peroxisomal function. Proc Natl Acad Sci USA 101: 1786–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolman BK, Yoder A, Bartel B (2000) Genetic analysis of indole-3-butyric acid responses in Arabidopsis thaliana reveals four mutant classes. Genetics 156: 1323–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]