

Abstract
Bone marrow failure is a disease characterized by a drastic decline in the marrow’s functional ability to produce mature blood cells. Aplastic anemia, a disease in which patients have essentially empty bone marrow accompanied by severe anemia, neutropenia, and thrombocytopenia, presents a paradigm for bone marrow failure. Damage to the marrow may first result from exposure to toxic chemicals, drug overdose, radiation, and viral infection; however, it is the extended immune-mediated reaction that causes massive destruction of hematopoietic cells and leads to marrow hypoplasia and peripheral pancytopenia.
In recent years, animal models of acquired bone marrow failure syndromes have helped to strengthen our understanding of the mechanisms causing bone marrow failure. In this review, animal models for bone marrow failure are summarized by two groups: 1) bone marrow failure induced by toxic chemicals and drugs such as benzene, busulfan, and chloramphenicol, and radiation, and 2) models developed by an immune-related mechanism such as viral infection or foreign lymphocyte infusion.
Keywords: Bone marrow diseases, Aplastic anemia, Animal models
Bone marrow (BM) failure is a disease syndrome characterized by a drastic decline in the marrow’s functional ability to produce mature blood cells. A paradigm of BM failure is the disease aplastic anemia, in which patients have essentially empty BM accompanied by severe anemia, neutropenia, and thrombocytopenia.1 , 2 This disease was first described by Ehrlich3 at the end of 19th century. Over the course of last century, it has become clear that some aplastic anemia and BM failure cases are secondary to the exposure of toxic chemicals, drugs, radiation, or infectious agents.4 However, the majority of cases remain idiopathic, meaning that the cause of disease could not be identified.1 , 2 Even when an exposure is confirmed, the identifiable factor or agent has been primarily circumstantial or inferential, since the pathogenesis from the causative agent through disease progression is unexplained.
Compelling clinical observations of therapeutic efficacy and systematic laboratory experiments have now formed a plausible model of the pathophysiology of the disease in which the immune system mediated an organ-specific destruction of BM cells.1 , 2 , 5 , 6 About 50% to 80% of patients can survive marrow failure when treated promptly with stem cell replacement or immunosuppressive drugs to alleviate pancytopenia and restore hematopoiesis.2 While more is still to be learned, clinical and laboratory observations have indicated that activated cytotoxic T cells producing type I cytokines are the effectors of an active process of stem cell destruction, mainly through Fas-mediated apoptosis.7 , 8 Experiments using animal models have certainly added strength to understanding the mechanisms for BM failure. This review summarizes animal models for BM failure into two major categories: 1) BM failure induced by toxic chemicals or drugs such as benzene, busulfan, and chloramphenicol, and 2) BM failure models developed by an immune-related mechanism such as viral infection and foreign lymphocyte infusion.
Benzene Induced BM Failure
Recognition of industry benzene as a causative agent for BM failure became well publicized in the 1920s and 1930s.4 In more recent years, animal models have been developed to understand the mechanism of benzene-induced BM failure. Administration of benzene through inhalation or injection produced hematopoietic damage in C57BL/6 (B6),9 , 10 Swiss,11 Kunming,12 CD1,13 and 129/Sv mice,14 as well as in rats.15
Velasco et al.13 subcutaneously injected 2 ml/kg (1940 mg/kg) benzene to CD1 male mice at either 5 days or 3 days per week for a total of 10, 15, and 20 injections, respectively. Mice that received 15 and 20 injections at 5 daily injections per week showed lethargy and irritability with 42% body weight loss and 68% spleen weight loss. Body and spleen weight losses were less severe (12% and 48%, respectively) in mice that received the same total injections but administrated at 3 daily injections per week. Decreases in hemoglobin, erythrocytes, leukocytes and BM cells ranged from 12% to 84%. While both injection schedules induced aplastic anemia, the disease was masked by spleen toxicity in the group receiving 5 daily injections per week.13 Inhalation of benzene vapor for 2.5 months induced aplastic anemia in Kunming mice with significant declines in erythroid progenitor cell counts and phosphoribosylpyrophosphate synthetase activity in colony-forming units (CFU)-erythroid. Treatment with a Chinese prescription, Sheng-Mai injection, brought erythroid progenitor cell counts and phosphoribosylpyrophosphate synthetase activity in CFU-erythroids to normal levels.12 Inhalation of various concentrations of benzene at 6 hours per day for 5 days caused the formation of phenylsulfate and phenylglucuronide to conjugate in the blood of Swiss mice. The reductions in spleen weight and white blood cell numbers correlated with the concentration of phenylsulfate in the blood.11
A specific hypothesis for benzene-induced aplastic anemia and BM failure is that benzene metabolites produce toxicity affecting BM stromal cells’ ability to produce sufficient cytokines needed to maintain normal hematopoietic cell survival and proliferation. When B6 mice were exposed to 600 or 800 mg/kg body weight of benzene for 2 days, their marrow cells were unable to process the 34 kDa pre-interleukin 1 alpha (IL1A) to the mature 17 kDa cytokine. Administration of recombinant murine IL1A to mice before benzene exposure completely prevented BM depression, suggesting that reduced stromal function to convert pre-IL1 to mature IL1 is the cause for benzene’s hematoxicity.10 Benzene at 600 mg/kg body weight administrated twice per day for 2 days caused a significant decline in total number of BM cells with net decreases in lymphocytic and erythroid cells.9
Because metabolism is required to produce benzene toxicity, Hoffmann et al.16 studied the effects of mouse strain and the route of benzene exposure to benzene toxicity. There was no obvious effect on benzene metabolism since seven major metabolites and unmetabolized benzene were detected in most samples at most time points, regardless of the route of exposure or strain of mouse. Benzene was absorbed more slowly after intradermal injection than after oral administration, and intradermally dosed mice produced more conjugates of hydroquinone than did the orally dosed mice.16
The two major metabolites related to benzene’s hematoxicity are hydroquinone and benzoquinone. Zhu et al.15 demonstrated that mice are much more susceptible to benzene toxicity than rats because mouse cells contain 2 times and 28 times less content (activity) of glutathione and quinone reductase, respectively. Treatment of both mouse and rat stromal cells with 1,2-dithiole-3-thione (D3T) to induce glutathione and quinone reductase produced protective effects against hydroquinone toxicity, while treatment with dicoumarol, an inhibitor of quinone reductase, enhanced hydroquinone toxicity.15 The toxic effect of benzoquinone is produced by forming protein and DNA adducts and by generating reactive oxygen species. Hematopoietic stem cells cultured in the presence of benzoquinone for 24 hours showed a dose-dependent cytotoxic response. RNA isolated from benzoquinone-treated hematopoietic stem cells and hematopoietic stem cells from mice exposed to benzene inhalation showed altered expression of genes regulating cell apoptosis, DNA repair, cell cycle, and cell growth control as compared to unexposed hematopoietic stem cells.14
Thus, benzene hematoxicity seems to be caused by its metabolites (hydroxyethylrutoside and quinone reductase) that affect growth, gene expression and apoptosis of hematopoietic cells. Benzene hematoxicity also affects stromal cells, hampering their ability to produce normal levels of cytokines to maintain normal hematopoietic cell growth and survival.
Busulfan, Chloramphenicol, and Irradiation Induced BM Failure
Busulfan is a chemical that has been widely used as a conditioning agent for allogenic BM transplantation for treatment of various malignancies. However, it is hematoxic and might cause marrow failure when used inappropriately. An early mouse model for busulfan-induced aplastic anemia was reported in 1974. Following a course of therapy, animals maintained normal blood counts and BM cellularity for one year before demonstrating pancytopenia and frank aplasia. Splenic CFU, representative of early hematopoietic progenitor cells, declined during this intervening period to very low numbers.17 Busulfan causes significant defects in hematopoietic stem cell proliferation in comparison to other cytotoxic agents (e.g., 5-fluorouracil) as measured by serial transplantation.18 C3H mice treated with 4 doses of 5 mg/kg busulfan produced a latent hematopoietic injury and showed persistent normal or near-normal peripheral blood counts and marrow cellularity with moderate to marked reduction in hematopoietic stem cells. Baseline splenic CFU culture showed no significant change. However, splenic CFU culture from latent mice increased only 7-fold, reaching its peak at day 3. In comparison, control mice peaked at day 10 with a 50-fold increase.19 , 20 In another study, busulfan therapy resulted in a chronic latent marrow aplasia characterized by normal peripheral blood neutrophil numbers, normal hematocrit and marrow cellularity, but reduced numbers of pluripotent hematopoietic stem cells and CFU-granulocyte-macrophage (CFU-GM).21 Inocula from busulfan-treated animals containing three to five times the stem cells and progenitor cells failed to establish long-term granulopoiesis in vitro, while small numbers of normal BM cells readily established and sustained long-term granulopoiesis in vitro. These results suggest that busulfan therapy produced a qualitative defect in either the hemopoietic stem cells, the stromal-forming elements, or both.21
Pugsley et al.22 studied busulfan-induced chronic hypoplastic marrow failure in an experimental murine model and found a 60% to 70% reduction in B lymphocytes and a 30% to 80% reduction in T lymphocytes. There was a two-thirds reduction in IgG and IgM antibody titer to sheep red blood cells and a dramatic decline in lymphocyte proliferative responses in vitro, indicating that the lesions underlying experimental marrow failure are not confined to the myeloid stem cell, but also involve cells of the lymphoid lineage. Ideriha et al.23 intramuscularly implanted devitalized bone matrix in 42 mice with busulfan-induced BM failure and in 42 control mice. The morphological volumetric estimation of these components in the hypoplastic mice showed a significant increase in cartilage and a decrease in hematopoietic BM as compared with control mice.
Several cases of aplastic anemia were induced by higher doses of busulfan. Gibson et al.24 treated BALB/c mice with 8 doses of busulfan at 0, 5.25 mg/kg, and 10.50 mg/kg over 23 days. Reductions in nucleated marrow cells, CFU-GM, CFU-erythroid, erythrocytes, leucocytes, platelets, and reticulocytes were noticeable at day 1 post treatment and continued until days 91 and 112. Treating A/J mice with 4 busulfan injections, three at 20 mg/kg and one (the last) at 10 mg/kg, at 2-week intervals resulted in a 75% decline in short-term adherent cell colonies per femur and a 90% decline in CFU-culture per femur. Marrow hypoplasia was associated with heightened endosteum and cortical bone thickening, but no stromal cell change was detected by electron microscopy at 40 weeks. These results suggest that busulfan-treated marrow stroma experiences morphological changes specifically involving endosteum, bone, adipocytes, and mast cells, and that the damages recover substantially with time.25 A single dose of busulfan injection at 35 mg/kg to 45 mg/kg to CBA mice resulted in 0% survival at 30 days with an average survival time of 15.8 days. A single dose of busulfan injection at 30 mg/kg caused a drastic decline in blood granulocytes at day 14.26
Chloramphenicol is a broad-spectrum antibiotic that has been widely used in the treatment of serious infections. However, this compound is also hematoxic to humans and can cause reversible anemia, aplastic anemia, and leukemia under various conditions.4 Administration of chloramphenicol had no detectable effect on the marrow of normal mice, but caused a progressive decline in the number of pluripotential stem cells and granulocytic progenitor cells in mice bearing residual marrow damage.27 Chloramphenicol is less toxic than its derivative, nitrosochloramphenicol. Chloramphenicol at concentrations >300 μM caused only reversible inhibition of DNA synthesis and CFU-culture growth without affecting marrow cell viability, while nitrosochloramphenicol at a concentration of 50 μM inhibited DNA synthesis and caused irreversible inhibition of CFU-culture growth and cell death.28 In a rapidly growing human lymphoid cell line, nitrosochloramphenicol caused an accumulation of cells in the G2M phase and an increase in cell death within the arrested population.28 The hematoxicity of chloramphenicol might be related to its effect on the hematopoietic inductive microenvironment. At concentrations of 10 μg/ml, 50 μg/ml, and 100 μg/ml, chloramphenicol suppressed the growth of granuloid-committed progenitor cells as well as fibroblast colonies.29 In a subcutaneous bone implantation experiment, implanted femora of chloramphenicol-treated mice (500 mg/kg/day x 6) had significantly decreased recovery of hematopoietic stem cells in comparison to the sham treated controls. This result further supports the observation from the in vitro study indicating that chloramphenicol toxicity affects the stromal environment of marrow.29
More recently, a series of studies failed to produce a chronic aplastic anemia mouse model by using chloramphenicol succinate.30 – 35 Administration of chloramphenicol succinate at 800 mg/kg to 2000 mg/kg for 7 days to CD1 mice,31 2000 mg/kg/day for 17 days to BALB/c mice,32 2000 mg/kg/day to 4000 mg/kg/day for 19 days to Wistar Hanover rats,31 and 2500 mg/kg/day to 3500 mg/kg/day for 9 days in guinea pigs35 all induced reversible anemia, but not irreversible aplastic anemia. In these animals, there were usually dose-related reductions in reticulocytes, erythrocytes, hematocrit, hemoglobin, CFU-erythroid, and CFU-GM, but these hematological parameters tend to return to normal without intervention.30 – 35 A significant difference between strains was observed in response to chloramphenicol succinate toxicity. Inbred C3H/He, CBA/Ca, BALB/c, and B6 mice were demonstrated to be much more susceptible to chloramphenicol succinate toxicity than outbred CD1 mice.33
Long-term bone marrow damage characterized by stem cell, progenitor cell, and stromal cell abnormalities is a frequent occurrence after cytotoxic treatments. The relative contributions of each of these components are difficult to analyze, especially in the case of patients who have received combined chemotherapy. The damage may be latent and not manifested in low numbers of mature functional cells in the blood, but may become apparent as a hypoplastic syndrome at later times.36 In addition to busulfan and chloramphenicol succinate, irradiation may also damage hematopoietic stem cells and progenitor cells. Residual radiation injury was demonstrated in long-term primary cultures of mouse BM. After irradiation with 0.5 Gy, 3 Gy, and 5.5 Gy, the accumulated postradiation cell production corresponded to an exponential dose-response relationship at any time after treatment.37 Both exposure of murine BM to 4 Gy ionizing radiation and incubation of BM cells with 30 μM busulfan caused significant inhibition of the frequency of various types of cobblestone area-forming cells. However, irradiation also induced apoptosis in hematopoietic stem cells, but a significant increase in apoptosis was not observed with busulfan treatment.38
BM Failure Following Viral Infection
While the early part of the 20th century research focused on chemically-induced aplastic anemia and BM failure, more recent studies seem to support an immune-mediated marrow destruction mechanism. In immune-mediated BM failure, an initial specific immune response expanded to an out-of-control state, resulting in nonspecific destruction of normal functional hematopoietic stem cells and progenitor cells. The effectiveness of immunosuppressive therapy to rescue 70% to 80% of aplastic anemia patients provides strong evidence to support an immune-mediated marrow failure.1 , 2 Data generated from studies using animal models also supports an immune-mediated marrow destruction theory.
A good example of immune-mediated BM failure is marrow failure following viral infection. It is well known that human parvovirus B19 infection causes human BM failure.39 , 40 When a strain of lymphocytic choriomeningitis virus failed to cause the fatal central nervous system syndrome in C3HeB/FeJ mice, investigators found that the affected animals had hematological abnormalities including pancytopenia, abnormal erythrocyte morphology, increased peripheral reticulocyte count, and marked erythroid hyperplasia in BM. The later development of leukopenia and thrombocytopenia could be traced to inhibition of granulocytes and megakaryocytes in BM.41 , 42 Chronic infection of perforin-deficient (P0/0) mice with lymphocytic choriomeningitis virus exhibited a vigorous T cell response with a progressive pancytopenia that eventually is lethal due to agranulocytosis and thrombocytopenia. Depletion of CD8+ T cells could prevent the disease, however, increasing the frequency of lymphocytic choriomeningitis virus-specific CD8+ T cells in T cell receptor transgenic mice accelerates the disease.43
Infection by human cytomegalovirus is often accompanied by transient neutropenia and thrombocytopenia probably due to one of the following: 1) alteration of accessory cell function by inducing the production of inhibitory cytokines, 2) perturbation of stromal cell function resulting in a decreased production of hematopoietic factors or by altering cell surface adhesion molecule expression, or 3) direct infection of the hematopoietic stem cells or progenitor cells.44 In a murine model of cytomegalovirus-induced aplastic anemia, functional integrity of the stroma was impaired and the expression of genes encoding the essential hemopoietin stem cell factor, granulocyte colony-stimulating factor, and interleukin-6 was markedly reduced despite the physical integrity of the stromal network and the lack of significant marrow cell loss.45
Infusion-Induced BM Failure
Infusion-induced BM failure is a subclass of marrow failure that directly addresses the issue of immune-mediated marrow destruction. There is a long history of producing mouse models for the study of BM failure using the immune approach. Early in 1967, Barnes and Mole transplanted 1 million to 10 million C3H/H lymph node cells into sublethally irradiated CBA/H mice. The investigators found that variable numbers of recipients with marrow aplasia eventually died within days or weeks.46 This lymph node cell infusion strategy was extended to C3H/He mice by injecting lymph node cells from B10.BR mice in which donor and recipient mice share the same H2k haplotype, but have different mixed lymphocyte reaction loci. Total body irradiation at 6 Gy plus injection of 10 million B10.BR lymph node cells produced fatal pancytopenia in C3H/He mice within 2 to 3 weeks and was accompanied by dramatic declines in splenic CFU and CFU-culture.47 The same approach using C3H/He lymph node cells, but not C3H/HeJ lymph node cells, induced aplastic anemia.48 Injecting lymph node cells from CBA/J donors to sublethally irradiated C3H/HeJ mice also induced BM failure that was not prevented by pre-administration of cyclosporin A (25 mg/kg).49 In a C3H/He to CBA/J lymph node-cell infusion model, Chiu and Knospe50 reported that cells from thymus but not spleen, peritoneal, or BM could induce aplastic anemia. Depletion of T cells, but not B cells, significantly reduces the potency to generate aplastic anemia. Plasma of affected mice contained a specific heat stable and non-dialyzable factor that can inhibit CFU-GM.51 This mouse model showed that T lymphocytes injure not only pluripotent hematopoietic stem cells but also marrow stromal cells to cause severe pancytopenia.52
Recently, we developed mouse models for BM failure syndromes and used these models to study the pathophysiological mechanisms of this disease.53 , 54 Infusion of lymph node cells from normal B6 mice to hybrid B6D2F1 and CByB6F1 recipients with53 or without54 a sublethal dose of irradiation induced severe BM failure showing fatal pancytopenia within 2 to 3 weeks. Marrow cavities were essentially empty in these affected mice in comparison to untreated control mice (figure 1 ). Residual BM contained a very large proportion of infiltrated/expanded donor T lymphocytes in BM failure mice; these residual cells were also apoptotic showing high efficiency in binding to Annexin V (figure 1 ). BM failure mice had an approximately 10-fold decline in total BM cells, 15-fold decline in BM Lin-Sca1+c−Kit+ cells, 100-fold decline in splenic CFU, and 100-fold decline in hematopoietic progenitor and stem cells as estimated by irradiation protection in vivo. Residual BM cells from affected BM failure mice were able to destroy normal BM cells as innocent bystanders in a co-transplantation experiment, thus preventing normal BM cells from engrafting lethally irradiated recipients (figure 2 ). Both hematopoietic progenitor/stem cells and marrow stromal cells were destroyed by this nonspecific mechanism.53 A characteristic feature of this model is the oligoclonal expansion of donor T lymphocytes in recipient BM with restricted expression of T cell receptor β variable regions.53 This observation is consistent with observations from human aplastic anemia and paroxysmal nocturnal hemoglobinuria patients in which clonal expansion of CD8 T lymphocytes were detected by Vβ analyses as well as by complementarity-determining-region-3 molecular analysis.6 , 55
Figure 1.
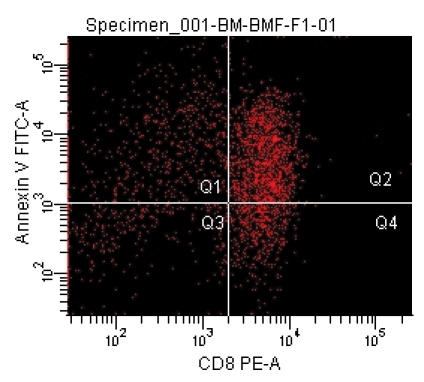
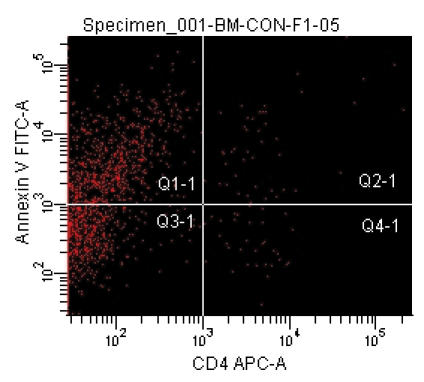


Marrow lymphocyte infiltration/expansion and marrow hypoplasia in a mouse model of infusion induced BM failure. CByB6F1 mice treated with 5 Gy total body irradiation and 5 x 106 BM cell infusion were euthanized at 14 days after cell infusion along with untreated control mice. BM cells from BM failure mice (A) and control mice (B) were analyzed by flow cytometry. Stern bones from BM failure (C) and control (D) mice were sectioned, HE stained and observed under a light microscope (100x). Marrow cavity was almost empty in BM failure mice (C) while residual BM cells were essentially infiltrated donor lymphocytes (A).
Figure 2.

Bystander destruction of marrow hematopoietic cells. BM cells from BM failure mice mixed with BM cells from normal B6-CD45.1 mice could not rescue lethally-irradiated B6D2F1 recipients (left) while B6-CD45.1 marrow cells alone (middle) or a mixture of normal B6D2F1 and B6-CD45.1 marrow cells (right) established effective engraftment and rescued recipients. This experiment shows that residual cells in the marrow of BM failure mice were able to destroy normal hematopoietic cells from B6-CD45.1 donors as bystanders.
In immune-mediated BM failure, damage to hematopoiesis may be caused by the excessive action of tumor necrosis factor-alpha and interferon-gamma (IFN-γ) secreted by activated CD8+ T cells.43 This is consistent with findings from human aplastic anemia patients in which activated cytotoxic T lymphocytes infiltrate aplastic BM and produce detectable amounts of IFN-γ mRNA.56 Murine marrow cells cultured in the presence of stromal cells transduced with a retroviral vector expressing murine IFN-γ had significantly less long-term repopulating stem cell activity in a competitive repopulation assay.57 IFN-γ inhibits long-term culture-initiating cells, CFU-GM, and erythroid burst-forming units. Continuous addition of relatively high IFN-γ concentrations (1,000 U/ml weekly or 200 U/ml every 2 days) was required for inhibition of secondary colony formation, a measure of long-term culture-initiating cell number and clonogenicity. To mimic local production of IFN-γ , human stromal cells were engineered to express a transduced IFN-γ gene by retroviral-mediated gene transfer. IFN-γ secreted by stromal cells was far more potent than exogenous IFN-γ in its effects on the long-term culture-initiating cell assay. Purified CD34+ cells cultured with IFN-γ-stroma had a dramatically reduced production of CFU-GM, erythroid burst-forming units, and numbers of secondary colonies.56
In conclusion, development of BM failure is a complicated matter. While many factors, such as toxic chemicals, drug overdose, and viral infection, can initially damage marrow cells, it is the extended immune reaction that causes massive destruction of hematopoietic cells leading to marrow hypoplasia and peripheral pancytopenia. Whether or not stromal cells are destroyed and how much stromal damage contributes to the overall marrow failure are questions yet to be answered. So far, no specific antigens have been identified for the initiation of immune attack. While the roles of class I cytokine IFN-γ and tumor necrosis factor-alpha have been identified, the exact mechanism for immune-mediated bystander marrow cell destruction is yet to be fully characterized. Studies using mice that are congenic at specific minor histocompatibility antigen loci will help to define the role of each specific antigen in the development of marrow failure. The recent finding that mutations in the telomerase complex caused BM failure in some patients provided fresh evidence indicating that acquired BM failure could actually be constitutional.58 Mice deficient in the telomerase gene products will be a good resource to study the pathogenesis of telomerase-related BM failure. In general, animal models will certainly provide platforms to test new prevention and treatment procedures for BM failure syndromes.
References
- 1. Young NS. Acquired aplastic anemia. Ann Intern Med 2002;136:534–546. [DOI] [PubMed] [Google Scholar]
- 2. Young NS. Aplastic anemia, myelodysplasia, and related bone marrow failure syndromes. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, eds. Harrison’s Principles of Internal Medicine. New York, NY: McGraw-Hill; 2005. 617–626.
- 3. Ehrich P. Ueber einen Fall von Anämie mit Bemerkungen über regenerative Veränderungen des Knochenmarks. Charite-Ann 1888;13:300–309. [Google Scholar]
- 4. Young NS. Drugs and chemicals. In: Young NS, Alter BP, eds. Aplastic Anemia: Acquired and Inherited. Philadelphia, PA: WB Saunders; 1994. 100–132.
- 5. Zeng W, Maciejewski JP, Chen G, Young NS. Limited heterogeneity of T cell receptor BV usage in aplastic anemia. J Clin Invest 2001;108:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet 2004;364:355–364. [DOI] [PubMed] [Google Scholar]
- 7. Maciejewski JP, Selleri C, Sato T, Anderson S, Young NS. Increased expression of Fas antigen on bone marrow CD34+ cells of patients with aplastic anaemia. Br J Haematol 1995;91:245–252. [DOI] [PubMed] [Google Scholar]
- 8. Maciejewski J, Selleri C, Anderson S, Young NS. Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood 1995;85:3183–3190. [PubMed] [Google Scholar]
- 9. Niculescu R, Kalf GF. A morphological analysis of the short-term effects of benzene on the development of the hematological cells in the bone marrow of mice and the effects of interleukin-1 alpha on the process. Arch Toxicol 1995;69:141–148. [DOI] [PubMed] [Google Scholar]
- 10. Renz JF, Kalf GF. Role for interleukin-1 (IL-1) in benzene-induced hematotoxicity: inhibition of conversion of pre-IL-1 alpha to mature cytokine in murine macrophages by hydroquinone and prevention of benzene-induced hematotoxicity in mice by IL-1 alpha. Blood 1991;78:938–944. [PubMed] [Google Scholar]
- 11. Wells MS, Nerland DE. Hematotoxicity and concentration-dependent conjugation of phenol in mice following inhalation exposure to benzene. Toxicol Lett 1991;56:159–166. [DOI] [PubMed] [Google Scholar]
- 12. Liu LP, Liu JF, Lu YQ. Effects of Sheng-Mai injection on the PRPP synthetase activity in BFU-es and CFU-es from bone marrows of mice with benzene-induced aplastic anemia. Life Sci 2001;69:1373–1379. [DOI] [PubMed] [Google Scholar]
- 13. Velasco Lezama R, Barrera Escorcia E, Munoz Torres A, Tapia Aguilar R, Gonzalez Ramirez C, Garcia Lorenzana M, Ortiz Monroy V, Betancourt Rule M. A model for the induction of aplastic anemia by subcutaneous administration of benzene in mice. Toxicology 2001;162:179–191. [DOI] [PubMed] [Google Scholar]
- 14. Faiola B, Fuller ES, Wong VA, Pluta L, Abernethy DJ, Rose J, Recio L. Exposure of hematopoietic stem cells to benzene or 1,4-benzoquinone induces gender-specific gene expression. Stem Cells 2004;22:750–758. [DOI] [PubMed] [Google Scholar]
- 15. Zhu H, Li Y, Trush MA. Differences in xenobiotic detoxifying activities between bone marrow stromal cells from mice and rats: implications for benzene-induced hematotoxicity. J Toxicol Environ Health 1995;46:183–201. [DOI] [PubMed] [Google Scholar]
- 16. Hoffmann MJ, Sinko PJ, Lee YH, Meeker RJ, Snyder R. Pharmacokinetic studies in Tg.AC and FVB mice administered [14C] benzene either by oral gavage or intradermal injection. Toxicol Appl Pharmacol 2001;174:139–145. [DOI] [PubMed] [Google Scholar]
- 17. Morley A, Blake J. An animal model of chronic aplastic marrow failure. I. Late marrow failure after busulfan. Blood 1974;44:49–56. [PubMed] [Google Scholar]
- 18. Botnick LE, Hannon EC, Hellman S. A long lasting proliferative defect in the hematopoietic stem cell compartment following cytotoxic agents. Int J Radiat Oncol Biol Phys 1979;5:1621–1625. [DOI] [PubMed] [Google Scholar]
- 19. Fitchen JH, Cline MJ. The effect of granulopoietic stress in mice with “latent” bone marrow failure. Exp Hematol 1980;8:788–794. [PubMed] [Google Scholar]
- 20. Fitchen JH, Deregnaucourt J, Cline MJ. An in vitro model of hematopoietic injury in chronic hypoplastic anemia. Cell Tissue Kinet 1981;14:85–90. [PubMed] [Google Scholar]
- 21. Boyd RL, Caro J, Halka KG, Erslev AJ. Granulopoiesis in long-term culture by marrow from mice with busulfan-induced chronic latent aplasia. Int J Cell Cloning 1986;4:357–367. [DOI] [PubMed] [Google Scholar]
- 22. Pugsley CA, Forbes IJ, Morley AA. Immunologic abnormalities in an animal model of chronic hypoplastic marrow failure induced by busulfan. Blood 1978;51:601–610. [PubMed] [Google Scholar]
- 23. Ideriha NM, Vugman I, Falcao RP. Ectopic bone marrow development in experimental busulfan-induced hypoplastic anemia in mice. Blut 1984;48:277–284. [DOI] [PubMed] [Google Scholar]
- 24. Gibson FM, Andrews CM, Diamanti P, Rizzo S, Macharia G, Gordon-Smith EC, Williams T, Turton J. A new model of busulphan-induced chronic bone marrow aplasia in the female BALB/c mouse. Int J Exp Pathol 2003;84:31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McManus PM, Weiss L. Busulfan-induced chronic bone marrow failure: changes in cortical bone, marrow stromal cells, and adherent cell colonies. Blood 1984;64:1036–1041. [PubMed] [Google Scholar]
- 26. Millar JL, Hudspith BN, Blackett NM. Reduced lethality in mice receiving a combined dose of cyclophosphamide and busulphan. Br J Cancer 1975;32:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morley A, Trainor K, Remes J. Residual marrow damage: possible explanation for idiosyncrasy to chloramphenicol. Br J Haematol 1976;32:525–531. [DOI] [PubMed] [Google Scholar]
- 28. Yunis AA, Miller AM, Salem Z, Corbett MD, Arimura GK. Nitroso-chloramphenicol: possible mediator in chloramphenicol-induced aplastic anemia. J Lab Clin Med 1980;96:36–46. [PubMed] [Google Scholar]
- 29. Nara N, Bessho M, Hirashima K, Momo H. Effects of chloramphenicol on hematopoietic inductive microenvironment. Exp Hematol 1982;10:20–25. [PubMed] [Google Scholar]
- 30. Holt DE, Andrews CM, Payne JP, Williams TC, Turton JA. The myelotoxicity of chloramphenicol: in vitro and in vivo studies: II: In vivo myelotoxicity in the B6C3F1 mouse. Hum Exp Toxicol 1998;17:8–17. [DOI] [PubMed] [Google Scholar]
- 31. Turton JA, Yallop D, Andrews CM, Fagg R, York M, Williams TC. Haemotoxicity of chloramphenicol succinate in the CD-1 mouse and Wistar Hanover rat. Hum Exp Toxicol 1999;18:566–576. [DOI] [PubMed] [Google Scholar]
- 32. Turton JA, Havard AC, Robinson S, Holt DE, Andrews CM, Fagg R, Williams TC. An assessment of chloramphenicol and thiamphenicol in the induction of aplastic anaemia in the BALB/c mouse. Food Chem Toxicol 2000;38:925–938. [DOI] [PubMed] [Google Scholar]
- 33. Festing MF, Diamanti P, Turton JA. Strain differences in haematological response to chloramphenicol succinate in mice: implications for toxicological research. Food Chem Toxicol 2001;39:375–383. [DOI] [PubMed] [Google Scholar]
- 34. Turton JA, Andrews CM, Havard AC, Robinson S, York M, Williams TC, Gibson FM. Haemotoxicity of thiamphenicol in the BALB/c mouse and Wistar Hanover rat. Food Chem Toxicol 2002;40:1849–1861. [DOI] [PubMed] [Google Scholar]
- 35. Turton JA, Andrews CM, Havard AC, Williams TC. Studies on the haemotoxicity of chloramphenicol succinate in the Dunkin Hartley guinea pig. Int J Exp Pathol 2002;83:225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Testa NG, Hendry JH, Molineux G. Long-term bone marrow damage in experimental systems and in patients after radiation or chemotherapy. Anticancer Res 1985;5:101–110. [PubMed] [Google Scholar]
- 37. Reincke U, Hannon EC, Hellman S. Residual radiation injury exhibited in long-term bone marrow cultures. J Cell Physiol 1982;112:345–352. [DOI] [PubMed] [Google Scholar]
- 38. Meng A, Wang Y, Van Zant G, Zhou D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res 2003;63:5414–5419. [PubMed] [Google Scholar]
- 39. Chisaka H, Morita E, Yaegashi N, Sugamura K. Parvovirus B19 and the pathogenesis of anaemia. Rev Med Virol 2003;13:347–359. [DOI] [PubMed] [Google Scholar]
- 40. Young NS, Brown KE. Parvovirus B19. N Engl J Med 2004;350:586–597. [DOI] [PubMed] [Google Scholar]
- 41. Broomhall KS, Morin M, Pevear DC, Pfau CJ. Severe and transient pancytopenia associated with a chronic arenavirus infection. J Exp Pathol 1987;3:259–269. [PubMed] [Google Scholar]
- 42. Stellrecht-Broomhall KA. Evidence for immune-mediated destruction as mechanism for LCMV-induced anemia in persistently infected mice. Viral Immunol 1991;4:269–280. [DOI] [PubMed] [Google Scholar]
- 43. Binder D, van den Broek MF, Kagi D, Bluethmann H, Fehr J, Hengartner H, Zinkernagel RM. Aplastic anemia rescued by exhaustion of cytokine-secreting CD8+ T cells in persistent infection with lymphocytic choriomeningitis virus. J Exp Med 1998;187:1903–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Almeida-Porada GD, Ascensao JL. Cytomegalovirus as a cause of pancytopenia. Leuk Lymphoma 1996;21:217–223. [DOI] [PubMed] [Google Scholar]
- 45. Mayer A, Podlech J, Kurz S, Steffens HP, Maiberger S, Thalmeier K, Angele P, Dreher L, Reddehase MJ. Bone marrow failure by cytomegalovirus is associated with an in vivo deficiency in the expression of essential stromal hemopoietin genes. J Virol 1997;71:4589–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barnes DW, Mole RH. Aplastic anaemia in sublethally irradiated mice given allogeneic lymph node cells. Br J Haematol 1967;13:482–491. [DOI] [PubMed] [Google Scholar]
- 47. Kubota K, Mizoguchi H, Miura Y, Kano S, Takaku F. Experimental hypoplastic marrow failure in the mouse. Exp Hematol 1978;6:791–800. [PubMed] [Google Scholar]
- 48. Knospe WH, Steinberg D, Speck B. Experimental immunologically mediated aplastic anemia (AA) in H-2k identical, Mls (M) locus different mice. Exp Hematol 1983;11:542–552. [PubMed] [Google Scholar]
- 49. Knospe WH, Steinberg D, Gratwohl A, Speck B. Experimental immunologically mediated aplastic anemia (AA) in mice: cyclosporin A fails to protect against AA. Int J Cell Cloning 1984;2:263–271. [DOI] [PubMed] [Google Scholar]
- 50. Chiu KM, Knospe WH. Immunologically mediated aplastic anemia in mice: effects of varying the source and composition of donor cells. Exp Hematol 1987;15:269–275. [PubMed] [Google Scholar]
- 51. Chiu KM, Knospe WH. Inhibitor of granulocyte-macrophage colony formation in plasma of mice rendered aplastic by allogeneic lymph node cells. Exp Hematol 1989;17:335–339. [PubMed] [Google Scholar]
- 52. Knospe WH, Husseini SG, Chiu KM, Fried W. Immunologically mediated aplastic anemia in mice: evidence of hematopoietic stromal injury and injury to hematopoietic stem cells. Exp Hematol 1994;22:573–581. [PubMed] [Google Scholar]
- 53. Chen J, Lipovsky K, Ellison FM, Calado RT, Young NS. Bystander destruction of hematopoietic progenitor and stem cells in a mouse model of infusion-induced bone marrow failure. Blood 2004;104:1671–1678. [DOI] [PubMed] [Google Scholar]
- 54. Bloom ML, Wolk AG, Simon-Stoos KL, Bard JS, Chen J, Young NS. A mouse model of lymphocyte infusion-induced bone marrow failure. Exp Hematol 2004;32:1163–1172. [DOI] [PubMed] [Google Scholar]
- 55. Risitano AM, Maciejewski JP, Muranski P, Wlodarski M, O’Keefe C, Sloand EM, Young NS. Large granular lymphocyte (LGL)-like clonal expansions in paroxysmal nocturnal hemoglobinuria (PNH) patients. Leukemia 2005;19:217–222. [DOI] [PubMed] [Google Scholar]
- 56. Selleri C, Maciejewski JP, Sato T, Young NS. Interferon-gamma constitutively expressed in the stromal microenvironment of human marrow cultures mediates potent hematopoietic inhibition. Blood 1996;87:4149–4157. [PubMed] [Google Scholar]
- 57. Yu JM, Emmons RV, Hanazono Y, Sellers S, Young NS, Dunbar CE. Expression of interferon-gamma by stromal cells inhibits murine long-term repopulating hematopoietic stem cell activity. Exp Hematol 1999;27:895–903. [DOI] [PubMed] [Google Scholar]
- 58. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med 2005;352:1413–1424. [DOI] [PubMed] [Google Scholar]