

Abstract
The catalytic asymmetric introduction of alkynyl functionality to α-amino acid derivatives was realized by the direct addition of terminal alkynes to α-imino ester in the presence of chiral Cu(I) complex under mild reaction conditions. Owing to the rich chemistry to which alkyne can be subjected, the present system provides a remarkably versatile tool for the construction of optically active α-amino acid derivatives. Good yields and enantiomeric excess values were achieved with an array of terminal alkynes and challenging, biologically active, unnatural α-amino acid derivatives could be conveniently obtained.
Keywords: asymmetric catalysis
Optically active nonproteinogenic α-amino acids are of exceptional and rapidly increasing popularity as important tools in protein engineering and peptide-based drug discovery due mainly to their implementation into nonscissile peptide mimics and peptide isosteres (1–7). Hence, intense research has been focused on the preparation of enantiomerically enriched unnatural α-amino acids, and, so far, several approaches, such as bioresolution routes (8–10) and the rhodium- or ruthenium-catalyzed asymmetric hydrogenation of dehydroamino acids derivatives (11–13), have shown much promise. Nevertheless, there is still a great demand for more efficient and, especially, technically feasible methods for convenient construction of various types of rational designed unnatural amino acid derivatives. In this regard, enantioselective nucleophilic addition to α-imino esters represents one of the most direct strategies (14), because a new chiral center and a new C C bond can be built in a single operation and an appropriately designed side chain can be introduced in the meantime. Previous work in this field mainly focused on the catalytic asymmetric alkylation of α-imino esters, in which enol silane (15–18), allyl-metal compounds (19, 20), trimethylsilyl nitronate (21), ketone (22), and nitroalkane
C bond can be built in a single operation and an appropriately designed side chain can be introduced in the meantime. Previous work in this field mainly focused on the catalytic asymmetric alkylation of α-imino esters, in which enol silane (15–18), allyl-metal compounds (19, 20), trimethylsilyl nitronate (21), ketone (22), and nitroalkane
Recently, we reported the successful alkynylation of α-imino ester by demonstrating the feasibility of direct addition of terminal alkynes to α-imino ester 1 in the presence of Ag(I) salts under mild reaction conditions (Scheme 1) (24). In this process, a new bond between an sp3 carbon and an sp carbon is formed. (23) have been used as nucleophiles.
Scheme 1.

Ag(I)-catalyzed alkynylation of α-imino ester.
Capitalizing on this method, we envisioned an attractive route to optically active α-amino acid derivatives via the direct asymmetric alkynylation of α-imino ester. The following considerations are particularly noteworthy:
The HC
 C moiety is an excellent two-carbon medium for the delivery of biologically interesting units into the γ-position of α-amino acids, because it could be easily hydrogenated to saturated status after completing the nucleophile role. In contrast, owing to the rich chemistry to which alkyne can be subjected, the presence of a CC bond in the desired targets offers a unique and highly valuable opportunity for further synthetic elaboration on the β,γ-positions of α-amino acids. For example, semireduction of the products could directly afford vinyl glycine derivatives, which are of increasing potential in therapeutic utility although their catalytic synthesis remains a challenge (25–28).
C moiety is an excellent two-carbon medium for the delivery of biologically interesting units into the γ-position of α-amino acids, because it could be easily hydrogenated to saturated status after completing the nucleophile role. In contrast, owing to the rich chemistry to which alkyne can be subjected, the presence of a CC bond in the desired targets offers a unique and highly valuable opportunity for further synthetic elaboration on the β,γ-positions of α-amino acids. For example, semireduction of the products could directly afford vinyl glycine derivatives, which are of increasing potential in therapeutic utility although their catalytic synthesis remains a challenge (25–28).This reaction is really atom economical as the nucleophile is generated directly. In previous study, the similar alkynylglycine skeleton was formed via coupling of α-haloglycinates with excessive reactive metal alkynilides (29, 30), which were prepared by deprotonation of terminal alkynes with stoichiometric BuLi or EtMgBr in a separate procedure.
A number of terminal alkynes are commercially available, and furthermore, there have been several routine procedures providing general and convenient accesses to a wide range of terminal alkynes with comprehensive substituents.
In this paper, we report the first catalytic asymmetric alkynylation of α-imino ester. Good to high yields and enantiomeric excess (ee) values were obtained when employing CuPF6·4MeCN/bis(oxazolidine)-pyridine (pybox) 9 (31) and CuOTf·0.5C6H6/pybox 9 as catalysts. As a simple illustration of the application of this methodology, one of the alkynylation products was conveniently transformed to synthetically challenging, biologically active, unnatural α-amino acid derivatives.
Experimental Procedures
General Information. 1H NMR and 13C NMR spectra were recorded in CDCl3 on a Varian AS 500 (500 and 125 MHz, respectively) NMR spectrometer at room temperature. Chemical shifts (δ) are expressed in ppm, and J values are given in Hz. High-resolution mass spectrometry (HRMS) was carried out by using the electrospray ionization (ESI) method on a Fisons VG platform or a MAT-95 spectrometer (Finnigan–MAT, San Jose, CA). HPLC analyses were performed by using a Waters 600 analytical liquid chromatography system with a Waters 486 UV detector. Optical rotations were measured on a PerkinElmer 341 polarimeter in a 10-cm cell. All reactions were conducted under a nitrogen atmosphere. All chemicals and solvents were used as received without further purification unless otherwise stated. CH2Cl2 was distilled from CaH2. Compound 1 (14) and pybox 9 (31) were synthesized according to established methods. Flash column chromatography was performed on silica gel (230–400 mesh).
General Procedure for Catalytic Asymmetric Alkynylation of α-Imino Ester. Pybox 9 (9.7 mg, 0.025 mmol) and CuOTf·0.5C6H6 (6.3 mg, 0.025 mmol) were added to a dried 10-ml round-bottom flask containing a magnetic stirring bar. CH2Cl2 (1.0 ml) was added, and the mixture was stirred at room temperature for 1 h. The solution was cooled to –10°C, and then α-imino ester 1 (52.3 mg, 0.25 mmol) in CH2Cl2 (400 μl), alkyne (0.5 mmol), and p-methoxyphenyl (PMP)-NH2 (3.2 mg, 0.025 mmol) in CH2Cl2 (100 μl) were sequentially added under vigorous stirring. The resulting solution was stirred at –10°C, and the reaction was monitored by TLC. Upon completion of the reaction (36–48 h), the mixture was then passed through a short plug of silica gel that was subsequently washed with EtOAc (10 ml). The combined solution was poured into a separatory funnel and diluted with EtOAc (25 ml) and H2O (5 ml). After mixing, the aqueous layer was discarded and the organic layer was washed with saturated aqueous brine (5 ml), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The purification of the residue by flash silica gel column chromatography (9:1 hexane/EtOAc as eluents) yielded the desired alkynylation product as a light yellow oil.
Ethyl-2-(p-methoyphenylamino)-6-phenyl-3-hexynoate. Compound 3a was obtained in 90% yield according to the general procedure. The ee value (85%) was determined by chiral HPLC analysis with a 25-cm × 4.6-mm Diacel Chemical Industries (Tokyo) CHIRALPAK AD column (eluent, 90:10 hexane:i-PrOH; flow rate, 1.0 ml/min; detection, 254-nm light): tR (major) = 11.48 min, tR (minus) = 16.75 min. [α]D20 –64.7° (c 0.5, CHC). 1H NMR (500 MHz, CDCl3): δ 7.28–7.25 (m, 2H), 7.20–7.16 (m, 3H), 6.80–6.78 (m, 2H), 6.67–6.65 (m, 2H), 4.69 (t, 1H, J = 2.3 Hz), 4.27–4.24 (q, 2H, J = 7.5 Hz), 3.76 (s, 3H), 2.80–2.77 (t, 2H, J = 7.3 Hz), 2.49–2.46 (dt, 2H, J = 7.3, 2.0 Hz), 1.31–1.28 (t, 3H, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3): δ 169.5, 153.7, 140.6, 137.6, 128.7, 128.6, 126.6, 116.5, 114.9, 84.8, 75.4, 62.5, 55.9, 50.5, 34.9, 21.1, 14.3. HRMS (ESI): calculated for C21H24NO3 [M + 1]+, 338.1756; found, 338.1782.
Ethyl-2-(p-methoyphenylamino)-5-phenyl-3-pentynoate. Compound 3b was obtained in 92% yield according to the general procedure. The ee value (83%) was determined by chiral HPLC analysis with a 25-cm × 4.6-mm Daicel Chemical Industries CHIRALPAK AD column (eluent, 90:10 hexane:i-PrOH; flow rate, 1.0 ml/min; detection, 254-nm light): tR (minor) = 21.49 min, tR (major) = 35.00 min. [α]D20 –38.1° (c 0.4, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.31–7.24 (m, 5H), 6.83–6.81 (m, 2H), 6.74–6.72 (m, 2H), 4.83 (t, 1H, J = 2.2 Hz), 4.32–4.28 (q, 2H, J = 7.2 Hz), 3.76 (s, 3H), 3.62 (d, 1H, J = 2.0Hz), 1.27 (t, 3H, J = 7.3 Hz). 13C NMR (125 MHz, CDCl3): δ 169.5, 153.6, 139.7, 136.3, 128.7, 128.1, 126.9, 116.4, 115.0, 82.9, 72.8, 62.5, 55.8, 50.5, 25.2, 14.3. HRMS (ESI): calculated for C20H22NO3 [M + 1]+, 324.1600; found, 324.1596.
Ethyl-2-(p-methoyphenylamino)-3-decynoate. Compound 3c was obtained in 89% yield according to the general procedure. The ee value (91%) was determined by chiral HPLC analysis with a 25-cm × 4.6-mm Daicel Chemical Industries CHIRALPAK AD column (eluent, 90:10 hexane:i-PrOH; flow rate, 1.0 ml/min; detection, 254-nm light): tR (major) = 9.96 min, tR (minus) = 12.63 min. [α]D20 –62.3° (c 0.4, CHCl3). 1H NMR (500 MHz, CDCl3): δ 6.79–6.77 (m, 2H), 6.70–6.68 (m, 2H), 4.70 (t, 1H, J = 2.3 Hz), 4.29–4.24 (q, 2H, J = 7.3 Hz), 3.75 (s, 3H), 2.19–2.15 (dt, 2H, J = 7.0, 2.3 Hz), 1.47–1.44 (m, 2H), 1.34–1.20 (m, 9H), 0.89–0.86 (t, 3H, J = 7.0 Hz). 13C NMR (125 MHz, CDCl3): δ = 169.6, 153.7, 139.3, 116.6, 114.9, 86.0, 75.0, 62.4, 55.8, 50.6, 31.5, 28.6, 28.5, 22.8, 18.9, 14.3, 14.2. HRMS (ESI): calculated for C19H28NO3 [M + 1]+, 318.2069; found, 318.2091.
Ethyl-2-(p-methoyphenylamino)-4-cyclopropyl-3-butynoate. Compound 3d was obtained in 92% yield according to the general procedure. The ee value (79%) was determined by chiral HPLC analysis with a 25-cm × 4.6-mm Daicel Chemical Industries CHIRALPAK AD column (eluent, 90:10 hexane:i-PrOH; flow rate, 1.0 ml/min; detection, 254-nm light): tR (major) = 11.08 min, tR (minus) = 19.46 min. [α]D20 –47.4° (c 0.7, CHCl3). 1H NMR (500 MHz, CDCl3): δ 6.78–6.74 (m, 2H), 6.66–6.62 (m, 2H), 4.64 (d, 1H, J = 2.3 Hz), 4.26–4.21 (q, 2H, J = 7.0), 3.17 (s, 3H), 1.26 (t, 3H, J = 7.0 Hz), 1.12–1.08 (m, 1H), 0.67–0.64 (m, 2H), 0.63–0.60 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 169.9, 153.7, 140.1, 116.4, 115.2, 88.8, 70.6, 62.6, 56.1, 50.7, 14.5, 8.8, 8.7. HRMS (ESI): calculated for C16H20NO3 [M + 1]+, 274.1443; found, 274.1453.
Ethyl-2-(p-methoyphenylamino)-5-(trimethylsilyl)-3-pentynoate. Compound 3e was obtained in 63% yield according to the general procedure. The ee value (77%) was determined by chiral HPLC analysis with a 25-cm × 4.6-mm Daicel Chemical Industries CHIRALPAK AD column (eluent, 90:10 hexane:i-PrOH; flow rate, 1.0 ml/min; detection, 254-nm light): tR (major) = 18.03 min, tR (minus) = 27.63 min. [α]D20 –36.3° (c 0.5, CHCl3). 1H NMR (500 MHz, CDCl3): δ 6.79–6.72 (m, 2H), 6.68–6.66 (m, 2H), 4.71 (t, 1H, J = 2.5Hz), 4.28–4.24 (q, 2H, J = 7.2 Hz), 3.74 (s, 3H), 1.46–1.45 (d, 2H, J = 3.0 Hz), 1.32–1.29 (t, 3H, J = 7.3 Hz), 0.04 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 169.8, 153.5, 139.6, 116.3, 114.9, 83.7, 73.9, 62.2, 55.9, 50.5, 14.3, 7.3, –1.9. HRMS (ESI): calculated for C17H26NO3Si [M + 1]+, 320.1682; found, 320.1711.
Ethyl-2-(p-methoyphenylamino)-4-(trimethylsilyl)-3-butynoate. Compound 3f was obtained in 55% yield according to the general procedure. The ee value (48%) was determined by chiral HPLC analysis with a 25-cm × 4.6-mm Daicel Chemical Industries CHIRALPAK AD column (eluent, 90:10 hexane:i-PrOH; flow rate, 1.0 ml/min; detection, 254-nm light): tR (major) = 6.97 min, tR (minus) = 9.15 min. [α]D20 –98.5° (c 0.3, CHCl3), 1H NMR (500 MHz, CDCl3): δ 6.65–6.63 (m, 2H), 6.55–6.53 (m, 2H), 4.58 (s, 1H), 4.15–4.11 (q, 2H, J = 7.0), 3.60 (s, 3H), 1.15 (t, 3H, J = 7.0 Hz), 0.04 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 169.0, 153.6, 139.5, 116.4, 114.9, 100.1, 90.0, 62.5, 55.8, 51.2, 14.2, –0.1. HRMS (ESI): calculated for C16H24NO3Si [M + 1]+, 306.1525; found, 306.1529.
Ethyl-2-(p-methoyphenylamino)-5-phenylpentanoate. A solution of compound 3b (37.6 mg, 0.12 mmol) in EtOAc (3 ml) was hydrogenated with 10% Pd/C (11 mg) at room temperature for 3 h. After filtering-off the solid catalyst, the solution was evaporated under reduced pressure. The residue was purified by a short silica gel column (hexane/EtOAc at 3:1) to afford compound 10 in quantitative yield (38.1 mg). [α]D20 +15.2° (c 0.5, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.21–7.07 (m, 5H), 6.69–6.66 (m, 2H), 6.53–6.51 (m, 2H), 4.08–4.04 (q, 2H, J = 7.0 Hz), 3.89 (t, 1H, J = 6.5 Hz), 3.64 (s, 3H), 2.58–2.55 (m, 2H), 1.78–1.66 (m, 4H), 1.14 (t, 3H, J = 7.0 Hz). 13C NMR (125 MHz, CDCl3): δ 173.2, 151.7, 140.7, 139.8, 127.4, 127.3, 124.8, 114.2, 113.8, 59.9, 56.8, 54.6, 34.4, 31.6, 26.3, 13.2. HRMS (ESI): calculated for C20H26NO3 [M + 1]+, 328.1913; found, 328.1903.
Ethyl-2-(p-methoyphenylamino)-5-phenyl-3-pentenoate. A solution of compound 3b (20.4 mg, 0.06 mmol) in EtOAc (3 ml) was hydrogenated in the presence of 5% Pd/BaSO4 (9 mg) at room temperature for 1 h. After filtering-off the solid catalyst, the solution was evaporated under reduced pressure. The residue was purified by a short silica gel column (hexane/EtOAc at 4:1) to afford compound 11 (19.5 mg, 95% yield). [α]D20 –36.9° (c 0.3, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.35–7.22 (m, 5H), 6.81–6.77 (m, 2H), 6.63–6.59(m, 2H), 5.91(dt, 1H, J = 10.5 Hz, 7.5 Hz), 5.45(dd, 1H, J = 10.5 Hz, 9.0 Hz), 4.91 (d, 1H, J = 9.0 Hz), 4.27–4.18 (q, 2H, J = 7.2 Hz), 3.75 (s, 3H), 3.66 (d, 2H, 7.5Hz), 1.25 (t, 3H, J = 7.2 Hz). 13C NMR (125 MHz, CDCl3): δ 172.7, 153.2, 140.7, 140.0, 134.3, 129.0, 128.9, 127.6, 126.6, 115.7, 115.2, 61.9, 56.2, 56.1, 34.7, 14.5. HRMS (ESI): calculated for C20H24NO3 [M + 1]+, 326.1756; found, 326.1759.
Ethyl-2-amino-5-phenylpentanoate. Ammonium cerium nitrate (0.22 g, 0.41 mmol) dissolved in H2O (1.5 ml) was added to a solution of compound 10 (35.4 mg, 0.11 mmol) in acetonitrile (3 ml) at 0°C under a nitrogen atmosphere. The reaction mixture was stirred for 30 min and then neutralized with 0.5 M NaOH aqueous and extracted with CH2Cl2 (3 × 4 ml). The organic layers were collected and concentrated in vacuo. The crude residue was purified by flash chromatography (hexane/EtOAc at 2:3) to give compound (R)-12 (18.2 mg, 76% yield). Alternatively, a modified procedure was performed under the same condition except by using crude compound 10 as starting material, which was concentrated from the filtered solution of the hydrogenation of compound 3b (37.1 mg, 0.11 mmol), to give compound (R)-12 (18.0 mg, 74% yield). [α]D20 –11.7° (c 0.4, CHCl3) for sample that is 83% ee [literature value: [α]D20 +14.5° (c 0.4, CHCl3) for optically pure compound (S)-12 (32)]. 1H NMR (500 MHz, CDCl3): δ 7.45–7.12 (m, 5H), 4.17 (q, 1H, J = 7.0 Hz), 3.43 (dd, 1H, J = 7.3, 5.1 Hz), 2.64–2.58 (m, 2H), 1.75–1.56 (m, 4H), 1.26 (t, 3H, J = 7.0 Hz). 13C NMR (125 MHz, CDCl3): δ 176.6, 142.6, 129.2, 129.1, 126.6, 61.4, 55.1, 36.3, 35.2, 28.2, 14.9. HRMS (ESI): calculated for C13H19NO2 [M + 1]+, 222.1494; found, 222.1497.
Results and Discussion
In the initial study, with our Ag(I)-catalyzed racemic process in hand, we envisioned that the corresponding asymmetric version could be realized by using an appropriate chiral ligand. The asymmetric addition of 4-phenyl-1-butyne to α-imino ester 1 was explored in the presence of AgOTf or AgNO3 and an additional ligand, respectively. However, it turned out unsuccessful when a series of chiral ligands, including aminophosphanes, diphosphanes, and pybox were subjected to this Ag(I)-catalyzed reaction. All of the reactions resulted in extremely low conversions, and no enantioselectivity was observed.
We then switched our efforts to other transition metals, such as Zn(II), Cu(I)/(II), Ir(I), and Sc(III) (Table 1), some of which have been reported to lead the formation of metal alkynilides (33–41). In the investigation of 4-phenyl-1-butyne 2a addition to α-imino ester 1, none of the target was essentially detected in the presence of IrCl·2COD, Zn(OTf)2, ZnCl2, and Sc(OTf)3 (entry 1). The desired product 3a was obtained in good yields when using CuPF6·4MeCN (entry 3) and CuOTf·0.5C6H6 (entry 4) as catalysts, whereas the other Cu complexes, including Cu(OTf)2 (entry 5), CuCl, CuBr (entry 2), CuO2, and CuOAc (entry 1), showed much lower or undetectable catalytic activity.
Table 1. 4-Phenyl-1-butyne addition to α-imino ester catalyzed by different metal salts.
 | ||
|---|---|---|
| Entry | Catalyst | Yield, % |
| 1 | IrCl·2COD, Zn(OTf)2, ZnCl2, Sc(OTf)3, CuO2, CuOAc | 0 |
| 2 | CuCl, CuBr | <10 |
| 3 | CuPF6·4MeCN | 72 |
| 4 | CuOTf·0.5C6H6 | 70 |
| 5 | Cu(OTf)2 | 35 |
Condition: Compounds 1 (0.25 mmol) and 2a (0.5 mmol) in CH2Cl2 (1.5 ml). Yield refers to isolated yields.
These interesting results led us to examine the effect of a variety of chiral ligands (Fig. 1, compounds 4–8a) in CuPF6·4MeCN-catalyzed asymmetric addition of 4-phenyl-1-butyne to α-imino ester 1 (Table 2). Encouragingly, although the reaction rates suffered noticeably with the use of 2,2′-bis(diphenyllphosphino)-1,1′-binaphthyl; Pybox, bis(oxazolidine)-pyridine (entry 1) and 1-(2-diphenylphosphino-1-naphthyl)isoquinoli (entry 3) as chiral ligands, different degrees of ligand acceleration were observed when employing compounds 5 and 7 and pybox (8a) as ligands (entries 2, 4, and 5), and the addition reaction in the presence of CuPF6·4MeCN/8a furnished target 3a with promising enantioselectivity (59% ee) and chemical yield (73% yield, entry 5).
Fig. 1.

Chiral ligands for screen.
Table 2. Asymmetric addition of 4-phenyl-1-butyne to α-imino ester.
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Yield, % | ee, % |
| 1 | CuPF6·4MeCN/4 | — | Trace | nd |
| 2 | CuPF6·4MeCN/5 | — | 92 | <5 |
| 3 | CuPF6·4MeCN/6 | — | Trace | nd |
| 4 | CuPF6·4MeCN/7 | — | 75 | <5 |
| 5 | CuPF6·4MeCN/8a | — | 73 | 59 |
| 6 | CuPF6·4MeCN/8a | A | Trace | nd |
| 7 | CuPF6·4MeCN/8a | B | Trace | nd |
| 8 | CuPF6·4MeCN/8a | C | 86 | 57 |
| 9 | CuPF6·4MeCN/8a | D | 90 | 57 |
| 10 | CuPF6·4MeCN/8b | D | 93 | 69 |
| 11 | CuPF6·4MeCN/9 | D | 91 | 77 |
| 12 | CuOTf·0.5C6H6/9 | D | 91 | 82 |
| 13* | CuOTf·0.5C6H6/9 | D | 90 | 85 |
| 14 | Cu(OTf)2/9 | D | 78 | 56 |
Condition: Compounds 1 (0.25 mmol) and 2a (0.5 mmol) in CH2Cl2 (1.5 ml) at 0°C. Yield refers to isolated yields. A, 0.5 equivalent of Et3N; B, 0.5 equivalent of i-Pr2NH; C, 0.5 equivalent of PMP-NH2; D, 0.1 equivalent of PMP-NH2; nd, not determined.
Reaction was conducted at — 10°C.
Further investigation involved the addition of amine bases and the use of other Cu sources and structurally different pybox ligands (Table 2). It is known that metal alkynilides, which were used in a couple of CC bond formation reactions, were generated in the presence of a base, such as Et3N (34, 35). Interestingly, in our alkynylation system, the reaction was markedly retarded by adding 0.5 equivalent of Et3N (entry 6) or i-Pr2NH (entry 7). In contrast, the utilization of 0.5 or 0.1 equivalent of PMP-NH2 as additives facilitated an enhancement in yields of compound 3a from 73% to 86% and 90%, respectively, with no diminution of enantioselectivities (entries 8 and 9 vs. entry 5). Subsequent optimization led to the preferred conditions, using CuOTf·0.5C6H6 as a metal source and confirmationally more restricted pybox (9) as a chiral ligand at –10°C, which afforded the desired product 3a in 90% yield and 85% ee (entry 13).
To establish the general utility of this methodology, the direct alkynylation of α-imino ester 1 with a spectrum of terminal alkynes were performed under the optimized conditions, and the representative results are summarized in Table 3. To our delight, in a like manner for the addition of 4-phenyl-1-butyne (entry 1), the addition reactions of 3-phenyl propyne (entry 2), 1-octyne (entry 3), and cyclopropylacetylene (entry 4) provided the corresponding alkynylation products in good yields and ee values, whereas alkynes with a bulky substituted group, such as trimethylsilyl group (entry 6), next to the triple bond led to a lower reaction rate and enantioselectivity. Noticeably, the present cyclopropylacetylene addition to α-imino ester 1 (entry 4) represents a direct and convenient access to α-amino acid derivatives containing conformationally constrained cyclopropane rings, which have recently attracted much attention because of their important biological activities (2, 42–46).
Table 3. Alkynylation of α-imino ester catalyzed by CuOTf·0.5C6H6/9.
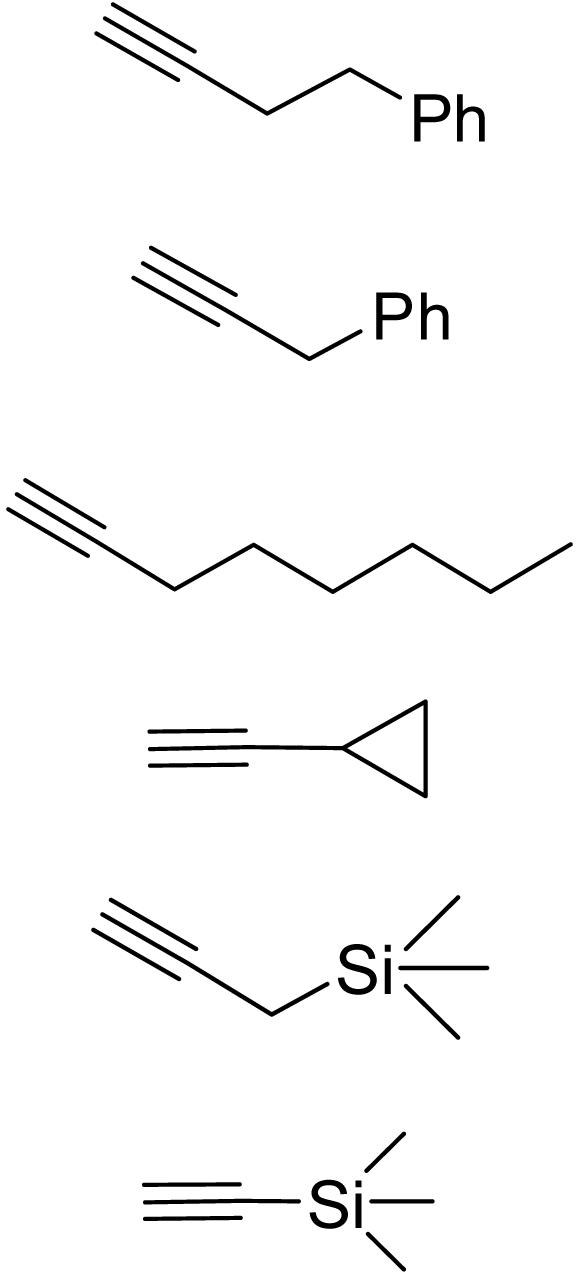 | ||||
|---|---|---|---|---|
| Entry | Alkyne | Product | Yield, % | ee, % |
| 1 |  |
3a | 90 | 85 |
| 2 | 3b | 92 | 83 | |
| 3 | 3c | 89 | 91 | |
| 4 | 3d | 92 | 79 | |
| 5 | 3e | 63 | 77 | |
| 6 | 3f | 55 | 48 | |
Yield refers to isolated yields.
A useful application of this catalytic methodology is illustrated by the modification of product 3b, which yielded bishomophenylalanine derivative (R)-12 (Scheme 2). Compound (R)-12 is a key intermediate of peptide-type growth hormone secretagogue, which can be used to promote the growth of food animals and to treat some human diseases, such as congestive heart failure and short stature in growth hormone-deficient children (47–50). Previous studies on the synthesis of compound 12 was focused on the bioresolution approach using d-aminoacylase as a resolution enzyme (47–50) and chiral auxiliaries-induced stoichiometric asymmetric synthesis (32). In this study, chemical catalytic synthesis of compound (R)-12 was achieved. The alkynylation product 3b was hydrogenated to compound 10 in quantitative yield. Subsequent treatment of compound 10 with cerium ammonium nitrate afforded the target molecule (R)-12 in 76% yield. The absolute configuration of compound 3b was thus determined to be R by inference [[α]D20 –11.7° (c 0.4, CHCl3) for (R)-12 (32), [α]D20 +14.5° (c 0.4, CHCl3) for its S enantiomer]. Furthermore, the semireduction of compound 3b in the presence of Lindlar catalyst (Pd/BaSO4) conveniently yielded (Z)-vinyl amino acid derivative 11. The construction of β,γ-vinyl amino acid derivatives has attracted considerable attention recently because of their significant mechanism-based enzyme inhibitory properties (51, 52), and, so far, several relatively long routes using chiral starting materials or auxiliaries have been developed for this aim (53–57). The present catalytic asymmetric alkynylation of α-imino ester 1, combined with semireduction, provides a catalytic introduction of the vinyl group to amino acid derivatives. Additionally, the hydrogenation of compound 11 using Pd/C also furnished intermediate 10 in quantitative yield.
Scheme 2.

Modification of compound 3b.
It has been suggested that the mechanism of the formation of “soft” metal alkynilides involves the deprotonation of the corresponding metal-alkyne π complex with a mild base (36, 58). Accordingly, a speculated mechanism of the catalytic alkynylation of α-imino ester is outlined in Scheme 3. The successive complexation of substrate 1 and alkyne 2 to the metal center produced the intermediate (13), in which substrate 1 acted as the base in the formation of active Cu(I) alkynilide and as a electrophile, through the activation by the Cu(I) ion and the terminal hydrogen of alkyne. Complex 13 underwent intramolecular proton and alkyne transfer to afford complex 14. Subsequent decomplexation of complex 14 delivered the free product 3 and concomitantly regenerated the catalyst. In this system, the reaction was retarded significantly by adding a tertiary amine [for Et3N, entry 6 (Table 2); an additional experiment using iPr2EtN gave the same result]. This finding was in contrast to those of previous studies that showed that tertiary amines were essential to produce Cu(I) alkynilides by deprotonation in some related CC formation processes (34, 35). These results could be explained as follows: The tertiary amine competed for the terminal proton of alkyne under the catalytic conditions (34, 35) and deprived the proton source, which was necessary for the activation of 1, resulting in the inhibition of the desired addition.
Scheme 3.

Proposed mechanism for the alkynylation of α-imino ester.
Conclusion
In conclusion, catalytic synthesis of enantiomerically enriched β,γ-alkynyl α-amino acid derivatives has been developed by realizing the direct asymmetric alkynylation of α-imino ester. The rich chemistry of alkynyl functionality makes the present method a powerful and versatile approach to a wide range of optically active α-amino acid derivatives. Taking into account the combination of desirable features, such as high potential to generate structural variety, truly atom-economic and readily available starting materials, simple experimental procedures, and mild reaction conditions, this catalytic system is expected to provide an excellent opportunity for applications in increasingly important protein engineering and peptide-based pharmaceutical research. Further work is necessary to elucidate the precise reaction mechanism and the utility of this reaction in the construction of complex unnatural amino acid derivatives.
Acknowledgments
We thank the University Grants Committee Areas of Excellence Scheme in Hong Kong (AoE P/10-01) and the Hong Kong Polytechnic University Area of Strategic Development Fund for financial support.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ee, enantiomeric excess; pybox, bis(oxazolidine)-pyridine; PMP, p-methoxyphenyl.
References
- 1.Barrett, G. C. (1985) Chemistry and Biochemistry of the Amino Acids (Chapman & Hall, London).
- 2.Williams, R. M. (1989) Synthesis of Optically Active α-Amino Acids (Pergamon, Oxford).
- 3.Duthaler, R. O. (1994) Tetrahedron 50, 1539–1650. [Google Scholar]
- 4.Hegedus, L. S. (1995) Acc. Chem. Res. 28, 299–305. [Google Scholar]
- 5.Chin, J. W., Cropp, T. A., Anderson, J. C., Mukherji, M., Zhang, Z. & Schultz, P. G. (2003) Science 301, 964–967. [DOI] [PubMed] [Google Scholar]
- 6.Wang, L. & Schultz, P. G. (2004) Angew. Chem. Int. Ed. 44, 34–66. [DOI] [PubMed] [Google Scholar]
- 7.Cropp, T. A. & Schultz, P. G. (2004) Trends Genet. 20, 625–630. [DOI] [PubMed] [Google Scholar]
- 8.Kamphuis, J., Boesten, W. H. J., Broxtermann, Q. B., Hermes, H. F. M., van Balken, J. A. M., Meijer, E. M. & Schoemaker, H. E. (1990) in Advances in Biochemical Engineering/Biotechnology, ed. Fiechter, A. (Springer, Berlin), Vol. 42, pp. 134–186. [DOI] [PubMed] [Google Scholar]
- 9.Scott, J. W. (1989) in Topics in Stereochemistry, eds. Eliel, E. L. & Wilen, S. H. (Wiley, New York), Vol. 19, pp. 209–226. [Google Scholar]
- 10.Kaptain, B., Boesten, W. H. J., Broxtermann, Q. B., Peters, P. J. H., Schoemaker, H. E. & Kamphuis, J. (1993) Tetrahedron: Asymmetry 4, 1113–1116. [Google Scholar]
- 11.Rossen, K. (2001) Angew. Chem. Int. Ed. 40, 4611–4613. [PubMed] [Google Scholar]
- 12.Noyori, R. & Ohkuma, T. (2001) Angew. Chem. Int. Ed. 40, 40–73. [PubMed] [Google Scholar]
- 13.Burk, M. J. (2000) Acc. Chem. Res. 33, 363–372. [DOI] [PubMed] [Google Scholar]
- 14.Taggi, A. E., Hafez, A. M. & Lectka, T. (2003) Acc. Chem. Res. 36, 10–19. [DOI] [PubMed] [Google Scholar]
- 15.Ferraris, D., Young, B., Dudding, T. & Lectka, T. (1998) J. Am. Chem. Soc. 120, 4548–4549. [Google Scholar]
- 16.Ferraris, D., Young, B., Cox, C., Dudding, T., Drury, W. J., III, Ryzhkov, L., Taggi, A. E. & Lectka, T. (2002) J. Am. Chem. Soc. 124, 67–77. [DOI] [PubMed] [Google Scholar]
- 17.Hagiwara, E., Fujii, A. & Sodeoka, M. (1998) J. Am. Chem. Soc. 120, 2474–2475. [Google Scholar]
- 18.Kobayashi, S., Hamada, T. & Manabe, K. (2002) J. Am. Chem. Soc. 124, 5640–5641. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura, K., Nakamura, H. & Yamamoto, Y. (1998) J. Org. Chem., 64, 2614–2615. [DOI] [PubMed] [Google Scholar]
- 20.Fang, X., Johannsen, M., Yao, S., Gathergood, N., Hazell, R. G. & Jörgensen, K. A. (1999) J. Org. Chem. 64, 4844–4849. [DOI] [PubMed] [Google Scholar]
- 21.Knudsen, K. R., Risgaard, T., Nishiwaki, N., Gothelf, K. V. & Jörgensen, K. A. (2001) J. Am. Chem. Soc. 123, 5843–5844. [DOI] [PubMed] [Google Scholar]
- 22.Cordova, A., Notz, W., Zhong, G., Betancort, J. M. & Barbas, C. F., III (2002) J. Am. Chem. Soc. 124, 1842–1843. [DOI] [PubMed] [Google Scholar]
- 23.Nishiwaki, N., Knudsen, K. R., Gothelf, K. V. & Jörgensen, K. A. (2001) Angew. Chem. Int. Ed. 40, 2992–2995. [DOI] [PubMed] [Google Scholar]
- 24.Ji, J.-X., Au-Yeung, T. T.-L., Wu, J., Yip, C.-W. & Chan, A. S. C. (2004) Adv. Synth. Catal. 346, 42–46. [Google Scholar]
- 25.Balsamini, C., Duranti, E., Mariani, L., Salvatori, A. & Spadoni, G. (1990) Synthesis 779–781.
- 26.Yim, A. M., Vidal, Y., Viallefont, P. & Martinez., J. (2002) Tetrahedron: Asymmetry 13, 503–510. [Google Scholar]
- 27.Pena, D., Minnaard, A. J., de Vries, A. H. M., de Vries, J. G. & Feringa, B. L. (2003) Org. Lett. 4, 475–478. [DOI] [PubMed] [Google Scholar]
- 28.Evans, D. A., Michael, F. E., Tedrow, J. S. & Campos, K. R. (2003) J. Am. Chem. Soc. 125, 3534–3543. [DOI] [PubMed] [Google Scholar]
- 29.Williams, R. M., Aldous, D. J. & Aldous, S. C. (1989) J. Org. Chem., 55, 4657–4663. [Google Scholar]
- 30.Castelhano, A. L., Horne, S., Taylor, G. J., Billedeau, R. & Krantz, A. (1988) Tetahedron 44, 5451–5466. [Google Scholar]
- 31.Davies, I. W., Gerena, L., Lu, N., Larsen, R. D. & Reider, P. J. (1996) J. Org. Chem. 61, 9629–9630. [Google Scholar]
- 32.Davis, F. A., Qu, J., Srirajan, V., Joseph, R. & Titus, D. D. (2002) Heterocycles 58, 251–258. [Google Scholar]
- 33.Miura, M., Enna, M., Okuro, K. & Nomura, M. (1995) J. Org. Chem. 60, 4999–5004. [Google Scholar]
- 34.Black, D. A. & Arndtsen, B. A. (2004) Org. Lett. 6, 1107–1110. [DOI] [PubMed] [Google Scholar]
- 35.Tornoe, C. W., Christensen, C. & Meldal, M. (2002) J. Org. Chem. 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- 36.Frantz, D. E., Fässler, R. & Carreira, E. M. (1999) J. Am. Chem. Soc. 121, 11245–11246. [Google Scholar]
- 37.Si, Y.-G. & Jiang, B. (2003) Tetrahedron Lett. 44, 6767–6768. [Google Scholar]
- 38.Wei, C. & Li, C. J. (2002) J. Am. Chem. Soc. 124, 5638–5639. [DOI] [PubMed] [Google Scholar]
- 39.Koradin, C., Polborn, K. & Knochel, P. (2002) Angew. Chem. Int. Ed. 41, 2535–2538. [DOI] [PubMed] [Google Scholar]
- 40.Sakaguchi, S., Kubo, T. & Ishii, Y. (2001) Angew. Chem. Int. Ed. 40, 2534–2536. [DOI] [PubMed] [Google Scholar]
- 41.Carreira, E. M. & Fischer, C. (2001) Org. Lett. 3, 4319–4321. [DOI] [PubMed] [Google Scholar]
- 42.Stammer, C. H. (1990) Tetrahedron 46, 2231–2254. [Google Scholar]
- 43.Burgess, K., Ho, K. K. & Moyesherman, D. (1994) Synlett, 575–583.
- 44.Salaün, J. & Baird, M. S. (1995) Curr. Med. Chem. 2, 511–542. [Google Scholar]
- 45.Salaün, J. (2000) Top. Curr. Chem. 207, 1–67. [Google Scholar]
- 46.Cativiela, C. & Diaz-de-Viellgas, M. D. (2000) Tetrahedron: Asymmetry 11, 645–732. [Google Scholar]
- 47.Combs, T. L. (2003) PCT Int. Appl., WO 2003/087036, Chem. Abstr. 139, 308017. [Google Scholar]
- 48.Dodge, J. A. & Lugar, C. W., III (2000) PCT Int. Appl., WO 2000/049037, Chem. Abstr. 133, 193495. [Google Scholar]
- 49.Kauffman, R. F. & Palkowitz, A. D. (1999) PCT Int. Appl., WO 99/08697, Chem. Abstr. 130, 209977. [Google Scholar]
- 50.Chen, M. H., Morriello, G. J., Nargund, R., Patchett, A. A. & Yang, L. (1995) PCT Int. Appl., WO 95/14666, Chem. Abstr. 123, 340951. [Google Scholar]
- 51.Rando, R. R. (1974) Nature 250, 586–587. [DOI] [PubMed] [Google Scholar]
- 52.Relyea, N. & Rando, R. R. (1975) Biochem. Biophys. Res. Commun. 67, 392–402. [DOI] [PubMed] [Google Scholar]
- 53.Mulzer, J. & Funk, G. (1995) Synthesis, 101–112.
- 54.Hallinan, K. O., Crout, D. H. G. & Errington, W. (1994) J. Chem. Soc., Perkin Trans. 1 24, 3537–3543. [Google Scholar]
- 55.Sibi, M. P. & Renhowe, P. A. (1990) Tetrahedron Lett., 31, 7407–7410. [Google Scholar]
- 56.Duthaler, R. O. (1991) Angew. Chem. Int. Ed. Engl. 30, 705–707. [Google Scholar]
- 57.Williams, R. M. & Zhai, W. (1988) Tetrahedron 44, 5425–5430. [Google Scholar]
- 58.Cotton, F. A. & Wilkinson, G. (1988) in Advanced Inorganic Chemistry (Wiley, New York), 5th Ed., p. 765.