

Abstract
C75, a synthetic inhibitor of FAS (fatty acid synthase), has both anti-tumour and anti-obesity properties. In this study we provide a detailed kinetic characterization of the mechanism of in vitro inhibition of rat liver FAS. At room temperature, C75 is a competitive irreversible inhibitor of the overall reaction with regard to all three substrates, i.e. acetyl-CoA, malonyl-CoA and NADPH, exhibiting pseudo-first-order kinetics of the complexing type, i.e. a weak non-covalent enzyme–inhibitor complex is formed before irreversible enzyme modification. C75 is a relatively inefficient inactivator of FAS, with a maximal rate of inactivation of 1 min−1 and an extrapolated KI (dissociation constant for the initial complex) of approx. 16 mM. The apparent second-order rate constants calculated from these values are 0.06 mM−1·min−1 at room temperature and 0.21 mM−1·min−1 at 37 °C. We also provide experimental evidence that C75 inactivates the β-ketoacyl synthase (3-oxoacyl synthase) partial activity of FAS. Unexpectedly, C75 also inactivates the enoyl reductase and thioesterase partial activities of FAS with about the same rates as for inactivation of the β-ketoacyl synthase. In contrast with the overall reaction, the β-ketoacyl synthase activity and the enoyl reductase activity, substrates do not protect the thioesterase activity of rat liver FAS from inactivation by C75. These results differentiate inactivation by C75 from that by cerulenin, which only inactivates the β-ketoacyl synthase activity of FAS, by forming an adduct with an active-site cysteine. Interference by dithiothreitol and protection by the substrates, acetyl-CoA, malonyl-CoA and NADPH, further distinguish the mechanism of C75-mediated inactivation from that of cerulenin. The most likely explanation for the multiple effects observed with C75 on rat liver FAS and its partial reactions is that there are multiple sites of interaction between C75 and FAS.
Keywords: C75, fatty acid synthase, mechanism of inactivation, obesity
Abbreviations: FAS, fatty acid synthase; DTT, dithiothreitol; ACP, acyl carrier protein
INTRODUCTION
A single, homodimeric, multifunctional FAS (fatty acid synthase; EC 2.3.1.38) is primarily responsible for the endogenous biosynthesis of palmitate in mammals [1]. It utilizes acetyl-CoA, malonyl-CoA and NADPH in a stepwise and sequential manner to build long acyl chains. Each subunit of the ∼260 kDa FAS polypeptide is composed of three N-terminal domains [containing the β-ketoacyl synthase (3-oxoacyl synthase), malonyl/acetyltransferase and dehydrase activities] and four C-terminal domains [containing the enoyl reductase, β-ketoacyl reductase (3-oxoacyl reductase), ACP (acyl carrier protein) and thioesterase activities]. (The nomenclature used for the partial activities of fatty acid synthase used in the remainder of the paper is that of [1,2]).
During each cycle of fatty acid chain elongation, malonate, covalently bound to ACP through a thioester bond, enters the active site, where acetate or the elongating fatty acid chain is covalently linked to a reactive cysteine residue. In the partial reaction catalysed by β-ketoacyl synthase, the reactive enolate anion, resulting from the decarboxylation of malonate, attacks the acyl-cysteine to form a transient tetrahedral oxyanion, which in turn decomposes rapidly to generate the resting state of β-ketoacyl synthase and the elongated β-ketoacyl (3-oxoacyl) intermediate bound to ACP [2]. Through a consecutive three-step reduction and dehydration catalysed by the β-ketoacyl reductase, β-hydroxyacyl dehydrase and enoyl reductase, the β-ketoacyl moiety is transformed into the saturated derivative. This elongated, saturated acyl derivative then serves as substrate in the next round of chain elongation until the acyl chain is 16 carbons long, a length that is required for optimal activity of the thioesterase that releases the ultimate product, free palmitic acid, from FAS [2]. Several of these partial reactions can be evaluated kinetically, independently of net palmitate synthesis, by use of alternative substrates [1,2].
C75 (see Figure 1C), an inhibitor of FAS [3], was initially designed to target the crucial condensation step catalysed by the β-ketoacyl synthase. This design was based on the structural similarity of C75, a member of the α-methylene-γ-butyrolactone chemical class, with cerulenin, a well studied FAS inhibitor that specifically targets the β-ketoacyl synthase partial activity. C75 has been suggested as the prototype for at least two aspects of therapeutic utility. First, C75 was proposed as an anti-tumour agent. In two breast cancer cell lines, SKBR3 and MCF7, where FAS was overexpressed, C75 caused apoptosis in a cancer cell-specific manner. Initiation of the apoptotic mechanism was proposed to be caused by the increase in malonyl-CoA levels as a result of FAS inhibition [4]. Secondly, C75 was proposed as an anti-obesity agent. When administered in rodents, C75 had a robust and sustained effect in reducing body weight [5]. Such weight loss effects are attributed to two metabolic consequences of C75 treatment. (1) In the central nervous system, it was hypothesized that C75 raised the malonyl-CoA concentration in the hypothalamus as a result of direct inhibition of FAS enzyme activity [6], and probably indirectly by inhibition of AMP-activated protein kinase in that region [7,8]. The elevation of the malonyl-CoA concentration in the hypothalamus, in turn, serves as a second messenger for nutrient status, which mediates appetite suppression [5,9]. (2) In peripheral tissues, C75 has another effect that is distinct from its action in the central nervous system. C75 acts as a malonyl-CoA analogue that antagonizes the allosteric inhibitory effect of malonyl-CoA on carnitine palmitoyltransferase-1, a rate-controlling enzyme for mitochondrial β-oxidation. As a result, C75 causes a constitutive stimulation of fatty acid β-oxidation in the liver, which also contributes to a robust weight loss effect in rodents [10].
Figure 1. Kinetics of the inactivation by C75 of the overall FAS reaction at room temperature (A) and at 37 °C (B).

FAS (0.25 mg/ml; ∼0.9 μM active sites) and C75 at concentrations of 0.25 mM (◆), 0.5 mM (▽), 1 mM (▲), 2 mM (□) and 4 mM (●) were incubated at room temperature or at 37 °C in a 100 μl inactivation reaction mixture containing 4% (v/v) DMSO in 0.1 M potassium phosphate buffer, pH 7.0, in one column of wells of a V-bottomed polypropylene 96-well plate. At different times of the inactivation reaction, a 4 μl aliquot from the reaction column was diluted 1:50 (v/v) into duplicate columns of assay mixtures in a clear polystyrene 96-well plate containing 100 μM NADPH, 60 μM malonyl-CoA and 20 μM acetyl-CoA in 0.1 M potassium phosphate buffer, pH 7.0, pre-equilibrated at room temperature. The overall FAS reactions were monitored as described in the Experimental section. Activity (% of control) was calculated relative to that in 4% (v/v) DMSO. Lines are fits of the data to a single exponential. The insets show replots of the observed rate constants against C75 concentration, with the line fitted to a hyperbolic equation (eqn 2) for (A) and to a linear equation for (B). (C) Structure of C75.
Although C75 has been shown to inhibit mammalian FAS in a time-dependent manner [3,11] and has been implicated in the therapeutic areas mentioned above, there have been limited studies characterizing the mechanism of C75-mediated inactivation of FAS at the enzyme level. To fill this void, we report here a detailed characterization of FAS inactivation by C75 through studies of the inhibition of the partial reactions catalysed by FAS and from studies of protection by the substrates.
EXPERIMENTAL
Materials
C75 (purity >95% by HPLC) was purchased from Calbiochem (La Jolla, CA, U.S.A.); acetyl-CoA, malonyl-CoA, NADPH, trans-1-decalone, DTT (dithiothreitol), crotonyl-CoA, EDTA disodium salt, cerulenin and palmitic acid were obtained from Sigma (St. Louis, MO, U.S.A.). [1-14C]Palmitoyl-CoA was supplied by NEN (Boston, MA, U.S.A.). All other reagents were of analytical grade.
Preparation of rat FAS
Purification of rat liver FAS followed the protocol described by Linn [12]. In the last purification step, Sephacryl 400 (Amersham Biosciences, Piscataway, NJ, U.S.A.) gel filtration chromatography was conducted in buffer A (100 mM potassium phosphate, pH 7.0, 1 mM MgCl2, 0.1 mM EDTA, 10% glycerol, 1 mM DTT). SDS/PAGE and subsequent Coomassie Blue staining of the final FAS preparation yielded a single band. During the experiments involving inactivation of FAS by C75, the concentration of DTT was kept below 0.25 mM, a concentration that has no effect on the inactivation of FAS by C75 (Figure 3). Aliquots of concentrated FAS in buffer A were stored at −80 °C until use.
Figure 3. Prevention by DTT of inactivation of FAS by C75.

FAS inactivation reactions were set up as described for Figure 1(A). In addition to 1 mM C75 in 4% DMSO (v/v), various concentrations of DTT (0, 0.1, 0.2, 0.4, 1, 2 and 4 mM) were added to each set of inactivation reactions. The C75 inactivation kinetics were analysed and the relative kinact plotted against DTT concentration (in this experiment at 0 mM DTT and 1 mM C75, kinact=0.07 min−1). Numbers in parentheses are the relative kinact expressed as a percentage of the control rate with no DTT at 1 mM C75.
Inhibition of the FAS overall reaction
A modification of the method described by Nepokroeff et al. [13] was used to measure the time course of inactivation of the overall reaction. For the inactivation reaction, FAS (0.25 mg/ml; ∼0.9 μM active sites; specific activity ∼300 nmol/min per mg) and the indicated concentrations of C75 (delivered in DMSO; final concentration 4%, v/v) and substrates were incubated at room temperature or 37 °C in 0.1 M potassium phosphate buffer, pH 7.0. Inactivation reactions were initiated at time zero by the simultaneous addition of aliquots of FAS. At different time points, 4 μl aliquots of the inactivation reaction mixtures were removed with a multi-channel pipette and diluted 1:50 (v/v) into duplicate wells of a clear polystyrene 96-well plate containing FAS overall reaction assay mixtures (100 μM NADPH, 60 μM malonyl-CoA and 20 μM acetyl-CoA in 0.1 M potassium phosphate buffer, pH 7.0) pre-equilibrated at room temperature. The release of the NADP+ product was followed spectrophotometrically for 10 min at 340 nm with a 96-well plate reader (Molecular Devices, Sunnyvale, CA, U.S.A.). Initial rates were used for the calculations.
Inhibition of FAS β-ketoacyl synthase activity
A modification of the assay described by Katiyar et al. [14] was used to measure the inactivation of the β-ketoacyl synthase reaction catalysed by FAS. This partial reaction monitors the formation of triacetic acid lactone at 283 nm from acetyl-CoA and malonyl-CoA in the absence of NADPH [15]. Although generation of triacetic acid lactone represents only ∼1% of the rate of synthesis of 3-oxobutyryl-CoA [16], the spectrophotometric assay was chosen for convenience and because detection of 3-oxobutyryl-CoA requires a radioactive chromatographic method, which is less practical for generating the number of data points needed for the detailed kinetic analysis. Due to the low sensitivity of this assay, a cuvette-based spectrophotometer and relatively large amounts of FAS were used. Inactivation reaction mixtures containing FAS (0.21 mg/ml enzyme and less than 50 μM DTT; ∼0.8 μM active sites; specific activity ∼3 nmol/min per mg) and the indicated concentrations of C75 (delivered in DMSO; final concentration 4%, v/v) and substrates were incubated at room temperature in 0.1 M potassium phosphate buffer, pH 7.0. At different time points, 18 μl aliquots of the inactivation mixtures were added to 382 μl of β-ketoacyl synthase assay mixtures containing 0.1 M potassium phosphate buffer, pH 7.0, acetyl-CoA (40 μM final) and malonyl-CoA (150 μM final) pre-equilibrated at room temperature in quartz, black, self-masking, 1-cm pathlength cuvettes. The formation of the lactone was monitored at 283 nm for 15 min in a UVIKON-940 spectrophotometer equipped with a multi-cell changer (Kontron Instruments, Basel, Switzerland).
Inhibition of FAS β-ketoacyl reductase activity
FAS β-ketoacyl reductase activity was assayed by adapting the method described by Joshi and Smith [17] to a 96-well-plate-based format similar to that described for the overall reaction. Inactivation reactions containing FAS (0.08 mg/ml and <80 μM DTT; ∼0.3 μM active sites; specific activity ∼900 nmol/min per mg) and the indicated concentrations of C75 and substrates were incubated at room temperature in 0.1 M potassium phosphate buffer, pH 7.0. At various time points, 7 μl aliquots of the inactivation reaction were diluted 1:30 (v/v) into duplicate columns of a clear polystyrene 96-well plate containing β-ketoacyl reductase assay mixtures (100 μM NADPH and 10 mM trans-1-decalone in 0.1 M potassium phosphate buffer, pH 7.0) pre-equilibrated at room temperature. The release of the NADP+ product was followed spectrophotometrically for 10 min at 340 nm with a 96-well plate reader. Initial rates were used for the calculations.
Inhibition of FAS enoyl reductase activity
The method described by Nixon et al. [15] was adapted to a 96-well-plate-based format similar to that described for the overall reaction. Inactivation reactions containing FAS (0.8 mg/ml and <200 μM DTT; ∼3 μM active sites; specific activity ∼25 nmol/min per mg) and the indicated concentrations of C75 and substrates were incubated at room temperature in 0.1 M potassium phosphate buffer, pH 7.0. At various time points, 10 μl aliquots of the inactivation reaction were diluted 1:20 (v/v) into duplicate enoyl reductase assay mixtures in a clear polystyrene 96-well plate containing 100 μM NADPH and 50 μM crotonyl-CoA in 0.1 M potassium phosphate buffer, pH 7.0, pre-equilibrated at room temperature. The release of the NADP+ product was followed spectrophotometrically for 10 min at 340 nm with a 96-well plate reader, and initial rates were used for the calculations.
Inhibition of FAS thioesterase activity
FAS thioesterase activity was assayed by measuring the hydrolysis of [1-14C]palmitoyl-CoA as described by Stoops and Wakil [18]. Inactivation reactions containing FAS (0.2 mg/ml and <50 μM DTT; ∼0.8 μM active sites; specific activity ∼25 nmol/min per mg) and the indicated concentrations of C75 and substrates were incubated at room temperature in 0.1 M potassium phosphate buffer, pH 7.0. At various time points, 100 μl aliquots were delivered to the thioesterase reaction mixture containing 100 mM potassium phosphate and 10 μM [1-14C]palmitoyl-CoA (3 Ci/mol; NEN) in a total volume of 500 μl. After incubation at room temperature for 2 min, the reactions were terminated by the addition of 0.1 ml of 10% (v/v) H2SO4. Carrier palmitate (0.62 mg) in ethanol was added and the mixture was extracted three times with 3 ml of pentane. The extracts were pooled and the pentane was evaporated to dryness under N2. The residue was redissolved in 0.2 ml of pentane and delivered to 5 ml of scintillation fluid, and radioactivity was counted.
Data analysis
Each inhibition reaction was normalized to a DMSO control [4% (v/v) final concentration] at each time point under conditions where C75 was in excess over FAS. The data for activity (% of control) against time were fitted to the equation for a single exponential decay (first order) to obtain the observed inactivation rate constant. For substrate protection experiments, the Kitz and Wilson analysis [19] was performed by fitting all of the observed inactivation rates at each substrate and C75 concentration to eqn (1) (see below) for competitive inhibition. For C75 saturation experiments in the absence of substrates, data were fitted to eqn (2), and for competition with substrates at a single C75 concentration, data were fitted to eqn (3) [20]. In these equations, kobs is the observed first-order rate of enzyme inactivation, kinact is the maximal rate of enzyme inactivation, KI is the dissociation constant for the initial (non-covalent) complex of inhibitor with FAS, [I] is the inactivator concentration, [A] is the concentration of protectant (here the substrates), and KA is the dissociation constant for the protectant.
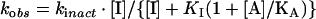 |
1 |
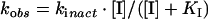 |
2 |
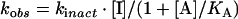 |
3 |
Graphs and fits of the data to non-linear least-squares equations were obtained using the programs XLfit (ID Business Solutions) add-in to Excel (Microsoft) or GraFit version 5.0 (Erithracus Software Ltd., Horley, Surrey, U.K.).
RESULTS
Inactivation of the FAS overall reaction by C75 is pseudo-first-order and saturable
Inactivation time courses were studied in detail by preincubation of various concentrations of C75 at a fixed concentration of rat liver FAS followed by a large dilution of an aliquot of the FAS/C75 mixture into either FAS overall assay mixtures or various FAS partial reaction mixtures at multiple time intervals. As shown in Figure 1, the mode of inactivation of the FAS overall reaction by C75 appears to be pseudo-first-order at room temperature and at 37 °C. Plots of log activity remaining (expressed as % of control) against preincubation time were linear. The observed rate constants were obtained from fits of the data for activity (% of control) against time to the equation for a single exponential decay. At 37 °C (Figure 1B, inset), the observed rate constant appeared to increase linearly with increasing concentration of C75, suggestive of either very weak initial inhibitor complex formation or a bimolecular reaction mechanism. However, at room temperature, over a 16-fold range of C75 concentrations, the concentration-dependence of the inactivation rate began to exhibit saturation, indicative of a weak initial inhibitor complex, but was limited by our ability to assay at higher inhibitor concentrations (inset to Figure 1A; hyperbolic fit of the observed rate constant against C75 concentration data to eqn 2). The results at room temperature are suggestive of the scheme shown in eqn (4), where an initial reversible complex is converted to the irreversibly inhibited enzyme:
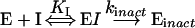 |
4 |
Another indication of this mechanism is shown in Figure 2(B), for data obtained in the absence of malonyl-CoA. The y-intercept of the Kitz and Wilson plot of 1/(observed kinact) against 1/[C75] is finite. The rate constant (kinact) at saturation and the dissociation constant for the initial complex (KI) were obtained from a hyperbolic fit of the data. Theoretical fitted lines shown in Figure 2(B) are to eqn (1); double-reciprocal graphs are shown for linear representations of the hyperbolic model.
Figure 2. Competition between C75 and malonyl-CoA, acetyl-CoA and NADPH.

(A) Protection of FAS by malonyl-CoA from inactivation by 1 mM C75. Aliquots of FAS were preincubated with 1 mM C75 as described in Figure 1(A). During the incubation, malonyl-CoA was added to the mixture at concentrations of 0 μM (○), 0.7 μM (■), 2.2 μM (△), 6.7 μM (▼) and 60 μM (◇). Lines are fits of the data to a single exponential. The inset shows a replot of the observed rate constants against malonyl-CoA concentration, with a fit of the data for competition at a single concentration of inactivator (eqn 3). In (B)–(D), for the Kitz–Wilson analyses, aliquots of FAS (0.25 mg/ml; ∼0.9 μM active sites) were mixed with various concentrations of C75 (0, 0.125, 0.25, 0.5, 1, 2 and 4 mM). To each of the C75/FAS inactivation reaction mixtures for competition analyses, various substrate concentrations were added as follows: (B) malonyl-CoA [0 μM (○), 0.74 μM (●), 2.22 μM (□), 6.67 μM (■)]; (C) acetyl-CoA [0 μM (○), 2.2 μM (●), 6.67 μM (□), 20 μM (■)]; (D) NADPH [0 μM (○), 3.3 μM (●), 10 μM (□), 100 μM (■)]. Lines in (B)–(D) are global fits of the data at multiple substrate and inhibitor concentrations to eqn (1).
The results of the theoretical fits are summarized in Table 1. For C75 at ambient temperature, kinact at saturation was approx. 1 min−1 and the KI for the initial complex formation was approx. 16 mM. These values are estimates, since the highest C75 concentration used was 4 mM. The estimated values of 16 mM for KI and 1 min−1 for kinact were used to calculate the apparent second-order rate constant, 0.06 min−1·mM−1, for inactivation at low concentrations of C75. At 37 °C an apparent second-order rate constant of 0.21 min−1·mM−1 was obtained from the slope of a linear fit to kobs against C75.
Table 1. Summary of substrate protection data from Kitz and Wilson analyses.
Substrates protect the FAS overall reaction from inactivation by C75 in a competitive manner
The effects of various substrates of FAS upon the rate of inactivation of the overall reaction were tested at multiple concentrations of both the substrates and C75 (Figure 2). Each of the three substrates, i.e. acetyl-CoA, malonyl-CoA and NADPH, diminished the rate of inactivation by C75 in a concentration-dependent and saturable manner. As a representative example, Figure 2(A) and the inset show that malonyl-CoA progressively lowered the observed rate of FAS inactivation by 1 mM C75 in a concentration-dependent manner, consistent with direct competition between C75 and malonyl-CoA.
The competitive nature of inactivation by C75 with malonyl-CoA is further illustrated in Figure 2(B), where the Kitz and Wilson plot of 1/(observed kinact) against 1/[C75] intersects on the y-axis. Similar results were obtained for acetyl-CoA (Figure 2C) and NADPH (Figure 2D). The results from the fits to eqn (1) are summarized in Table 1. kinact at saturation and KI for the initial complex formation obtained for each of the three substrates were in close agreement. Also, the apparent dissociation constants (KA) for each of the protectants are in good agreement with their reported Km values (Table 1).
High concentrations of DTT interfere with the inactivation of FAS by C75
To investigate potential effects of DTT carried over from the storage buffer on the inactivation of FAS by C75, which may obscure the interpretation of kinetic data, we designed experiments to systematically address the effects of DTT. We found that DTT does prevent inactivation of the overall reaction by 1 mM C75. The dose–response relationship of the interference appears to be steep, with little effect at or below 0.4 mM, but a substantial effect at or above 1 mM (Figure 3). The apparent DTT effect may be attributed to two potential mechanisms. (1) Since it is known that cerulenin forms a covalent adduct with the active cysteine in the condensing enzyme [1,2], C75 may also inactivate FAS by such a covalent interaction. DTT may protect this cysteine from the formation of such an adduct. (2) Alternatively, DTT could react directly with C75 chemically, destroying its ability to inactivate FAS. To differentiate between these two possibilities, we first pretreated 5 mM C75 with 5 mM DTT for 45 min. After 10-fold dilution with FAS and buffer, the observed rate of inactivation by this pretreated C75 was then determined and compared with that by untreated C75. Of note, in the subsequent inactivation reaction following the treatment, the DTT concentration was 0.5 mM, a concentration that was shown to have little effect on the inactivation of FAS by C75 (Figure 3). Pretreatment of C75 with DTT completely abolished its ability to inactivate FAS. This experiment establishes that high concentrations of DTT interfere with the inactivation of FAS by C75.
Malonyl-CoA and acetyl-CoA protect the FAS β-ketoacyl synthase reaction from inactivation by C75
Inactivation of the β-ketoacyl synthase partial reaction of FAS by C75 was characterized in a manner similar to that for the overall reaction, except that NADPH was not present in the assay (see the Experimental section). Similar to that observed for the overall reaction, inactivation of the β-ketoacyl synthase activity was concentration-dependent, pseudo-first-order, and prevented by both acetyl-CoA and malonyl-CoA (Figure 4). Since carry-over of even a small amount of NADPH from the preincubation mixture interfered with this assay for monitoring the β-ketoacyl synthase partial activity, the protectant effects of this substrate were not studied.
Figure 4. Inactivation of the β-ketoacyl synthase partial activity of FAS by C75, and protection by malonyl-CoA and acetyl-CoA.

(A) Inactivation reactions containing FAS (0.21 mg/ml enzyme and <50 μM DTT; ∼0.8 μM active sites) and various concentrations of C75 [0.5 mM (○), 1 mM (■) and 2 mM (△)] were set up in 0.1 M potassium phosphate buffer, pH 7.0. At different times, 18 μl of a mixture of acetyl-CoA (40 μM final) and malonyl-CoA (150 μM final) was added to 382 μl of inactivation reaction mixture pre-equilibrated at room temperature in five quartz 1-cm pathlength cuvettes, and the formation of the lactone was monitored at 283 nm to assess β-ketoacyl synthase activity. For substrate protection experiments, inactivation reactions and β-ketoacyl synthase assays were carried out as described in (A), except that malonyl-CoA (B) or acetyl-CoA (C) was added to the inactivation reactions at a concentration of 0 μM (○), 2.2 μM (■), 6.7 μM (△) or 20 μM (◆). Activity (% of control) was calculated relative to that in 4% (v/v) DMSO. Lines shown are fits of the data to a single exponential. The insets to (B) and (C) show replots of the observed rate constants against protectant concentration, with lines fitted to eqn (3) for competition at a single concentration of inactivator.
The observed rates of inactivation of the β-ketoacyl synthase were about 1.5 times higher than those observed for the overall reaction at the same concentrations of C75 (Table 2). At the same concentrations of either acetyl-CoA or malonyl-CoA, the protection, measured by the relative kinact at 1 mM C75, was about 2-fold lower for the β-ketoacyl synthase than for the overall reaction (Table 2). From the fits of the data to eqn (3), as shown in the insets to Figures 4(B) and 4(C), it appears that both acetyl-CoA and malonyl-CoA protect the β-ketoacyl synthase activity of FAS from inactivation by C75 in a competitive manner. The dissociation constants obtained for these two protectants from fits of the data to eqn (3) were 2.8 μM for acetyl-CoA and 4.5 μM for malonyl-CoA, in good agreement with the values obtained for the overall reaction (Table 1).
Table 2. Summary of C75-mediated inactivation and substrate protection of FAS partial reactions.
The concentration of C75 used was 1 mM. The concentration of protectant was 20 μM for malonyl-CoA and acetyl-CoA, and 100 μM for NADPH. Values in parentheses are kinact (min−1). For thioesterase, values are relative to the zero time point. ND, not determined, due to interference with the assay.
| FAS reaction | Protectant | Relative kinact (% of control) |
|---|---|---|
| Overall | None | 100 (0.056 min−1) |
| Malonyl-CoA | 11.1 | |
| Acetyl-CoA | 13.3 | |
| NADPH | 19.4 | |
| β-Ketoacyl synthase | None | 100 (0.082 min−1) |
| Malonyl-CoA | 21.1 | |
| Acetyl-CoA | 22.0 | |
| NADPH | ND | |
| β-Ketoacyl reductase | None | 100 (0.008 min−1*) |
| Malonyl-CoA | ND | |
| Acetyl-CoA | 100 (0.011 min−1*) | |
| NADPH | 0 | |
| Enoyl reductase | None | 100 (0.082 min−1) |
| Malonyl-CoA | 35.0 | |
| Acetyl-CoA | 17.6 | |
| NADPH | 23.9 | |
| Thioesterase | None | 100 (0.13 min−1) |
| Malonyl-CoA | 85 | |
| Acetyl-CoA | 101 | |
| NADPH | 120 |
* After a 60 min lag.
Inactivation by C75 of the FAS β-ketoacyl reductase reaction is not a simple first-order process and is not reversed by acetyl-CoA
Preincubation/dilution was also used to characterize the inactivation of the β-ketoacyl reductase partial activity of FAS by C75. Unlike the overall reaction or the other partial reactions studied, there was a lag before the C75-mediated inactivation of the β-ketoacyl reductase by an apparent pseudo-first-order process (Figure 5). The extent of the lag was inversely dependent on the concentration of C75: at 1 mM C75, the lag period extended to nearly 1 h before the loss of β-ketoacyl reductase activity was observed; at 2 mM C75 the lag was reduced to about 20 min; and at 4 mM C75 the lag was reduced further to <5 min (Figure 5). No loss of β-ketoacyl reductase activity was observed at 0.5 mM C75 with up to 120 min of preincubation.
Figure 5. Inactivation of the β-ketoacyl reductase partial activity of FAS by C75.

FAS (0.8 mg/ml and <20 μM DTT; ∼3 μM active sites) and C75 at concentrations of 0.5 mM (○), 1 mM (■), 2 mM (△) and 4 mM (◆) were incubated at room temperature in a 100 μl inactivation reaction mixture containing 4% (v/v) DMSO in 0.1 M potassium phosphate buffer, pH 7.0, in one column of wells of a V-bottomed polypropylene 96-well plate. At different times of the inactivation reaction, a 7 μl aliquot from the reaction column was diluted 1:30 (v/v) into duplicate columns of assay mixtures for the β-ketoacyl reductase reactions, as described in the Experimental section. Plots of activity as % of control (no C75) against preincubation time are shown in linear–linear format using the average of duplicate determinations to emphasize the lag phase. Activity (% of control) was calculated relative to that in 4% (v/v) DMSO. Lines are fits of the data after the lag period to a single exponential.
We do not yet understand the origin of the lag observed when monitoring this partial reaction. However, inactivation of the β-ketoacyl reductase activity at 4 mM C75 was pseudo-first-order, and the observed kinact was 0.056 min−1, approx. 3.4 times slower than the observed kinact for the overall reaction at 4 mM C75 (0.19 min−1). At a lower concentration of C75 in the preincubation (1 mM), neither the lag period nor the rate of inactivation was affected by acetyl-CoA (Table 2). At 100 μM NADPH and 1 mM C75, the lag period was >120 min, and no inactivation was observed up to that time point, indicating that NADPH was either preventing or delaying further the onset of inactivation by C75 (Table 2). Malonyl-CoA interfered with the β-ketoacyl reductase assay and could not be studied as a protectant.
Substrates protect the FAS enoyl reductase reaction from inactivation by C75
Inactivation by C75 of the enoyl reduction catalysed by FAS was probed using crotonyl-CoA instead of malonyl-CoA and acetyl-CoA in the assay post-preincubation. As observed for the overall reaction and the β-ketoacyl synthase partial reaction, C75-mediated inactivation of enoyl reduction was concentration-dependent, followed pseudo-first-order kinetics and was prevented by all three substrates, i.e. NADPH, acetyl-CoA and malonyl-CoA (Figure 6). Unlike the unusual inactivation kinetics of the β-ketoacyl reductase activity, no lag periods were observed for the inactivation of the enoyl reductase by C75. The observed kinact values were comparable with those for the β-ketoacyl synthase at the same C75 concentrations and about 1.5 times higher than the inactivation rates for the overall reaction (Table 2). Also, similar to the results with the β-ketoacyl synthase activity, at the same concentrations of acetyl-CoA and malonyl-CoA, relatively less protection was observed for the enoyl reductase than for the overall reaction, while 100 μM NADPH afforded about the same protection for both reactions (Table 2). Protection by acetyl-CoA, malonyl-CoA and NADPH also appeared to be concentration-dependent and competitive with C75 for the enoyl reductase partial reaction (Figure 6, insets). Dissociation constants obtained for these three protectants from fits of the data to eqn (3) were 1.2 μM for acetyl-CoA, 4.9 μM for malonyl-CoA and 6.4 μM for NADPH, in good agreement with the values obtained for the overall reaction (Table 1) and the β-ketoacyl synthase activity (see above).
Figure 6. Inactivation of the enoyl reductase partial activity of FAS by C75, and protection by substrates.

(A) Inactivation reactions containing rat liver FAS (0.8 mg/ml enzyme and <200 μM DTT; ∼3 μM active sites) and various concentrations of C75 [0.5 mM (○), 1 mM (■) and 4 mM (△)] were incubated at room temperature in a 100 μl inactivation reaction mixture containing 4% (v/v) DMSO in 0.1 M potassium phosphate buffer, pH 7.0, in one column of wells of a V-bottomed polypropylene 96-well plate. At different times of the inactivation reaction, a 10 μl aliquot from the reaction column was diluted 1:20 (v/v) into duplicate columns of assay mixtures for the enoyl reductase reactions, as described in the Experimental section. For substrate protection experiments, inactivation reactions and enoyl reductase assays were carried out as described in (A), except that malonyl-CoA (B), acetyl-CoA (C) or NADPH (D) was added to the inactivation reactions at concentrations of 0 μM (○), 2.2 μM (■), 6.7 μM (△) and 20 μM (◆) for the CoA substrates, and 0 μM (○), 3.3 μM (■), 10 μM (△) and 100 μM (◆) for NADPH. Activity (% of control) was calculated relative to that in 4% (v/v) DMSO. Lines shown are to fits of the data to a single exponential. The insets to (B)–(D) are replots of the observed rate constants against the protectant concentration, with lines fitted to eqn (3) for competition at a single concentration of inactivator.
Substrates do not protect the FAS thioesterase reaction from inactivation by C75
The thioesterase activity of FAS was monitored using a radio-chemical assay. As shown in Figure 7 and Table 2, inactivation of the thioesterase was pseudo-first-order at 1 mM C75, and the observed rate of inactivation was 0.13 min−1, i.e. 2.4 times faster than inactivation of the overall reaction and about 1.6 times faster than inactivation of the β-ketoacyl synthase and the enoyl reductase activities at the same concentration of C75. In sharp contrast with the overall reaction, the β-ketoacyl synthase reaction and the enoyl reductase reaction, acetyl-CoA, malonyl-CoA and NADPH did not protect the thioesterase from inactivation by C75.
Figure 7. Inactivation of the thioesterase reaction by 1 mM C75.

Inactivation reactions containing FAS (0.2 mg/ml and <50 μM DTT; ∼0.8 μM active sites) and the indicated concentrations of C75 and substrates were incubated at room temperature in 0.1 M potassium phosphate buffer, pH 7.0. At various time points, 100 μl aliquots were delivered to the thioesterase reaction mixture containing 100 mM potassium phosphate and 10 μM [1-14C]palmitoyl-CoA (3 Ci/mol; NEN) in a total volume of 500 μl. After incubation at room temperature for 2 min, the reactions were terminated by the addition of 0.1 ml of 10% (v/v) H2SO4 and analysed as described in the Experimental section. Data are for the addition of no substrate (○), 20 μM malonyl-CoA (□), 20 μM acetyl-CoA (△) or 100 μM NADPH (▽) at 1 mM C75. Activity (% of control) is calculated relative to the zero time data.
DISCUSSION
The experiments described here were conducted in order to clarify the detailed mechanism of inhibition of mammalian FAS by C75. The rat liver enzyme was inactivated by C75 in a concentration-dependent manner which obeyed pseudo-first-order kinetics. Inactivation kinetics were saturable, but relatively inefficient, with a kinact at saturation of 1 min−1 and a KI for initial complex formation of approx. 16 mM. The apparent second-order rate constants of 0.06 mM−1·min−1 calculated from these values at room temperature and of 0.21 mM−1·min−1 at 37 °C calculated from the slope of kobs against [C75] are similar in magnitude to the value reported for iodoacetamide of 0.19 mM−1·min−1 with chicken liver FAS at 25 °C [18], but are considerably lower than those of the well-studied, specific FAS inactivator cerulenin (5.8 mM−1·min−1 for yeast FAS at 0 °C) [21], and the reported value for C75-mediated inactivation of chicken liver FAS at 37 °C (1.3 min−1·mM−1) [11]. In our own experiments, we observed that 1 mM cerulenin inactivated rat liver FAS activity completely within 30 s at room temperature (results not shown). This rapid inactivation of rat FAS by cerulenin is consistent with the large difference between the kinetic parameters for C75 with mammalian FAS and those reported for cerulenin at lower temperatures with yeast FAS.
The present study represents the first detailed report of the effects of C75 on various partial reactions catalysed by FAS. Although we provide experimental evidence that C75 inactivates the β-ketoacyl synthase activity, as long speculated in the literature [3], to our surprise C75 also exhibited clear inactivation of other partial activities, such as the enoyl reductase and the thioesterase. Of note, the relative kinact values for these three partial activities were similar to each other and about the same or higher than that of the overall reaction. Therefore, if the inactivation of these partial activities is independent, then inactivation of any one of these activities could contribute significantly to inactivation of the overall reaction. In contrast, the onset of inactivation of the β-ketoacyl reductase was delayed and much slower than inactivation of the overall reaction, indicative of an inactivation mechanism that may not involve the normal functions of the enzyme. This feature of inactivation of multiple partial activities of FAS by C75 is in sharp contrast with the effects of cerulenin, which was shown to inactivate only the β-ketoacyl synthase [21,22].
The differences between cerulenin and C75 were highlighted further in substrate protection experiments. Protection from inactivation by C75 was afforded by both acetyl-CoA and malonyl-CoA for the overall reaction and the β-ketoacyl synthase and enoyl reductase partial reactions, but not for the thioesterase activity. However, only the β-ketoacyl synthase is inactivated by cerulenin, with protection by only acetyl-CoA and not malonyl-CoA [21]. Also in contrast with inactivation by cerulenin, NADPH protected the overall reaction and the enoyl reductase from inactivation by C75.
With respect to the overall FAS reaction, C75 is a competitive irreversible inhibitor with regard to all three substrates, with the kinetics of the complexing type, i.e. a weak non-covalent enzyme–inhibitor complex is formed before irreversible enzyme modification. In addition, protection of the β-ketoacyl synthase and enoyl reductase activities by malonyl-CoA and acetyl-CoA appears to be competitive with C75, as does NADPH protection of the enoyl reductase activity. Although the differences could be due to a large temperature or species effect or both, the protection results with rat liver FAS and C75 at ambient temperatures differ from those reported by Wang and Tian [11] for chicken liver FAS at 37 °C. With chicken liver FAS, the overall reaction was protected from C75-mediated inactivation by malonyl-CoA, but not significantly by acetyl-CoA or NADPH [11]. Also, in contrast with the saturation shown here at room temperature, but similar to our observations at 37 °C, Wang and Tian [11] observed a linear relationship between the observed kinact and the concentration of C75 at 37 °C, perhaps because KI was elevated at the higher temperature to an extent not detected in their experiments. Finally, they also observed biphasic inactivation kinetics at the higher temperature, with the slower phase having an apparent second-order rate constant (∼0.13 mM−1·min−1) much closer to the value for rat liver FAS (∼0.21 mM−1·min−1 at 37 °C and 0.06 mM−1·min−1 at room temperature). Both differences may be explained by the different experimental temperatures [20].
The lower inactivation rates and delay in the onset of inactivation for the β-ketoacyl reductase activity could be due to non-specific and/or non-active-site-directed effects of C75, in contrast with its effects on the β-ketoacyl synthase, enoyl reductase or overall reactions. Partitioning of the initial reversible EI complex between inactive enzyme and a slowly interconverting, reversible conformation (FI) that cannot directly form inactive enzyme could explain the lag prior to the inactivation of the β-ketoacyl reductase (eqn 5):
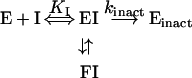 |
5 |
In this mechanism, FI is either a very weakly inhibitory complex or a non-inhibitory complex, such that dilution into the β-ketoacyl reductase assay results in full activity. Alternatively, FI could reversibly release and recombine with C75 during the preincubation. It is also interesting to note that the β-ketoacyl reductase assay does not use a substrate with a CoA thioester, which may explain the unusual kinetics for C75-mediated inactivation. C75 may interfere more rapidly with either the cysteine or ACP thiol groups involved in thioester exchange with the CoA substrates, but not immediately affect the reduction of trans-decalone. Much slower interaction with a different enzyme nucleophile may account for the slower C75-mediated inactivation kinetics for the β-ketoacyl reductase. Consistent with these findings, Wang and Tian [11] did not observe inactivation of chicken liver β-ketoacyl reduction using a different substrate, ethyl acetoacetate, which also lacked a CoA thioester.
We also observed that, at concentrations of ≥1 mM, DTT blocked the inactivation of rat FAS by C75, but this is most probably due to a reaction in solution to form an ineffective DTT–C75 adduct. This feature of C75 is another difference from cerulenin, in that DTT, cysteine and 2-mercaptoethanol did not protect yeast FAS from inactivation by cerulenin [21]. One potential consequence of the apparent reactivity of C75 with nucleophiles in solution is that its efficacy in vivo may be diminished by reaction with endogenous thiols such as glutathione.
The present study is also the first report on the inactivation of the thioesterase activity of a mammalian FAS by C75. The inability of malonyl-CoA to protect the thioesterase activity from inactivation by C75 implies that the mechanism of inactivation may be very different from that for the β-ketoacyl synthase or the enoyl reductase activities. It is difficult to explain all the observed effects of C75 by postulating a single site of interaction. However, additional studies are needed to fully elucidate whether C75 has a single site of action resulting in multiple effects on the partial activities of FAS, or has multiple sites of action with similar inactivation kinetics leading to the different effects on FAS reactions.
The specific activities of the rat FAS preparation used in the present study were lower than the best values reported in the literature, when assessed either for the overall reaction or for the various partial activities [12,23]. These discrepancies could be due to different methods for measuring protein concentration, or to different assay temperatures (ambient temperature was employed for the present study), or to the possibility that the assay conditions were not optimized for maximal velocity at room temperature. However, the relative order of the activities, i.e. β-ketoacyl reductase (∼900 nmol/min per mg)>overall reaction (∼300 nmol/min per mg)>thioesterase (∼25 nmol/min per mg)=enoyl reductase (∼25 nmol/min per mg)>β-ketoacyl synthase (∼3 nmol/min per mg), was the same as that observed for chicken liver FAS [18] and for human FAS [24]. Also, because the rates of C75-mediated inactivation of the β-ketoacyl synthase, enoyl reductase and thioesterase partial activities are comparable with that for the overall reaction, we believe that the conclusions from the current work are valid.
FAS has been discussed as a potential target for anti-obesity intervention therapy [5]. However, the relatively weak ability of C75 to inactivate FAS as reported in the present paper raises questions about the relevance in vivo of inhibition of FAS by C75. In addition to inhibition of FAS, C75 has been shown to have a variety of biological activities, including activation of carnitine palmitoyltransferase-1, which is a rate-limiting enzyme for β-oxidation [10]. C75 has also been shown to decrease AMP-activated protein kinase activity in the brain [7,8], which may conceivably result in an anorectic effect [25]. These biological activities of C75 offer alternative explanations to inhibition of FAS for the robust weight loss effect elicited by C75 [5]. Recent studies of the pharmacokinetic features of C75 and comparisons of the anorectic level of C75 in vivo with the potency of FAS inhibition in vitro provide support for the alternative explanations [26].
Acknowledgments
We thank Rupalie Meegalla, Cathy Kieras and Milton Hillman for excellent technical assistance, Jon Hangeland for useful discussions, and Mark Harpel for a critical reading of the manuscript.
References
- 1.Wakil S. J. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. 1989;28:4523–4530. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- 2.Smith S., Witkowski A., Joshi A. K. Structure and functional organization of the animal fatty acid synthase. Prog. Lipid Res. 2003;42:289–317. doi: 10.1016/s0163-7827(02)00067-x. [DOI] [PubMed] [Google Scholar]
- 3.Kuhajda F. P., Pizer E. S., Li J. N., Mani N. S., Frehywot G. L., Townsend C. A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. U.S.A. 2000;97:3450–3454. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pizer E. S., Thupari J., Han W. F., Pinn M. L., Chrest F. J., Frehywot G. L., Townsend C. A., Kuhajda F. P. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000;60:213–218. [PubMed] [Google Scholar]
- 5.Loftus T. M., Jaworsky D. E., Frehywot G. L., Townsend C. A., Ronnett G. V., Lane M. D., Kuhajda F. P. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288:2379–2381. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 6.Hu Z., Cha S. H., Chohnan S., Lane M. D. Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc. Natl. Acad. Sci. U.S.A. 2003;100:12624–12629. doi: 10.1073/pnas.1834402100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim E.-K., Miller I., Aja S., Landree L. E., Pinn M., McFadden J., Kuhajda F. P., Moran T. H., Ronnett G. V. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J. Biol. Chem. 2004;279:19970–19976. doi: 10.1074/jbc.M402165200. [DOI] [PubMed] [Google Scholar]
- 8.Landree L. E., Hanlon A. L., Strong D. W., Rumbaugh G., Miller I. M., Thupari J. N., Connolly E. C., Huganir R. L., Richardson C., Witters L. A., et al. C75, a fatty acid synthase inhibitor, modulates AMP-activated protein kinase to alter neuronal energy metabolism. J. Biol. Chem. 2004;279:3817–3827. doi: 10.1074/jbc.M310991200. [DOI] [PubMed] [Google Scholar]
- 9.Kumar M. V., Shimokawa T., Nagy T. R., Lane M. D. Differential effects of a centrally acting fatty acid synthase inhibitor in lean and obese mice. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1921–1925. doi: 10.1073/pnas.042683699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thupari J. N., Landree L. E., Ronnett G. V., Kuhajda F. P. C75 increases peripheral energy utilization and fatty acid oxidation in diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9498–9502. doi: 10.1073/pnas.132128899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X., Tian W. Green tea epigallocatechin gallate: a natural inhibitor of fatty-acid synthase. Biochem. Biophys. Res. Commun. 2001;288:1200–1206. doi: 10.1006/bbrc.2001.5923. [DOI] [PubMed] [Google Scholar]
- 12.Linn T. C. Purification and crystallization of rat liver fatty acid synthetase. Arch. Biochem. Biophys. 1981;209:613–619. doi: 10.1016/0003-9861(81)90320-9. [DOI] [PubMed] [Google Scholar]
- 13.Nepokroeff C. M., Lakshmanan M. R., Porter J. W. Fatty-acid synthase from rat liver. Methods Enzymol. 1975;35:37–44. doi: 10.1016/0076-6879(75)35136-7. [DOI] [PubMed] [Google Scholar]
- 14.Katiyar S. S., Briedis A. V., Porter J. W. Synthesis of fatty acids from malonyl-CoA and NADPH by pigeon liver fatty acid synthetase. Arch. Biochem. Biophys. 1974;162:412–420. doi: 10.1016/0003-9861(74)90199-4. [DOI] [PubMed] [Google Scholar]
- 15.Nixon J. E., Putz G. R., Porter J. W. Synthesis of triacteic acid lactone by the pigeon liver fatty acid synthetase complex. J. Biol. Chem. 1968;243:5471–5478. [PubMed] [Google Scholar]
- 16.Witkowski A., Joshi A. K., Lindqvist Y., Smith S. Conversion of a beta-ketoacyl synthase to a malonyl decarboxylase by replacement of the active-site cysteine with glutamine. Biochemistry. 1999;38:11643–11650. doi: 10.1021/bi990993h. [DOI] [PubMed] [Google Scholar]
- 17.Joshi A. K., Smith S. Construction, expression, and characterization of a mutated animal fatty acid synthase deficient in the dehydrase function. J. Biol. Chem. 1993;268:22508–22513. [PubMed] [Google Scholar]
- 18.Stoops J. K., Wakil S. J. Animal fatty acid synthetase. A novel arrangement of the β-ketoacyl synthetase sites comprising domains of the two subunits. J. Biol. Chem. 1981;256:5128–5133. [PubMed] [Google Scholar]
- 19.Kitz R., Wilson I. B. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 20.Silverman R. B. Mechanism–Based Enzyme Inactivation: Chemistry and Enzymology, vol. 1. Boca Raton, FL: CRC Press; 1988. Introduction. [Google Scholar]
- 21.Kawaguchi A., Tomoda H., Nozoe S., Omura S., Okuda S. Mechanism of action of cerulenin on fatty acid synthetase. Effect of cerulenin on iodoacetamide-induced malonyl-CoA decarboxylase activity. J. Biochem. (Tokyo) 1982;92:7–12. doi: 10.1093/oxfordjournals.jbchem.a133933. [DOI] [PubMed] [Google Scholar]
- 22.Funabashi H., Kawaguchi A., Tomoda H., Omura S., Okuda S., Iwasaki S. Binding site of cerulenin in fatty acid synthetase. J. Biochem. (Tokyo) 1989;105:751–755. doi: 10.1093/oxfordjournals.jbchem.a122739. [DOI] [PubMed] [Google Scholar]
- 23.Witkowski A., Joshi A. K., Smith S. Fatty acid synthase: in vitro complementation of inactive mutants. Biochemistry. 1996;35:10569–10575. doi: 10.1021/bi960910m. [DOI] [PubMed] [Google Scholar]
- 24.Jayakumar A., Tai M., Huang W., Al-Feel W., Hsu M., Abu-Elheiga L., Chirala S. S., Wakil S. J. Human fatty acid synthase: properties and molecular cloning. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8695–8699. doi: 10.1073/pnas.92.19.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minokoshi Y., Alquier T., Furukawa N., Kim Y. B., Lee A., Xue B., Mu J., Foufelle F., Ferre P., Birnbaum M. J., et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature (London) 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 26.Rohrbach K. W., Han S., Gan J., O'Tanyi E. J., Zhang H., Chi C. L., Taub R., Largent B. L., Cheng D. Disconnection between the early onset anorectic effects by C75 and hypothalamic fatty acid synthase inhibiiton in rodents. Eur. J Pharmacol. 2005 doi: 10.1016/j.ejphar.2005.01.034. in the press. [DOI] [PubMed] [Google Scholar]
- 27.Kumar S., Dorsey J. A., Muesing R. A., Porter J. W. Comparative studies of the pigeon liver fatty acid synthetase complex and its subunits. Kinetics of partial reactions and the number of binding sites for acetyl and malonyl groups. J. Biol. Chem. 1970;245:4732–4744. [PubMed] [Google Scholar]