

Abstract
We cloned a novel β-1,3-xylanase gene, consisting of a 1728-bp open reading frame encoding 576 amino acid residues, from a marine bacterium, Vibrio sp. strain AX-4. Sequence analysis revealed that the β-1,3-xylanase is a modular enzyme composed of a putative catalytic module belonging to glycoside hydrolase family 26 and two putative carbohydrate-binding modules belonging to family 31. The recombinant enzyme hydrolysed β-1,3-xylan to yield xylo-oligosaccharides with different numbers of xylose units, mainly xylobiose, xylotriose and xylotetraose. However, the enzyme did not hydrolyse β-1,4-xylan, β-1,4-mannan, β-1,4-glucan, β-1,3-xylobiose or p-nitrophenyl-β-xyloside. When β-1,3-xylo-oligosaccharides were used as the substrate, the kcat value of the enzyme for xylopentaose was found to be 40 times higher than that for xylotetraose, and xylotriose was extremely resistant to hydrolysis by the enzyme. A PSI-BLAST search revealed two possible catalytic Glu residues (Glu-138 as an acid/base catalyst and Glu-234 as a nucleophile), both of which are generally conserved in glycoside hydrolase superfamily A. Replacement of these two conserved Glu residues with Asp and Gln resulted in a significant decrease and complete loss of enzyme activity respectively, without a change in their CD spectra, suggesting that these Glu residues are the catalytic residues of β-1,3-xylanase. The present study also clearly shows that the non-catalytic putative carbohydrate-binding modules play an important role in the hydrolysis of insoluble β-1,3-xylan, but not that of soluble glycol-β-1,3-xylan. Furthermore, repeating a putative carbohydrate-binding module strongly enhanced the hydrolysis of the insoluble substrate.
Keywords: β-1,3-xylan; β-1,3-xylanase; carbohydrate-binding module; glycoside hydrolase; marine bacteria; site-directed mutagenesis
Abbreviations: CBM, carbohydrate-binding module; GH, glycoside hydrolase; IPTG, isopropyl β-D-thiogalactoside; ORF, open reading frame; pNP, p-nitrophenyl; rXYL4, recombinant AX-4 β-1,3-xylanase; TBS, Tris-buffered saline; T-TBS, TBS containing 0.02% Tween 20; X1–X5, β-1,3-xylan substrates containing 1–5 xylose units respectively
INTRODUCTION
β-1,3-Xylan, which is found mainly in the cell walls of red and green algae [1], is a homopolysaccharide composed of β-1,3-linked D-xylose. β-1,3-Xylanase (1,3-β-D-xylan xylanohydrolase; EC 3.2.1.32) is an enzyme that is capable of hydrolysing the internal β-1,3-xylosidic linkages of the polysaccharide to produce β-1,3-xylo-oligosaccharides with different xylose units. Algal polysaccharides and oligosaccharides have various biological activities, e.g. signalling and defence reactions [2], inhibition of bacterial adhesion [3,4], inhibition of complement activation [5], anticoagulant activity [6] and antitumour activity [7,8]. Therefore β-1,3-xylanase is an effective tool for the preparation of β-1,3-xylo-oligosaccharides from β-1,3-xylan in order to investigate their physiological activity, as well as for the structural analysis of algal cell walls. The enzyme can be also used, in combination with other glycosidases, for the preparation of protoplasts of algae [9], making possible the cell fusion and gene manipulation of algae.
Plant polysaccharides such as cellulose (β-1,4-glucan) and β-1,4-xylan are the most abundant organic macromolecules in the biosphere. The microbial degradation of these polysaccharides is very important for the recycling of photosynthetically fixed carbon, and thus enzymes responsible for their degradation have been well studied. Generally, polysaccharide-degrading enzymes such as cellulase (β-1,4-glucanase) and β-1,4-xylanase possess a modular structure, consisting of a catalytic module and one or several non-catalytic CBMs (carbohydrate-binding modules) [10,11]. On the basis of the similarity of their amino acid sequences, GHs (glycoside hydrolases) and CBMs are now classified into 99 and 42 families respectively (current information on GHs and CBMs is available at the CAZy website: http://afmb.cnrs-mrs.fr/CAZY/) [10,12].
Compared with those of cellulase and β-1,4-xylanase, the structure and functions of β-1,3-xylanase are not well understood, although several β-1,3-xylanase genes have been cloned [13,14]. We report here the cloning and characterization of a novel β-1,3-xylanase which possesses two putative CBMs at the C-terminus. The enzyme was expressed in a marine bacterium, Vibrio sp. strain AX-4, in the presence of β-1,3-xylan or cell walls containing β-1,3-xylan [15], suggesting that this bacterium serves to degrade β-1,3-xylan in the marine environment.
In the present study, we first show that two conserved glutamate residues in β-1,3-xylanase, Glu-138 and Glu-234, are essential for enzyme activity, and probably function as an acid/base and a nucleophile respectively. This paper also reports that the presence of two putative CBMs strongly enhanced the hydrolysis of insoluble β-1,3-xylan by the enzyme, although putative CBMs were not necessary for the hydrolysis of soluble glycol-β-1,3-xylan.
EXPERIMENTAL
Materials
Pyrobest DNA polymerase was purchased from Takara Shuzo (Shiga, Japan). Plasmids pET23a, pET23b and pBluescript II SK(+) were obtained from Novagen (Madison, WI, U.S.A.) and Stratagene (La Jolla, CA, U.S.A.) respectively. The restriction endonucleases and T4 DNA ligase were obtained from Wako Pure Chemical Industries (Osaka, Japan). Silica gel 60 TLC plates were purchased from Merck (Darmstadt, Germany). β-1,3-Xylan was prepared from the green alga Caulerpa racemosa var. laete-virens according to the method of Iriki et al. [1], and β-1,4-mannan was prepared from Codium fragile according to the method of Love and Percival [16]. Avicel, β-1,4-xylan and pNP (p-nitrophenyl)-glycosides were purchased from Sigma (St. Louis, MO, U.S.A.). β-1,3-Xylo-oligosaccharide was prepared by partial hydrolysis of β-1,3-xylan with trifluoroacetic acid and purified according to the method of Aoki et al. [17], except that gel filtration with Sephadex G-15 was used instead of charcoal chromatography. Glycol-β-1,3-xylan was prepared according to the method of Yamaura et al. [18]. All other reagents were of the highest purity available.
Bacterial strains
Vibrio sp. strain AX-4 was grown as described previously [15] and used as the source of genomic DNA. Escherichia coli strains JM109 and BL21(DE3)pLysS were purchased from Takara Shuzo.
Molecular cloning and DNA sequencing
General cloning techniques were essentially as described by Sambrook et al. [19]. Nucleotide sequences were determined by the dideoxynucleotide chain termination method using a BigDye Terminator Cycle Sequencing Ready Reaction Kit (PE Biosystems) and a DNA Sequencer (model 377A; PE Biosystems).
Sequence analysis
Nucleotide and amino acid sequences were determined using DNASIS software (Hitachi Software Engineering). The search of amino acid sequences was performed with BLAST [20] or PSI-BLAST [21]. The alignment of amino acid sequences was performed with CLUSTAL W [22].
PCR amplification of a partial fragment of the β-1,3-xylanase gene
For cloning of the β-1,3-xylanase gene of Vibrio sp. strain AX-4, PCR primers were designed based on the sequence of the β-1,3-xylanase gene of Vibrio sp. XY-214 (txyA) [13]. The sense primer AX4U1 (5′-TTCGATAGGCCTGTTTATTTACG-3′) and the antisense primer AX4L2 (5′-CTATCCCAGTCAGCATTAATGTA-3′) were used for PCR with the genomic DNA of AX-4 as a template. The PCR was conducted in a GeneAmp PCR System 9700 (PE Biosystems) for 40 cycles (each consisting of denaturation at 95 °C for 20 s, annealing at 55 °C for 30 s and extension at 72 °C for 30 s) using AmpliTaq Gold (PE Biosystems). Amplified PCR products were extracted from agarose gels with a Sephaglas BandPrep Kit (Amersham Pharmacia Biotech) and subcloned into the pGEM T-Easy vector (Promega). The resulting plasmid, designated pGEMU1L2, was then sequenced.
Isolation of genomic DNA clones encoding β-1,3-xylanase
Genomic DNA (10 μg) was digested with various restriction endonucleases, and the digests were fractionated by 0.7% (w/v) agarose gel electrophoresis using the standard method [19]. DNA was transferred from agarose gels on to nylon membranes (Hybond N+; Amersham Pharmacia Biotech) according to the protocol of the manufacturer. Probe I, which was the digest of pGEMU1L2 obtained with EcoRI, was labelled with [α-32P]dCTP using a Ready-To-Go DNA labelling kit (Amersham Pharmacia Biotech) and used for hybridization. Hybridization was performed in 0.5 M phosphate buffer, pH 7.0, containing 1 mM EDTA and 7% (w/v) SDS at 65 °C for 16 h. After hybridization, the membrane was washed three times with 40 mM phosphate buffer, pH 7.0, containing 1% SDS at 65 °C and exposed on an imaging plate, which was then examined after several hours using a BAS 1500 imaging analyser (Fuji Film). Judging from the Southern blots of the EcoRI digest using probe I, only the 4.3 kb fragment was found to contain the β-1,3-xylanase gene. For cloning of this gene, a digest with EcoRI was prepared using 10 μg of genomic DNA. Restriction fragments of the genomic DNA of AX-4 were fractionated by preparative 0.7% (w/v) agarose gel electrophoresis, and fragments of 4.3 kb were extracted from the gel. The EcoRI fragments were ligated to the EcoRI site of pBluescript II SK(+) (Stratagene). The recombinant plasmids thus obtained were used to transform E. coli strain JM109, which was employed for the preparation of a gene library enriched with the β-1,3-xylanase gene. Colony hybridization was performed by the standard procedure using probe I [19]. One clone was selected, and the plasmid in the clone was designated pSKAERI. Probe II, which was approx. 350 bp long, was prepared by digestion of pSKAERI with EcoRI and HincII. The probe, which contained the 5′-end of the β-1,3-xylanase gene in pSKAERI, was labelled with [α-32P]dCTP. HincII fragments (1.1 kb) from the genomic DNA of AX-4 were ligated to the HincII site of pBluescript II SK(+) and used to transform E. coli strain JM109, which was used for the preparation of a gene library enriched with the 5′-end of the β-1,3-xylanase gene. The library was screened by colony hybridization with probe II, and one clone was selected. The plasmid in the selected clone was designated pSKAHCII. pSKAERI and pSKAHCII were sequenced and overlapped, and then the nucleotide sequence of the β-1,3-xylanase gene of AX-4 (xyl4) was determined.
Construction of expression plasmid with the β-1,3-xylanase gene (xyl4)
The following primers were used for PCR: AETFLU (5′-GGGATCCATGAAACGAACCTATTTGT-3′) and AXG-L (5′-GTAAGCTTAATGATGATGATGATGATGTTGAGCGCAACTAGTATTTA-3′). AETFLU and AXG-L contain a BamHI site (underlined) and a HindIII site (double underlined) respectively. PCR was performed in a GeneAmp PCR System 9700 (PE Biosystems) for 30 cycles (each consisting of denaturation at 95 °C for 20 s, annealing at 52 °C for 1 min and extension at 72 °C for 1.5 min) using Pyrobest DNA polymerase and genomic DNA of AX-4 as a template. After gel purification, the amplified products were digested with BamHI and HindIII. The BamHI/HindIII fragments were cloned into BamHI/HindIII-digested pET23a. The recombinant plasmid was designated pETXYL4.
Site-directed mutagenesis of β-1,3-xylanase
Site-directed mutagenesis was performed by the PCR overlap extension method [23], with AETFLU, AXG-L and the following oligonucleotide primers: E138D-S, 5′-GGGCATATGACGTTGATGGAC-3′; E138D-AS, 5′-GTCCATCAACGTCATATGCCC-3′; E138Q-S, 5′-CTGGGCATATCAGGTTGATGG-3′; E138Q-AS, 5′-CCATCAACCTGATATGCCCAG-3′; E234D-S, 5′-TCTTAAATGACTCAACTCCTC-3′; E234D-AS, 5′-GAGGAGTTGAGTCATTTAAGA-3′; E234Q-S, 5′-ATTCTTAAATCAATCAACTCC-3′; E234Q-AS, 5′-GGAGTTGATTGATTTAAGAAT-3′; E268Q-S, 5′-GCTGTGGGATCAATGGTTTGC-3′; E268Q-AS, 5′-GCAAACCATTGATCCCACAGC-3′ (underlining shows the locations of the mutations). PCR products were extracted from a 1.0% (w/v) agarose gel and digested with BamHI and HindIII. The BamHI/HindIII fragments were cloned into BamHI/HindIII-digested pET23a. The plasmids of recombinant mutants in which Glu-138, Glu-234 and Glu-268 were mutated were designated pETE138D, pETE138Q, pETE234D, pETE234Q and pETE268Q. These mutant enzymes were expressed and purified by the methods described below.
Construction of deletion mutants of β-1,3-xylanase
For the construction of deletion mutants, AETFLU, AXG-L and the following oligonucleotide primers were used for PCR: AXD-U1 (5′-GGGATCCTTTAGGCTATGGCTCAACT-3′), AXD-L1 (5′-GTAAGCTTAATGATGATGATGATGATGAGTTGAGCCATAGCCTAACG-3′) and AXD-L2 (5′-GTAAGCTTAATGATGATGATGATGATGAGGTGTGCCAGTAGAGCCAC-3′). AXD-U1 contains a BamHI site (underlined), and AXD-L1 and AXD-L2 each contain a HindIII site (double underlined) following the histidine tag. PCR was performed in a GeneAmp PCR System 9700 (PE Biosystems) for 30 cycles (each consisting of denaturation at 95 °C for 20 s, annealing at 52 °C for 1 min and extension at 72 °C for 1.5 min) using Pyrobest DNA polymerase, pETXYL4 as a template, and the following sets of primers: AETFLU and AXD-L1 for the construction of pETCM, AETFLU and AXD-L2 for the construction of pETCM-BM-1, and AXD-U1 and AXG-L for the construction of pETBM-1-2. After gel purification, the products amplified using the primer sets AETFLU/AXD-L1 and AETFLU/AXD-L2 were digested with BamHI/HindIII and cloned into a pET23a vector digested with the same restriction enzymes. The amplified product obtained using AXD-U1/AXG-L was digested with BamHI/HindIII and cloned into pET23b digested with the same restriction enzymes. These mutants were expressed and their enzyme activities were measured by the method described below.
Expression and purification of rXYL4 (recombinant β-1,3-xylanase)
E. coli strain BL21(DE3)pLysS cells transformed with pETXYL4 were grown at 25 °C for 12 h in 10 ml of medium A (Luria–Bertani medium supplemented with 100 μg/ml ampicillin and 35 μg/ml chloramphenicol) with shaking. The culture was then transferred to 100 ml of medium A and incubated until the A600 reached approx. 0.5. Then IPTG (isopropyl β-D-thiogalactoside) was added to the culture at a final concentration of 0.1 mM to cause transcription, and cultivation was continued for an additional 6 h at 25 °C. After cultivation, cells were harvested by centrifugation (8000 g for 10 min), and suspended in extraction solution (deionized water containing 5 μg/ml leupeptin and pepstatin A). The cell suspension was sonicated and cell debris was removed by centrifugation (8000 g for 10 min). The supernatant obtained was loaded on to a HiTrap Q HP column (Amersham Pharmacia Biotech) for ion-exchange chromatography. The enzyme was absorbed on the column, which was washed with 50 mM sodium phosphate buffer, pH 7.5, containing 0.2 M NaCl. The enzyme was eluted with 50 mM sodium phosphate buffer, pH 7.5, containing 0.3 M NaCl, and then the fractions showing β-1,3-xylanase activity were pooled. The pooled enzyme solution was applied to a HiTrap Chelating HP column (Amersham Pharmacia Biotech) which was chelated with Ni2+, then the column was washed with 20 mM sodium phosphate buffer, pH 7.5, containing 0.15 M NaCl and 10 or 30 mM imidazole. The enzyme was eluted from the column with 20 mM sodium phosphate buffer, pH 7.5, containing 0.15 M NaCl and 50 mM imidazole. The purified enzyme was dialysed against deionized water and used for the subsequent experiments.
Enzyme assay
The activity of β-1,3-xylanase was measured as follows. A reaction mixture containing 0.5% β-1,3-xylan and an appropriate amount of enzyme in 300 μl of 50 mM sodium phosphate buffer, pH 7.5, was incubated at 37 °C for 10 min. After incubation, the reaction mixture was centrifuged and 250 μl of the supernatant was recovered. The reducing sugars liberated by hydrolysis of the substrate in the supernatant were determined by the Somogyi–Nelson method [24]. One unit of enzyme activity was defined as the amount of enzyme that liberates 1 μmol of xylose per min under the conditions described. To determine the activity of the deletion mutants, glycol-β-1,3-xylan was used as a substrate and the incubation period was changed to 30 min.
Characterization of the general properties of rXYL4
To investigate the optimal pH of rXYL4, activity was measured using β-1,3-xylan as substrate by the method described above, except that a 50 mM sodium acetate buffer (pH range 3–6), 50 mM sodium phosphate buffer (pH range 6–8) or 50 mM Tris/HCl buffer (pH range 7–9) was used. The optimal temperature of rXYL4 was measured in the range 0–100 °C at intervals of 10 °C. The effects of metal ions on enzyme activity were assayed by adding various metal ions (Na+, Ca2+, Mg2+, Mn2+, Ni2+, Pb2+, Cu2+, Zn2+ and Hg2+, or EDTA) to the reaction mixture at 1 mM. Exoglycosidase activities of rXYL4 were measured using various pNP-glycosides (pNP-α- and -β-glucosides, pNP-α- and -β-galactosides, pNP-α- and -β-mannosides, pNP-α- and -β-xylosides, and pNP-α- and -β-fucosides) as substrate at 1 mM. Substrate specificity for other polysaccharides was investigated using β-1,4-xylan, Avicel (microcrystalline β-1,4-glucan) and β-1,4-mannan as substrate at a final concentration of 0.5% (w/v), and analysing the digestion products by TLC as described below.
TLC analysis of the products of digestion by β-1,3-xylanase
The digestion products of various polysaccharides and β-1,3-xylo-oligosaccharides after β-1,3-xylanase treatment were analysed by the following methods.
Method I: polysaccharide was used as a substrate. The reaction mixture containing 0.5% β-1,3-xylan and 150 ng of enzyme in 30 μl of 50 mM sodium phosphate buffer, pH 7.5, was incubated at 37 °C for a given period. After incubation, 2 vol. of ethanol was added to the mixture. Then the mixture was centrifuged and the supernatant obtained was evaporated with a Speed Vac concentrator. The dried material was dissolved in water and spotted on a Silica Gel 60 TLC plate, which was then developed with n-butanol/acetic acid/water (10:5:1, by vol.). After development, the TLC plate was sprayed with diphenylamine/aniline/phosphate reagent [25] and heated at 100 °C for 10 min to visualize the digestion products.
Method II: β-1,3-xylo-oligosaccharide was used as a substrate. The reaction mixture containing 1 mM of each β-1,3-xylo-oligosaccharide and an appropriate amount of enzyme in 10 μl of 10 mM sodium phosphate buffer, pH 7.5, was incubated at 37 °C for a given period. The subsequent procedure was the same as described for Method I.
The digestion products on the TLC plate were then analysed and quantified with a Shimadzu CS-9300PC chromatoscanner (reflection 540 nm). The extent of hydrolysis was calculated as follows:
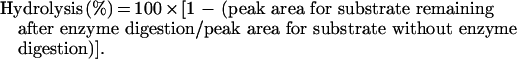 |
Protein assay
Protein content was determined by the bicinchoninic acid protein assay with BSA as the standard (Pierce).
SDS/PAGE and Western blotting
SDS/PAGE was carried out according to the method of Laemmli [26]. Proteins were separated by SDS/12.5%-PAGE and stained with Coomassie Brilliant Blue. A low-molecular-mass SDS/PAGE calibration kit (Amersham Pharmacia Biotech) was used as the standard. To perform Western blotting, proteins separated by SDS/12.5%-PAGE were transferred on to a nitrocellulose membrane using a semi-dry blotter (Bio-Rad, Hercules, CA, U.S.A.). Then the membrane was blocked with 3% (w/v) skimmed milk in TBS (Tris-buffered saline) for 1 h and washed three times with T-TBS (TBS containing 0.02% Tween 20). The membrane was incubated with anti-His (C-term) antibodies (Invitrogen Japan, Tokyo, Japan) for 2 h at room temperature and washed three times with T-TBS. Then the membrane was incubated with horse-radish peroxidase-conjugated anti-(mouse IgG) antibodies for 1.5 h at room temperature. After washing three times with T-TBS, the membrane was stained with a peroxidase stain kit (Nacalai Tesque, Kyoto, Japan) according to the manufacturer's instructions.
Measurement of CD spectra
CD spectra in the far-UV range (200–250 nm) were measured at room temperature on a JASCO J-720 spectropolarimeter (Japan Spectroscopic Co., Tokyo, Japan). The path length of the cells used was 1 mm, and samples were prepared at 1.5 mg/ml in 10 mM sodium acetate buffer, pH 6.0, by the measurement of absorbance at 280 nm using BSA as the standard.
RESULTS
Molecular cloning and sequencing of the β-1,3-xylanase gene of Vibrio sp. strain AX-4
A 460 bp PCR product was obtained from an AX-4 genomic DNA library with PCR primers which were designed using the sequence of the β-1,3-xylanase gene (txyA) from Vibrio sp. strain XY-214 [13]. The PCR product was labelled with 32P and used as probe I for Southern blotting. We found that a 4.3 kb band was hybridized with probe I when the AX-4 genomic DNA was digested with EcoRI. A clone, pSKAERI, containing a 4.3 kb insert was isolated from the gene library enriched with the AX-4 β-1,3-xylanase gene. The nucleotide sequence of pSKAERI contained the putative ORF (open reading frame), but lacked the N-terminal region compared with the ORF of txyA. We thus screened the genomic library to find the missing N-terminal sequence using probe II prepared from pSKAERI. Southern blotting with 32P-labelled probe II of the HincII digest gave a 1.1 kb band. A clone, pSKAHCII, containing a 1.1 kb insert was isolated from the library enriched with the 5′-end of the β-1,3-xylanase gene and sequenced. The deduced amino acid sequence was found to contain the N-terminal sequence. The 5′-end sequence of the β-1,3-xylanase gene in pSKAERI was also found in pSKAHCII, indicating that the sequences of the two clones overlapped.
DNA and deduced amino acid sequences of the AX-4 β-1,3-xylanase (XYL4)
We cloned and sequenced the two contiguous clones pSKAERI and pSKAHCII, and found an ORF of the β-1,3-xylanase gene (xyl4). The xyl4 gene consists of 1728 nucleotides, encoding a putative protein of 576 amino acid residues. The translated product of the xyl4 gene, designated XYL4, had a predicted molecular mass of 62587 Da and pI of 4.2. As shown in Figure 1(A), the putative catalytic module of XYL4 showed high identity with that of other β-1,3-xylanases, i.e. 71.3% identity with TxyA of Vibrio sp. strain XY-214 (GenBank accession no. AB029043), 70.5% identity with TxyA of Alcaligenes sp. strain XY-234 (AB039953), and 39.5% identity with xylC of Thermotoga neapolitana (U58632). A BLAST search showed that the sequence corresponding to residues 104–238 of XYL4 was similar to the conserved regions of other β-1,3-xylanases and β-mannanases (Figure 1B), both of which are classified into GH family 26 (GH26), suggesting that this region is a catalytic module of XYL4.
Figure 1. Sequence alignments of catalytic modules of β-1,3-xylanases.

(A) Catalytic modules of β-1,3-xylanases with different origins were aligned using CLUSTAL W [22]; (B) parts of catalytic modules of β-1,3-xylanases were aligned with Pseudomonas cellulosa mannanase 26A (PcMan26A) using PSI-BLAST [21] and CLUSTAL W. Identical and similar amino acid residues are shown by white letters on a black background and black letters on a grey background respectively. Four conserved Glu residues (Glu-138, Glu-218, Glu-234 and Glu-268 in XYL4) are indicated by arrowheads; putative catalytic residues (Glu-138 and Glu-234) are indicated by black arrowheads, and other conserved Glu residues (Glu-218 and Glu-268) by white arrowheads. The numbers at the start of the respective lines indicate the amino acid number from Met-1. Gaps inserted into the sequences are shown by dots. Abbreviations used are as follows: Vib.XYL4, XYL4 of Vibrio sp. strain AX-4 (present study); Vib.TxyA, TxyA of Vibrio sp. strain XY-214; Alc.TxyA, TxyA of Alcaligenes sp. strain XY-234; The.xylC, xylC (ManA) of Thermotoga neapolitana.
The C-terminal region (residues 349–576) of XYL4 contains a repeating sequence (residues 349–458 and 459–576). The sequence of the repeating unit is similar to that of CBM31, suggesting that this C-terminal region is the substrate-binding module of XYL4. All β-1,3-xylanases have a similar catalytic module in their N-terminal region, whereas they show clear differences in their C-terminal region. TxyA of Vibrio sp. XY-214 and TxyA of Alcaligenes sp. XY-234 have a single CBM31, whereas xylC of Thermotoga neapolitana does not have a CBM (Figure 2). In this context, XYL4 seems to be a novel β-1,3-xylanase, because it possesses two putative CBMs, which belong to family 31.
Figure 2. Modular architecture of β-1,3-xylanases.

The predicted modular architecture of β-1,3-xylanases is shown as a schematic representation. Open and closed boxes indicate catalytic modules and signal peptides respectively. Putative CBMs are shown by hatched boxes. Black bars indicate glycine-rich linkers that connect the modules. Abbreviations used are as follows: Vib.XYL4, XYL4 of Vibrio sp. strain AX-4 (present study); Vib.TxyA, TxyA of Vibrio sp. strain XY-214; Alc.TxyA, TxyA of Alcaligenes sp. strain XY-234; The.xylC, xylC (ManA) of Thermotoga neapolitana.
Expression and purification of rXYL4
The expression plasmid pETXYL4 was constructed by insertion of a fragment of the coding sequence of xyl4 into the BamHI and HindIII sites of plasmid pET23a. E. coli strain BL21(DE3)pLysS cells were transformed with pETXYL4 and cultured in a medium containing 0.1 mM IPTG. The lysate of the harvested cells was subjected to successive column chromatographies with HiTrap Q HP and HiTrap Chelating HP. The purified rXYL4 gave a single protein band corresponding to a molecular mass of 66 kDa on SDS/PAGE when stained with Coomassie Brilliant Blue (see Figure 5A, lane 1) and anti-His (C-term) antibody (see Figure 5B, lane 1), which was very similar to that deduced from the putative amino acid sequence. The specific activity of the purified rXYL4 was determined to be 4.7 units/mg when β-1,3-xylan was used as a substrate.
Figure 5. Site-directed mutagenesis of XYL4 for analysis of catalytic amino acid residues.

Wild-type and mutant β-1,3-xylanases were purified and then examined by SDS/PAGE. (A) Coomassie Brilliant Blue staining; (B) Western blotting using anti-His tag (C-term) antibody. Lane M, low-molecular-mass SDS/PAGE calibration kit; lane 1, wild-type (rXYL4); lane 2, E138D; lane 3, E138Q; lane 4, E234D; lane 5, E234Q; lane 6, E268Q. Wild-type and all mutant enzymes are indicated by arrowheads. (C) Time courses for hydrolysis of β-1,3-xylan by wild-type and mutant enzymes. The reaction mixtures containing 0.5% β-1,3-xylan and 1 μg of wild-type, 1 μg of E268Q mutant, or 100 μg of other mutant enzymes in 300 μl of 50 mM sodium phosphate buffer, pH 7.5, were incubated at 37 °C for the periods indicated. After incubation, the reaction mixtures were centrifuged and the reducing sugars generated in the supernatants were quantified by the Somogyi–Nelson method [24]. (D) CD spectra of the wild-type and mutant β-1,3-xylanases. Purified wild-type and mutant enzymes were dissolved in 10 mM sodium acetate buffer, pH 6.0, at a concentration of 1.5 mg/ml and their CD spectra were measured at room temperature on a JASCO J-720 spectropolarimeter.
General properties of rXYL4
The optimal pH was found to be 7.0–7.5. It is noted that rXYL4 hydrolysed β-1,3-xylan most efficiently at 37 °C, but did not hydrolyse the substrate above 60 °C at pH 7.5. The activity was completely inhibited by Hg2+, and 40–90% inhibition was observed with Mn2+, Cu2+ and Pb2+ at 1 mM. On the other hand, Ca2+, Mg2+ and EDTA had no significant effects on activity at the same concentration.
Specificity and kinetics of rXYL4
The patterns of hydrolysis of β-1,3-xylan during incubation with purified rXYL4 were analysed by TLC. As shown in Figure 3, disaccharide (X2), trisaccharide (X3) and tetrasaccharide (X4) were detected as major products throughout the course of the incubation, whereas the generation of monosaccharide (X1), pentasaccharide (X5) and oligosaccharides with more than five xylose units was delayed and the quantities were small under the conditions used.
Figure 3. Hydrolysis of β-1,3-xylan by rXYL4.

The pattern for the hydrolysis of β-1,3-xylan by rXYL4 was analysed by TLC. The reaction mixture containing 0.5% (w/v) β-1,3-xylan and 150 ng of enzyme in 30 μl of 50 mM sodium phosphate buffer, pH 7.5, was incubated at 37 °C for the periods indicated. After incubation, the reaction mixtures were centrifuged and the supernatants were analysed by TLC as described in the Experimental section.
Next, we investigated the kinetics and Km values of the enzyme using soluble β-1,3-xylo-oligosaccharides as substrates. It was revealed that rXYL4 hydrolysed β-1,3-xylo-oligosaccharides with different numbers of xylose units, i.e. X3 was converted into X1 and X2 (Figure 4B), X4 was mainly converted into X1 and X3 (Figure 4C), and X5 was converted into X2 and X3 (Figure 4D). A small amount of X2 (Figure 4C) was also detected on hydrolysis of X4 by the enzyme. However, it is noteworthy that the enzyme did not hydrolyse X2 (Figure 4A). The time course of the hydrolysis of X2, X3, X4 and X5 by increasing concentrations of rXYL4 were also examined. At low concentrations of the enzyme (0.5 μg/ml and 5 μg/ml in Figures 4E and 4F respectively), rXYL4 hydrolysed X5 much faster than X4, whereas X3 seemed to be completely resistant to the enzyme. At the highest concentration of the enzyme (2.5 mg/ml), X3 was hydrolysed slowly, but even at this concentration X2 was not hydrolysed at all (Figure 4G). Kinetic parameters of rXYL4 for X4 and X5 were calculated according to Hanes–Woolf plots (Table 1). Interestingly, the Km value for X4 was almost equivalent to that for X5 (7.4×10−3 M and 7.5×10−3 M respectively), but the kcat value for X5 (1.4×104 min−1) was 40-fold higher than that for X4 (3.5×102 min−1).
Figure 4. Hydrolysis of β-1,3-xylo-oligosaccharides by rXYL4.

The patterns for the hydrolysis of β-1,3-xylo-oligosaccharides by rXYL4 were analysed by TLC. The reaction mixture containing 1 mM substrate and an appropriate amount of enzyme in 10 μl of 10 mM sodium phosphate buffer, pH 7.5, was incubated at 37 °C for the periods indicated. (A) Xylobiose (X2); (B) Xylotriose (X3) with 2.5 mg/ml enzyme; (C) xylotetraose (X4) with 5 μg/ml enzyme; (D) xylopentaose (X5) with 500 ng/ml enzyme. The time courses for the hydrolysis of β-1,3-xylo-oligosaccharide with various concentrations of rXYL4 were also examined. The reaction mixture containing 1 mM substrate (●, X2; ◆, X3; ■, X4; ▲, X5) and an appropriate amount of enzyme in 10 μl of 10 mM sodium phosphate buffer, pH 7.5, was incubated at 37 °C for the periods indicated; enzyme concentrations used were (E) 500 ng/ml, (F) 5 μg/ml and (G) 2.5 mg/ml.
Table 1. Kinetic parameters of rXYL4 for the hydrolysis of β-1,3-xylotetraose and β-1,3-xylopentaose.
The reaction mixture containing various concentrations of substrate (0.25, 0.5, 1, 2, 3, 4 and 5 mM) and 50 ng of rXYL4 in 10 μl of 10 mM sodium phosphate buffer, pH 7.5, was incubated at 37 °C for 1 h (for X4) or 5 min (for X5). After incubation, the reaction mixtures were centrifuged and the supernatants were analysed by TLC as described in the Experimental section. The kinetic parameters of rXYL4 were calculated using Hanes–Woolf plots.
| Substrate | Km (M) | kcat (min−1) | kcat/Km (min−1·M−1) |
|---|---|---|---|
| Xylotetraose (X4) | 7.4×10−3 | 3.5×102 | 4.7×104 |
| Xylopentaose (X5) | 7.5×10−3 | 1.4×104 | 1.9×106 |
The substrate specificity of rXYL4 was examined further using other polysaccharides and pNP-glycosides. It was found that the enzyme did not hydrolyse β-1,4-xylan, Avicel (microcrystalline β-1,4-glucan), β-1,4-mannan or various pNP-glycosides (pNP-α- and -β-glucosides, pNP-α- and -β-galactosides, pNP-α- and -β-mannosides, pNP-α- and -β-xylosides, and pNP-α- and -β-fucosides) under the conditions described in the Experimental section. Conclusively, therefore, XYL4 is an endo-type β-1,3-xylanase (EC 3.2.1.32).
Determination of catalytic amino acid residues of XYL4
The deduced amino acid sequence of XYL4 was analysed using PSI-BLAST [21] to search for similar sequences in other glycohydrases. Part of the putative catalytic module of XYL4 (residues 101–234) showed weak but significant similarity (identity 19.0%) with mannanase 26A (residues 181–320) of Pseudomonas cellulosa (PcMan26A; GenBank accession no. X82179), which is classified into GH26 (Figure 1B), although XYL4 specifically hydrolysed β-1,3-xylan but not β-1,4-mannan. Site-directed mutagenesis and a crystal analysis revealed that Glu-212 and Glu-320 of PcMan26A function as an acid/base catalyst and a nucleophile respectively [27,28]. The PSI-BLAST search revealed that Glu-138 and Glu-234 in the putative catalytic module of XYL4 are aligned with these two catalytic glutamates. Moreover, these two Glu residues were found to be conserved in all β-1,3-xylanases cloned (black arrowheads in Figures 1A and 1B). To address whether these Glu residues function as catalytic residues of β-1,3-xylanases, Glu-138 and Glu-234 of XYL4 were replaced with Asp (E138D, E234D) or Gln (E138Q, E234Q) by site-directed mutagenesis. As a control, another Glu residue (Glu-268), which is conserved but is not predicted to be a catalytic residue, was also replaced by a Gln residue (E268Q). These five mutant and wild-type enzymes were expressed separately in E. coli strain BL21(DE3)pLysS, and then purified (Figures 5A and 5B). The time courses for the hydrolysis of β-1,3-xylan by the enzymes indicated that E138Q and E234Q, in which the acidic Glu residue was replaced by a neutral Gln residue, completely lost their enzyme activities. On the other hand, E138D and E234D, in which the acidic Glu residue was replaced by a similar acidic Asp residue, showed extremely low but reliable activity when β-1,3-xylan was incubated with a 100-fold quantity of enzyme (Figure 5C). The activity of E268Q was decreased by 30% in comparison with that of the wild-type enzyme. It was confirmed that the CD spectrum of each mutant was virtually identical with that of the wild-type enzyme (Figure 5D), indicating that the point mutation did not affect the secondary structure of the mutant enzymes. These results clearly indicate that Glu-138 and Glu-234 of XYL4 are crucial for the hydrolysis of β-1,3-xylan, and strongly suggest that these glutamates function as catalytic residues in β-1,3-xylanases, as they do in PcMan26A.
Function of putative CBMs of XYL4
XYL4 seems to be a modular enzyme composed of a putative catalytic module in the N-terminal region and putative CBMs belonging to family 31 at the C-terminus, the latter of which are composed of a repeating unit, tentatively designated CBM31-1 and CBM31-2, showing 55.0% identity. To investigate the function of the repeating putative CBM unit at the C-terminus, we constructed three deletion mutants: CM, which has a catalytic module but lacks the repeating putative CBM unit; CM-BM-1, which has a catalytic module and CBM31-1, but lacks CBM31-2; and BM-1-2, which has two putative CBMs but lacks the catalytic module of XYL4 (Figure 6A). These three mutants and the wild-type enzyme were expressed separately in E. coli strain BL21(DE3)pLysS and their activities were assayed using insoluble β-1,3-xylan (Figure 6B). In comparison with the wild type, CM-BM-1 showed relatively weak activity towards β-1,3-xylan, possibly because it lacks CBM31-2. The activity of CM, which lacks two putative CBMs, was much lower than that of CM-BM-1. As expected, BM-1-2, which lacks a catalytic module, did not show any enzyme activity, although the expression was compatible with that of other mutant and wild-type enzymes at the protein level (inset of Figure 6B). Next, the function of putative CBMs in the hydrolysis of soluble substrates by the enzyme was investigated using glycol-β-1,3-xylan (Figure 6C). Interestingly, no effects were observed on the hydrolysis of soluble glycol-β-1,3-xylan by the enzyme even upon removal of the two putative CBMs, indicating that the putative CBM is not necessary for the hydrolysis of soluble substrates by the enzyme (Figure 6C). However, the extent of hydrolysis of β-1,3-xylan by the enzyme was strongly affected by the presence and number of putative CBMs (Figures 6B and 6C). In summary, putative CBMs in the C-terminal region of XYL4 are shown to be very important for the hydrolysis of insoluble β-1,3-xylan, but not of soluble glycol-β-1,3-xylan.
Figure 6. Construction of rXYL4 deletion mutants and examination of their activities with insoluble and soluble substrates.

(A) Modular architectures of rXYL4 deletion mutants. Open and closed boxes indicate catalytic modules and signal peptides respectively. Putative CBMs are shown by hatched boxes. Putative CBMs at the N- and C-termini are designated CBM31-1 and CBM31-2 respectively. Black bars indicate linkers that connect modules. CM has a catalytic module, but lacks putative CBMs. CM-BM-1 and BM-1-2 lack CBM31-2 and the catalytic module respectively. (B) Time courses for hydrolysis of β-1,3-xylan with wild-type and mutant enzymes. The reaction mixtures containing 0.5% β-1,3-xylan and 0.01 unit of wild-type or mutant enzyme in 300 μl of 50 mM sodium phosphate buffer, pH 7.5, were incubated at 37 °C for the periods indicated. After incubation, the reaction mixtures were centrifuged, and the reducing sugars generated in the supernatants were quantified by the Somogyi–Nelson method [24]. Expression of wild-type and mutant enzymes was analysed by Western blotting (inset). Lane 1, wild type; lane 2, CM; lane 3, CM-BM-1; lane 4, BM-1-2. (C) Effect of CBMs on hydrolysis of soluble substrate. The reaction mixtures containing 0.5% β-1,3-xylan (□) or glycol-β-1,3-xylan (■) and 0.01 unit of wild-type or mutant enzyme in 300 μl of 50 mM sodium phosphate buffer containing 50 mM NaCl, pH 7.5, were incubated at 37 °C for 30 min.
DISCUSSION
In general, GHs have a modular structure, consisting of a catalytic module linked to one or more non-catalytic CBMs. Judging from its putative amino acid sequence, XYL4 seems to consist of three modules: an N-terminal catalytic module and two C-terminal CBMs. The putative catalytic module of XYL4 shows high identity with other β-1,3-xylanases and should be classified into GH26. The two putative CBMs of XYL4 show significant similarity with the members of CBM31. In contrast with other β-1,3-xylanases reported so far, XYL4 contains two putative CBMs linked in tandem, and thus is the first enzyme possessing a repeating unit of a putative CBM belonging to family 31. The presence of the putative CBMs leads to enhanced hydrolysis of β-1,3-xylan, possibly due to an increase in the binding of the enzyme to the insoluble substrate, although it did not affect the activity towards the soluble substrate (Figure 6). It should be emphasized that repeating a putative CBM strongly enhanced the hydrolysis of β-1,3-xylan by the enzyme.
The present study revealed that XYL4 hydrolysed X5 much faster than X4 and X3, and did not hydrolyse X2. This suggests that a certain degree of polymerization of xylose by β-1,3 linkage is required for an efficient reaction. Xylose was also detected after prolonged incubation, but is unlikely to be produced directly from xylan. Alternatively, it seems to be generated from xylo-oligosaccharides such as X3 and X4.
Family 26 GHs belong to the superfamily clan GH-A, in which a (β/α)8-barrel architecture with eight repeating β-strands and α-helices is conserved. Two catalytic Glu residues are typically conserved at the end of β-strand 4 and β-strand 7 of GH-A enzymes. A PSI-BLAST search indicated that two possible catalytic Glu residues are conserved in the catalytic module of XYL4 (Glu-138 and Glu-234), although XYL4 showed only 19.0% identity with the β-mannanase PcMan26A, whose crystal structure and site-directed mutagenesis were used for the identification of catalytic Glu residues [27,28]. In the present study, site-directed mutagenesis was employed to investigate the role of these Glu residues of XYL4. The replacement of Glu-138 and Glu-234 with Gln resulted in a complete loss of enzyme activity, and replacement with Asp resulted in a marked loss of activity. On the other hand, CD spectra of these mutants were not changed by the site-directed mutagenesis, indicating that the complete loss or significant decrease of enzyme activity was not caused by disruption of the secondary structure. Therefore we conclude that the conserved Glu residues at positions 138 and 234 are the catalytic residues of XYL4, and probably function as an acid/base catalyst and a nucleophile respectively. The reason why replacement of the non-catalytic Glu-268, which seems to be located at α-helix 7, with Gln reduced the activity by 30% is unknown at present. Some pivotal amino acid residues in the active-site region of PcMan26A are also found in XYL4, i.e. Arg-208 (Arg-134 in XYL4) and Trp-217 (Trp-143 in XYL4) are aligned, and His-211 and Tyr-285 in PcMan26A seem to be replaced by Tyr-137 and Phe-206 respectively in XYL4. The residue that coincides with Trp-360 in PcMan26A is not found in XYL4. These results suggest that the structure of the active site of PcMan26A is similar but not identical to that of XYL4, resulting in the different substrate specificity of the enzymes, i.e. the former is active towards the internal β-1,4-mannosidic linkage and the latter towards the internal β-1,3-xylosidic linkage. Recently, crystallization of the catalytic module of XYL4 (CM in Figure 6A) was accomplished and its X-ray crystal structure is under investigation [29]. This will provide further insights into the structure–function relationship of the enzyme.
The present study showed that the catalytic module of XYL4 lacking putative CBMs hydrolysed a soluble glycol-β-1,3-xylan as efficiently as did the wild-type enzyme, whereas the putative CBM-deleted mutant barely hydrolysed an insoluble β-1,3-xylan. Furthermore, the mutant lacking one putative CBM (CM-BM-1 in Figure 6A) hydrolysed β-1,3-xylan much more slowly than did the wild-type enzyme. These results clearly indicate that the putative CBMs at the C-terminus of XYL4 play an important role in the hydrolysis of insoluble substrates, but not soluble substrates. A preliminary study indicated that the putative CBMs of XYL4 (BM-1-2 in Figure 6A) specifically bind to β-1,3-xylan, but not β-1,4-xylan, β-1,4-mannan or Avicel (results not shown), suggesting that the specificity of the putative CBMs also contributes to the efficiency of hydrolysis of insoluble β-1,3-xylan by XYL4.
Acknowledgments
We thank Dr M. Kimura (Kyushu University, Fukuoka, Japan) for measurement of CD spectra. This work was supported in part by Grants-in-Aid for Scientific Research B (15380073) from the Ministry of Education, Science and Culture of Japan.
References
- 1.Iriki Y., Suzuki T., Nisizawa K., Miwa T. Xylan of siphonaceous green algae. Nature (London) 1960;187:82–83. doi: 10.1038/187082a0. [DOI] [PubMed] [Google Scholar]
- 2.Potin P., Bouarab K., Kupper F., Kloareg B. Oligosaccharide recognition signals and defence reactions in marine plant-microbe interactions. Curr. Opin. Microbiol. 1999;2:276–283. doi: 10.1016/S1369-5274(99)80048-4. [DOI] [PubMed] [Google Scholar]
- 3.Saeki Y., Kato T., Naito Y., Takazoe I., Okuda K. Inhibitory effects of funoran on the adherence and colonization of mutans streptococci. Caries Res. 1996;30:119–125. doi: 10.1159/000262147. [DOI] [PubMed] [Google Scholar]
- 4.Guzman-Murillo M. A., Ascencio F. Anti-adhesive activity of sulphated exopolysaccharides of microalgae on attachment of red sore disease-associated bacteria and Helicobacter pylori to tissue culture cells. Lett. Appl. Microbiol. 2000;30:473–478. doi: 10.1046/j.1472-765x.2000.00751.x. [DOI] [PubMed] [Google Scholar]
- 5.Blondin C., Fischer E., Boisson-Vidal C., Kazatchkine M. D., Jozefonvicz J. Inhibition of complement activation by natural sulfated polysaccharides (fucans) from brown seaweed. Mol. Immunol. 1994;31:247–253. doi: 10.1016/0161-5890(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 6.Nishino T., Kiyohara H., Yamada H., Nagumo T. An anticoagulant fucoidan from the brown seaweed Ecklonia kurome. Phytochemistry. 1991;30:535–539. doi: 10.1016/0031-9422(91)83722-w. [DOI] [PubMed] [Google Scholar]
- 7.Itoh H., Noda H., Amano H., Zhuaug C., Mizuno T., Ito H. Antitumor activity and immunological properties of marine algal polysaccharides, especially fucoidan, prepared from Sargassum thunbergii of Phaeophyceae. Anticancer Res. 1993;13:2045–2052. [PubMed] [Google Scholar]
- 8.Ren D. L., Wang J. Z., Noda H., Amano H., Ogata S. The effects of an algal polysaccharide from Gloiopeltis tenax on transplantable tumors and immune activities in mice. Planta Medica. 1995;61:120–125. doi: 10.1055/s-2006-958029. [DOI] [PubMed] [Google Scholar]
- 9.Araki T., Hayakawa M., Tamaru Y., Yoshimatu K., Morishita T. Isolation and regeneration of haploid protoplasts from Bangia atropurpurea (Rhodophyta) with marine bacterial enzymes. J. Phycol. 1994;30:1040–1046. [Google Scholar]
- 10.Coutinho P. M., Henrissat B. Carbohydrate-active enzymes: an integrated database approach. In: Gilbert H. J., Davies G., Henrissat B., Svensson B., editors. Recent Advances in Carbohydrate Bioengineering. Cambridge: The Royal Society of Chemistry; 1999. pp. 3–12. [Google Scholar]
- 11.Tomme P., Warren R. A., Gilkes N. R. Cellulose hydrolysis by bacteria and fungi. Adv. Microbiol. Physiol. 1995;37:1–81. doi: 10.1016/s0065-2911(08)60143-5. [DOI] [PubMed] [Google Scholar]
- 12.Coutinho P. M., Henrissat B. The modular structure of cellulases and other carbohydrate-active enzymes: an integrated database approach. In: Ohmiya K., Hayashi K., Sakka K., Kobayashi Y., Karita S., Kimura T., editors. Genetics, Biochemistry and Ecology of Cellulose Degradation. Tokyo: Uni Publishers Co.; 1999. pp. 15–23. [Google Scholar]
- 13.Araki T., Hashikawa S., Morishita T. Cloning, sequencing, and expression in Escherichia coli of the new gene encoding β-1,3-xylanase from a marine bacterium, Vibrio sp. strain XY-214. Appl. Environ. Microbiol. 2000;66:1741–1743. doi: 10.1128/aem.66.4.1741-1743.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okazaki F., Tamaru Y., Hashikawa S., Li Y. T., Araki T. Novel carbohydrate-binding module of β-1,3-xylanase from a marine bacterium, Alcaligenes sp. strain XY-234. J. Bacteriol. 2002;184:2399–2403. doi: 10.1128/JB.184.9.2399-2403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araki T., Aoki T., Kitamikado M. Isolation and identification of a β-1,3-xylanase-producing bacterium. Nippon Suisan Gakkaishi. 1987;53:2077–2081. [Google Scholar]
- 16.Love J., Percival E. The polysaccharides of green seaweed Codium fragile. Part III. A β-1,4-linked mannan. J. Chem. Soc. 1964;1964:3345–3350. [Google Scholar]
- 17.Aoki T., Araki T., Kitamikado M. Purification and characterization of endo-β-1,3-xylanase from Vibrio sp. Nippon Suisan Gakkaishi. 1988;54:277–281. [Google Scholar]
- 18.Yamaura I., Matsumoto T., Funatsu M., Mukai E. Purification and some properties of endo-1,3-β-D-xylanase from Pseudomonas sp. PT-5. Agric. Biol. Chem. 1990;54:921–926. [PubMed] [Google Scholar]
- 19.Sambrook H., Fritsch E. F., Maniatis T. 2nd. edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 20.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 21.Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomopson J. D., Higgins D. G., Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho S. N., Hunt H. D., Horton R. N., Pullen J. K., Pease L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 24.Somogyi M. Notes on sugar determination. J. Biol. Chem. 1952;195:19–23. [PubMed] [Google Scholar]
- 25.Bailey R. W., Bourne E. J. Colour reactions given by sugars and diphenylamine-aniline spray reagents on paper chromatograms. J. Chromatogr. 1960;4:206–213. [Google Scholar]
- 26.Laemmli U. K. Cleavage of structural proteins during the asssembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 27.Bolam D. N., Hughes N., Virden R., Lakey J. H., Hazlewood G. P., Henrissat B., Braithwaite K. L., Gilbert H. J. Mannanase A from Pseudomonas fluorescens ssp. cellulosa is a retaining glycosyl hydrolase in which E212 and E320 are the putative catalytic residues. Biochemistry. 1996;35:16195–16204. doi: 10.1021/bi961866d. [DOI] [PubMed] [Google Scholar]
- 28.Hogg D., Woo E. J., Bolam D. N., McKie V. A., Gilbert H. J., Pickersgill R. W. Crystal structure of mannanase 26A from Pseudomonas cellulosa and analysis of residues involved in substrate binding. J. Biol. Chem. 2001;276:31186–31192. doi: 10.1074/jbc.M010290200. [DOI] [PubMed] [Google Scholar]
- 29.Sakaguchi K., Kiyohara M., Watanabe N., Yamaguchi K., Ito M., Kawamura T., Tanaka I. Preparation and preliminary X-ray analysis of the catalytic module of β-1,3-xylanase from a marine bacterium Vibrio sp. AX-4. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1470–1472. doi: 10.1107/S0907444904013411. [DOI] [PubMed] [Google Scholar]