

Abstract
We have serendipitously established a mouse that expresses an N-terminal human huntingtin (htt) fragment with an expanded polyglutamine repeat (≈120) under the control of the endogenous human promoter (shortstop). Frequent and widespread htt inclusions occur early in shortstop mice. Despite these inclusions, shortstop mice display no clinical evidence of neuronal dysfunction and no neuronal degeneration as determined by brain weight, striatal volume, and striatal neuronal count. These results indicate that htt inclusions are not pathogenic in vivo. In contrast, the full-length yeast artificial chromosome (YAC) 128 model with the identical polyglutamine length and same level of transgenic protein expression as the shortstop demonstrates significant neuronal dysfunction and loss. In contrast to the YAC128 mouse, which demonstrates enhanced susceptibility to excitotoxic death, the shortstop mouse is protected from excitotoxicity, providing in vivo evidence suggesting that neurodegeneration in Huntington disease is mediated by excitotoxic mechanisms.
Keywords: Huntington disease, mouse models, excitotoxicity, aggregates, fragment
Huntingtin (htt), the protein product encoded by the gene mutated in Huntington disease (HD), forms aggregates and inclusion bodies in the presence of a pathogenic expanded polyglutamine (polyQ) repeat. Htt protein inclusions are a hallmark of HD and are present in brains of human patients (1), in HD mouse models (2, 3), and in cell culture models of HD (4). It is still controversial whether htt inclusions are pathogenic (2), benign biomarkers (5), or neuroprotective (4, 6). The distinction between these hypotheses is clinically relevant, because much therapeutic research has focused on screening compounds for their ability to inhibit inclusion formation (7, 8). A decrease in inclusion formation has been interpreted as a positive outcome in preclinical therapeutic trials with mouse models (9, 10).
Increasing evidence in vitro in cell culture models supports the hypothesis that htt inclusions are not pathogenic (5, 11). In a recent study, Arrasate et al. (4) discovered that in their cell culture system, neurons with inclusions had an increased likelihood of survival compared with neurons without inclusions. However, because these results were obtained in a cell culture system, the question of whether htt inclusions are toxic in vivo during the lifespan of an organism and therefore clinically relevant for patients with HD remains unanswered.
Examinations of inclusions in brains from HD patients are limited due to the inability to sample inclusions over the natural history of the disease, and, therefore, studies of mouse models of HD can be useful in determining the role of htt inclusions in vivo. The yeast artificial chromosome (YAC) 128 model of HD, which expresses full-length mutant htt, forms intranuclear inclusions 12 months after the onset of behavioral changes measured by rotarod and 6 months after striatal neuronal degeneration (3).
During the development of the full-length YAC mouse models, a mouse expressing a short fragment (exons 1 and 2 of 67) of human htt with an expanded polyQ repeat was serendipitously established (shortstop). We could therefore investigate in vivo the effects of the expression of a truncated fragment of htt on both inclusion formation and neuropathology. In addition, the establishment of the shortstop mouse allowed the ability to characterize in parallel the full-length and truncated HD mouse models, which share all aspects of the transgene, including the promoter, but differ in the length of htt. The absence of any behavioral abnormalities or evidence for neurodegeneration indicates that htt inclusions are nontoxic in vivo.
Materials and Methods
Generation of Mice. YAC mutagenesis was performed as described in ref. 12 by using a construct containing 120 CAG repeats. The shortstop transgene likely arose from truncation or recombination of the full-length YAC during multiplication of the YAC in yeast before injection. Mice were maintained on the FVB/N strain (Charles River Laboratories). Mice were genotyped by using primers HD 344, 5′-CCTTCGAGTCCCTCAAGTCCTTC-3′, and HD 482, 5′-GGCTGAGGAAGCTGAGGAG-3′, as described in ref. 13, with a 6-carboxyfluorescein-labeled 5′ primer used for CAG repeat sizing. Products were run on an Applied Biosystems 3100 Genetic Analyzer (Amersham Pharmacia Biosciences) and visualized by using genescan analysis 3.7.1 software (Applied Biosystems). Mice were housed and tested following University of British Columbia animal protocol A00-0254.
Protein Analysis. Protein lysates were prepared from whole mouse brain or dissected cortex, striatum, hippocampus, and cerebellum and run on 7.5% polyacrylamide gels as described in ref. 3. Blots were probed with anti-actin (Chemicon), polyclonal BKP1, and 1C2 (Chemicon). quantity one imaging software (Bio-Rad) and nih image software were used for quantification.
PCR and Bioinformatics Characterization of YAC Shortstop Truncation Sites. Primer sequences include 9343F, 5′-TGCAACCTCATTGGCATTTACAG-3′; 9617R, 5′-TTCCCTTTTGACTTCACTTCTGACC-3′; 9680R, 5′-CAGCACCCCACAAGTTTAGAAATC-3′; and M476R, 5′-TTCATCTTTGCTGGAAACAGTGC3-′.
For inverse PCR, 1 μg of shortstop genomic DNA was digested with MspI or AluI (New England Biolabs) and purified. The digested DNA was then religated in a dilute reaction. For the first round of PCR, primers 9234R and 9271F were used to amplify 1 μl of the ligation reaction using Taq. For the nested round of PCR, 2 μl of the primary reaction product was used for amplification with primers 9229R and 9343F (see Fig. 5, which is published as supporting information on the PNAS web site, for primer location). A 750-bp band from the MspI-digested DNA was purified and sequenced with primer 9229R and identified as spanning the junction between HD intron 2 and mouse genomic DNA.
The genomic location and gene context of derived DNA sequences were identified by using the human and mouse Ensembl Genome Browsers (www.ensembl.org) and by using blastn with default parameters at the National Center for Biotechnology Information web site (www.ncbi.nlm.nih.gov/blast). Poly(A) addition signals were predicted by searching for “AATAAA” motifs by using the program patmatdb within the emboss suite of bioinformatics programs (http://emboss.sourceforge.net).
Quantitative Analyses. Mice were terminally anesthetized, and the brains were removed and stored as described in ref. 3. Transgenic and WT mice were matched based on age and sex, and littermates were used when possible.
Quantitative analyses were performed blind with respect to genotype. Vibratome-cut coronal sections (25 μm) spaced 200 μm apart throughout the striatum were stained with NeuN antibody (Chemicon) at 1:100 dilution. Biotinylated secondary antibodies (Vector Laboratories) at 1:200 were used before signal amplification with the Elite ABC kit (Vector Laboratories) and detection with diaminobenzidine (Pierce). Striatal volume and neuronal count were quantified as described in ref. 3.
Aggregates and Aggregation Foci (AF) Assessment. Brain sections (25 μm) throughout the striatum were stained for the presence of inclusions. Sections were immunostained as described in ref. 1 by using polyclonal EM48 antibody at 1:1,000 and using diaminobenzidine as the chromogen (Pierce). Sections were also costained with EM48 1:2,000, visualized with goat anti-rabbit-Alexa Fluor 488 (Molecular Probes) and NeuN 1:1,000 and goat anti-mouse-Alexa Fluor 594 (Molecular Probes). The percentage of aggregates was determined by counting a minimum of 300 neurons.
For polyQ recruitment reactions that detect AF, brains perfused in situ with 4% paraformaldehyde were embedded in gelatin blocks, postfixed, and sectioned at 40 μm, using proprietary MultiBrain technology at Neuroscience Associates (Concord, TN). The synthetic peptide bPEGQ30 was prepared at the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University (New Haven, CT) (http://info.med.yale.edu/wmkeck) and was composed of 30 glutamine residues flanked by two lysyl residues at each end to enhance solubility and with an N-terminal glutamine residue with a biotinylated polyethylene glycol moiety added at the γ-amide position. The peptide was disaggregated and purified as described in ref. 14. The protocol for polyQ recruitment used a previously published method (15) that avoided the use of proteinaceous blocking reagents by using sodium borohydride pretreatment and high concentrations of Triton X-100 (0.4%). Free-floating sections were incubated for 18 h at room temperature in biotinylated polyQ peptide, bPEGQ30 (10-25 nM), and biotin was detected after a single cycle of biotinylated tyramide enhancement using the Elite ABC kit and nickel-enhanced diaminobenzidine (Pierce) as the chromogen.
Rotarod Analysis. Mice were singly housed in microisolator cages. All behavioral testing was executed in the light in a behavioral testing suite. Mice were tested on a rotarod (Ugo Basile, Varese, Italy), which accelerated from 0 to 40 rpm over 300 s. Mice were initially trained on the accelerating rotarod for 3 days with three trials per day. Testing occurred over 1 day with 1.5 h of rest in between tests. Mice were tested at 12 months of age and were naïve (i.e., had never been tested before).
Quinolinic Acid (QA) Injections. QA (Sigma) was dissolved into 0.1 M PBS (pH 7.4). Mice were anesthetized with isofluorane and received bilateral intrastriatal injections of 6 nmol QA. Seven days postinjection, mice were terminally anesthetized as described above and analyzed as described in ref. 16.
Striatal Neuronal Cultures. Anterior striata from postnatal day-0-1 mice were dissected, and neurons were dissociated as described in ref. 16. Neurons were cultured for 9 days and then exposed for 10 min to varying concentrations of NMDA (100 and 500 μM). Twenty-four hours later, cultures were fixed with 4% paraformaldehyde and neurons (determined by morphology) were assessed for apoptotic death by using TUNEL staining (Roche Applied Science, Indianapolis). All treatments were done in triplicate, with a minimum of 1,000 neurons counted.
Statistics. All statistics were carried out by using an unpaired Student's t test or one-way ANOVA with Tukey's posttest for multiple comparisons. P values, SEM, means, and SDs were calculated by using graphpad prism 3.0 or excel 2002 (Microsoft).
Results
Discovery of the Shortstop Mouse. During generation of additional full-length YAC mice, it was noted that one of the generated lines was missing the left YAC arm (LYA) by PCR screening (data not shown). The loss of the LYA, which lies 3′ to the htt gene, and the presence of an expanded CAG tract in this line of mice suggested that the YAC had a truncation 3′ of the CAG tract. Western blots of brain lysates from the truncated mouse line revealed an ≈75-kDa band immunostained with BKP1, an antibody specific to the N terminus of htt (Fig. 1A). Stripping the blots and reprobing with 1C2, an antibody specific to expanded polyQ, demonstrated an identical band at 75 kDa (Fig. 1A). These results indicated that the truncated mouse line was expressing a short fragment of htt from a premature stop and was therefore given the name “shortstop.” Accurate sizing of fragments PCR-amplified across the repeat tract revealed that both the full-length YAC128 and the shortstop mouse had identical polyQ tract lengths of ≈120 CAG (Fig. 1C).
Fig. 1.

Characterization of shortstop fragment and comparison with full-length YAC128. (A) Western blot showing protein expression of shortstop fragment (arrowhead) probed with the htt-specific antibody BKP1 and the polyQ-specific antibody 1C2. Full-length, mouse-endogenous htt is shown (asterisk). (B) PCRs on genomic mouse DNA from shortstop (SS), YAC128 (53), and WT mice using primers within intron 2 of human htt before (9343F/9617R) and after (9343F/9680R) the shortstop breakpoint reveal the presence of part of intron 2 in shortstop. PCRs with primers from human intron 2 (9343F) and mouse chromosome 4 (m476R) reveal a band in shortstop, indicating integration into mouse chromosome 4. (C) Fragment analysis of PCR products amplified by using human-specific primers that border the CAG tract (including 69 bp of flanking sequence) demonstrate identical CAG size in YAC128 and shortstop mice. (D) Western blots with 1C2 show similar protein expression in shortstop and YAC128 mice across brain regions. (E) Western blots with 1C2 show similar levels of transgenic protein in shortstop and YAC128 cortex (n = 3). (F) Accompanying densitometry analysis of a shorter exposure of blot in E (in the linear range of detection) reveals that shortstop mice express 1.46 times the protein of YAC128 mice.
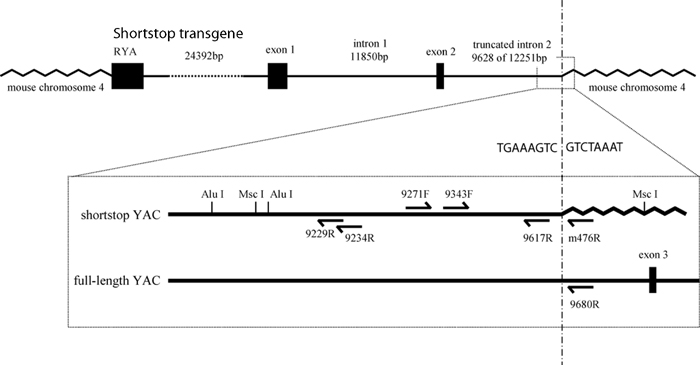
The truncation site of shortstop was localized to intron 2 through successive PCRs, and the precise site of insertion into the mouse genome was identified by performing inverse PCR. A 750-bp product obtained after two rounds of nested PCR was sequenced and was found to comprise HD intron 2 truncated at base 9628 and joined to mouse chromosome 4. Additional evidence for the truncation of the YAC at base 9628 of HD intron 2 was provided by testing shortstop genomic DNA for PCR amplification with primer 9343F paired with either primer 9617R (predicted to be positive) or primer 9680R (predicted to be negative) (see Figs. 1B and 5).
According to the inverse PCR product, the insertion site of the YAC occurred at base 63804 of the bacterial artificial chromosome (BAC) clone RP23-123E1, which has been mapped to band B3 of mouse chromosome 4. This region of the mouse genome is gene poor, and, according to Ensembl Mouse Genome Assembly NCBI Build 33 (updated May 27, 2004), the nearest known centromeric gene is located 566 kb proximal, and the nearest known telomeric gene is located 627 kb distal to the BAC. This mapping provides assurance that any phenotypes observed in the shortstop are due to the effects of the transgene and are not the result of the disruption of an endogenous murine gene.
There are five potential poly(A) addition signals within the portion of intron 2 present in the shortstop YAC. The first codon within HD intron 2 is a “TAA” stop codon; therefore, there are no additional amino acids in the shortstop protein. The predicted protein sequence translated from the shortstop YAC is identical to that translated from HD exons 1 and 2 and should express a protein from amino acids 1-117 in the human sequence. The shortstop protein runs above the predicted size of ≈24 kDa (Fig. 1A). This phenomenon has been reported previously in the R6/2 short fragment model of HD (an expected size of 23 kDa, but the protein band runs at ≈80 kDa) and is consistent with aberrant migration due to the polyQ stretch (17).
Shortstop and YAC128: Identical Pattern of Transgenic Protein Expression. Protein expression analysis with 1C2 revealed similar transgenic protein distribution in YAC128 and shortstop brain regions, including the striatum, cortex, hippocampus, and cerebellum (Fig. 1D). Protein lysates from the cortices of YAC128 and shortstop mice (n = 3) were run on a gel and probed with 1C2 (Fig. 1E). Densitometric analysis revealed that the shortstop mice expressed ≈1.46 times the amount of transgenic protein compared with the full-length YAC128 mice (Fig. 1F). Identical CAG size and tissue distribution, combined with a similar level of transgenic protein expression in the full-length YAC128 and shortstop mice, allows the comparison between a suitably matched and controlled full-length and truncated htt mouse model.
Inclusion Formation in Shortstop Mice. To investigate the distribution of neuronal inclusions (NIs) in the shortstop mice, brain sections from 18-month-old animals were immunostained with EM48, an antibody specific for htt aggregates and inclusions (1). NIs in the shortstop mouse at 18 months could be visualized in >95% of striatal neurons (Fig. 2B), 90% of cortical neurons (Fig. 2D), and ≈100% of neurons of the hippocampus (Fig. 2F). In comparison, the YAC128 at 18 months demonstrated fewer NIs, with ≈30% of striatal neurons (Fig. 2A), <5% of cortical neurons (Fig. 2C), and no NIs in the hippocampus (Fig. 2E), a brain region less frequently affected in HD. To determine the natural history of NI formation, brain sections from 3-, 6-, 9-, 12-, and 18-month-old shortstop and YAC128 mice were immunostained with EM48 and scored for the presence of NIs as well as localization of htt to the nucleus (Tables 1 and 2, which are published as supporting information on the PNAS web site), a marker of htt nuclear translocation. Neuronal nuclear htt staining is accelerated in the shortstop mice and exhibits regional differences compared with the full-length YAC128 (Table 1). In addition, NI formation is accelerated and more widespread both in neuronal numbers affected and tissue distribution in the shortstop compared with the YAC128 (Table 2). The comparison of the time course of shortstop and YAC128 NI formation therefore reveals a much earlier onset of NIs in the shortstop mouse, accompanied by a more widespread tissue distribution in vivo.
Fig. 2.
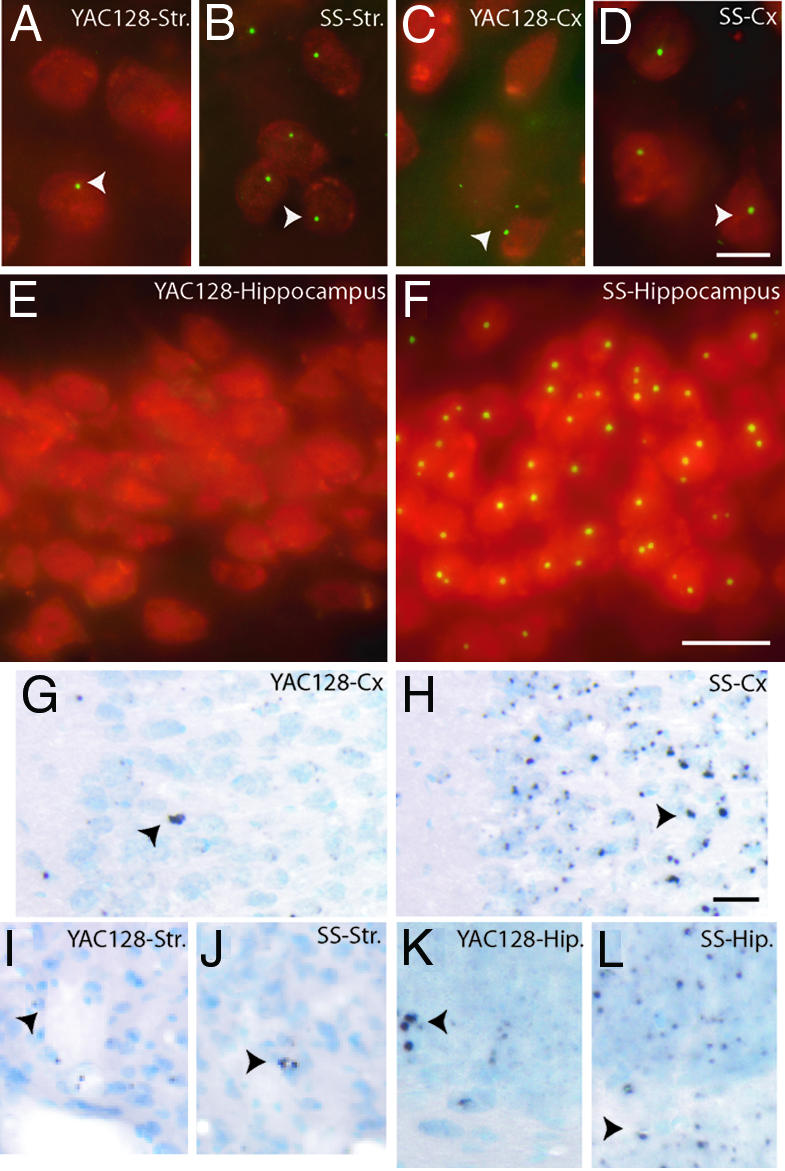
Htt inclusions in shortstop and YAC128 mice at 18 months and AF at 12 months. EM48 inclusions labeled with FITC (green) are present in NeuN-stained neurons labeled with Texas red in the striatum (A) and cortex (C) of YAC128 mice, but not in the hippocampus (E). Inclusions are present in most cells in the striatum (B), cortex (D), and hippocampus (F) in the shortstop (SS) mouse. At 12 months of age, YAC128 cortex (G) and hippocampus (K) demonstrate less labeling of AF compared with shortstop cortex (H) and hippocampus (L). Few AF are present in the striatum of the YAC128 (I) or shortstop (J) mice. Inclusions are indicated with white arrowheads; AF are indicated with black arrowheads. (Scale bar in D, which applies to A-D:10 μm; scale bar in F, which apples to E and F: 20 μm; scale bar in H, which applies to G-L: 20 μm.)
AF in Shortstop and YAC128 Mice. Tissues from the shortstop and YAC128 mouse were also examined for AF, a precursor of htt inclusions. AF are intracellular sites that demonstrate polyQ recruitment activity and have been identified in the cortex of patients with HD as putative precursors of neuropil aggregates (unpublished data). AF are visualized and defined by their ability to recruit synthetic biotinylated polyQ peptides and occur early in the development of htt inclusions.
AF are diffusely present in shortstop mouse brain at 12 months of age in the cortex (Fig. 2H) and hippocampus (Fig. 2L), indicating that this aggregation precursor is present in the shortstop mouse. AF are similarly present, although at a lesser density in the YAC128 mouse at 12 months of age in the cortex (Fig. 2G) and hippocampus (Fig. 2K). AF are present at low frequency in the striatum of the shortstop (Fig. 2 J) and YAC128 mice (Fig. 2I).
Shortstop Does Not Exhibit a Rotarod Deficit. Cell culture experiments have demonstrated that decreasing the size of the N-terminal htt fragment increases the toxicity of the fragments in vitro (18, 19). We hypothesized that the shortstop mouse would manifest an earlier and more severe neuronal dysfunction and degenerative phenotype compared with the full-length YAC128 mice. Previous studies of the full-length YAC128 mouse demonstrated a severe, progressive rotarod deficit (3, 20). We compared the shortstop mice at 12 months of age with YAC128 and WT mice. Surprisingly, the shortstop mice exhibited no deficit on the rotarod compared with WT and performed significantly better than the full-length YAC128 mice (P < 0.001; ANOVA, P = 0.0002; Fig. 3A). These results were replicated in a separate cohort of animals that demonstrated the shortstop mice performing similarly to WT at 12 months of age, whereas the YAC128 mice were significantly worse (P < 0.01; ANOVA, P = 0.01).
Fig. 3.

Shortstop mice do not manifest the neuronal dysfunction or degeneration of the YAC128 model. (A) Shortstop do not exhibit a deficit on an accelerating rotarod, whereas YAC128 mice demonstrate a deficit compared with WT littermates (P < 0.001). (B-D) The shortstop mice at 12 months do not exhibit significant brain weight decrease (B), striatal volume decrease (C), or striatal neuronal count decrease (D) when compared with WT mice, whereas YAC128 mice demonstrate significant decreases. (E and F) At 18 months, there is no significant decrease in brain weight (E) or striatal volume (F) of shortstop mice compared with WT mice; YAC128 mice exhibit significant deficits. Mean ± SD is shown in B-F, and mean ± SEM is shown in A. Data were analyzed by a one-way ANOVA, and P values between groups were calculated by using Tukey's posttest.
Shortstop Does Not Exhibit Neuronal Degeneration. Brain weight and striatal volume loss accompanied by striatal neuronal loss are manifest in the full-length YAC128 mice by 12 months of age, recapitulating the neurodegeneration present in HD patients. Interestingly, shortstop brain weight at 12 months of age was not significantly decreased compared with WT, whereas YAC128 mouse brains weighed 8% less than those of their WT littermates (P < 0.01; ANOVA, P = 0.006; Fig. 3B).
Striatal volume and neuronal count were quantified, and, surprisingly, striatal volume in the shortstop mice was no different from that in WT controls (ANOVA, P = 0.02; Fig. 3C); YAC128 striatal volume demonstrated a 15% decrease (P < 0.05). Shortstop mice revealed no difference in striatal neuronal count compared with WT mice, whereas YAC128 neuronal count was decreased (P < 0.05; ANOVA, P = 0.01; Fig. 3D). These results reveal that shortstop mice at 12 months of age do not manifest the HD-related phenotypes that are evident in the full-length YAC128 mice at the same age.
To examine whether these phenotypes are present in older shortstop mice, an 18-month cohort was examined. Shortstop mice did not demonstrate significant brain weight or striatal volume decrease compared with WT; full-length YAC128 exhibited a significant decrease in brain weight (ANOVA, P = 0.03) and striatal volume (ANOVA, P = 0.003) compared with WT littermates, suggesting that even at an older age, the shortstop mice are resistant to the HD-related neurodegenerative phenotypes present in the full-length YAC128 mice (Fig. 3 E and F).
Shortstop Is Resistant to NMDA-Induced Excitotoxicity. YAC72 transgenic mice with 72 CAG repeats (12) were previously shown to be significantly more susceptible to excitotoxic cell death compared with WT (16). Zeron et al. (39) in 2001 found that cells transfected with full-length mutant htt were much more susceptible to NMDA-mediated excitotoxicity than cells transfected with a truncated 548-aa N-terminal fragment of htt, suggesting the necessity for full-length htt or a longer fragment in increased susceptibility to excitotoxicity. This finding led to the hypothesis that differential susceptibility to excitotoxicity may underlie the difference in susceptibility to HD-related neurodegeneration in the YAC128 and shortstop models.
To test this hypothesis, cohorts of 6-month-old YAC128 and shortstop mice underwent bilateral striatal injections of the NMDA receptor agonist QA. Fluorojade, a fluorescent marker of dying neurons, was used to visualize excitotoxic injury. The YAC128 mice demonstrated significantly larger excitotoxic lesions compared with age-matched shortstop mice with the same dose of QA (P < 0.05; Fig. 4A) as visualized in Fig. 4B. DARPP-32 (dopamine- and cAMP-regulated phosphoprotein of Mr 32,000) staining was used to identify medium spiny neurons (MSNs), the neuronal cell type predominantly targeted in HD, and revealed a greatly reduced number of surviving MSNs in the YAC128 mice compared with the shortstop mice after QA injection (data not shown), again demonstrating that the YAC128 mice are more susceptible to excitotoxic injury compared with the shortstop mouse.
Fig. 4.

Shortstop mice demonstrate resistance to excitotoxicity compared with the YAC128 model. (A and B) Intrastriatal injection of the NMDA agonist QA in 6-month-old mice revealed larger fluorojade-stained (apoptotic) lesions in coronal sections of YAC128 mice compared with shortstop mice (B). Lesion volume was quantified and found to be significantly less in shortstop mice compared with YAC128 mice by Student's t test (A). (C) Primary striatal neuronal cultures established from YAC128 and SS postnatal day-0 pups were treated with NMDA. Apoptotic neurons were stained and quantified by using TUNEL and propidium iodide (PI). (D) YAC128 mice revealed a significantly increased percentage of apoptotic neurons compared with shortstop mice (C) by one-way ANOVA with Tukey's posttest. MK-801 reduces the level of apoptotic death in the YAC128 neurons to baseline. Mean ± SD is shown.
To further investigate excitotoxicity, cultures of MSNs from postnatal day-0-1 pups were established and assessed for susceptibility to excitotoxicity through treatment with NMDA in a procedure described in ref. 16 using TUNEL staining to distinguish apoptotic neurons. Treatment of primary striatal neuronal cultures with 500 μM NMDA resulted in 37% apoptotic death in YAC128 mice compared with only 10% apoptotic death in WT (P < 0.001) or shortstop (P < 0.001; ANOVA, P < 0.0001; n = 3; Fig. 4 C and D). Treatment with MK-801, an NMDA antagonist, reduced the YAC128 apoptotic levels to the baseline WT levels, revealing that the increased susceptibility to excitotoxicity in the YAC128 mice resulted at least in part from increased NMDA receptor activation (Fig. 4C).
Discussion
Since the initial discovery of htt inclusions, there has been continued controversy over the role of htt aggregates in the pathogenesis of the disease. The shortstop mice reveal widespread htt NIs both in tissue distribution and percentage of affected neurons. Despite this widespread inclusion burden, these mice do not manifest a behavioral, HD-related phenotype as assessed by rotarod or decreases in brain weight, striatal volume, and striatal neuronal count at 12 or 18 months of age. The shortstop mouse therefore illustrates in vivo the inability of htt inclusions to have a toxic effect over the lifespan of an organism, clearly demonstrating that htt inclusions are not toxic. These data are supported by in vitro htt studies (4, 5, 11) and in vivo studies in mouse models of other polyQ disorders (21-23).
Despite the increasing evidence that htt inclusions are not toxic, aggregation assays are used to identify potential therapeutic compounds (8, 24), and inhibition of inclusion formation is viewed as a positive outcome in therapeutic preclinical mouse trials (10, 25). The presence of inclusions without pathology in the shortstop mouse suggests that primary screening for compounds that selectively inhibit inclusions may not identify agents that will be beneficial in clinical trials.
The formation of visible htt NIs has been hypothesized to be the last stage of a complex, linear process beginning with a critical concentration of aggregate precursors and ending with the production of a visible aggregate (NIs) (26). The toxicity of aggregate precursors has been offered as an explanation for those mouse models that manifest inclusions after pathology and behavioral changes, including the YAC128 mouse model (3). However, if the formation of inclusions is a linear process, with visible inclusions as the last stage in this process, then the shortstop mouse should at one time express each of the “toxic” aggregation precursors, yet it does not present with a neurodegenerative phenotype. This finding would suggest that other components of the aggregation pathway are not toxic or that the kinetics of inclusion formation are altered such that fragments of htt spend little time in a toxic precursor state. However, it is also possible that multiple different pathways of inclusion formation and maturation exist, and inclusions in the shortstop mice could be formed through a nontoxic inclusion pathway.
The purpose of this work was to examine the shortstop model and compare and contrast it with the full-length YAC128. However, questions arise regarding the differences in phenotype between the shortstop and the R6/2 short fragment mouse model. The R6/2 model expresses the first exon of htt with 144 (17) to 205 (27) polyQ repeats (compared with 120Q in the shortstop) and demonstrates rotarod deficits (28), nonselective brain weight decreases, and an early death phenotype (17). It is surprising that shortstop and R6/2 mice share no phenotypic features. There are, however, significant differences between the two models. The transgenic htt fragment in the shortstop mouse is expressed under 24 kb of upstream human promoter and regulatory elements compared with only 1 kb of human promoter in the R6/2 (Fig. 6, which is published as supporting information on the PNAS web site). Bioinformatic analysis reveals evolutionarily conserved elements within the first 5 kb of upstream regulatory sequence and within intron 1 of human htt. The shortstop contains this 5 kb of upstream sequence and intron 1, whereas the R6/2 model only contains the first 1 kb of upstream sequence, suggesting that the transgenic protein is likely expressed most appropriately (i.e., closest to normal human htt regulation) in the shortstop. It is also possible that inappropriate expression and/or overexpression of the transgene in the R6/2 model is responsible for the phenotypes observed in this model that are not present in the shortstop.
The study of the shortstop mouse in parallel with a full-length YAC128 mouse allows the controlled comparison of a short fragment of htt with the full-length protein in vivo. The toxic fragment hypothesis proposes that toxic, N-terminal fragments of mutant htt cause alterations in transport, and/or endocytosis, and/or transcription, leading to increased susceptibility to excitotoxicity and increased proteolysis, which, in turn, produces more toxic N-terminal fragments and results in a steady amplification of this cycle until the neuron is ultimately committed to apoptosis (29). The lack of a neurodegenerative phenotype in the shortstop mouse could be interpreted to negate the toxic fragment hypothesis. Alternatively, this finding might suggest that there is specificity in the size of the N-terminal fragment that can initiate the toxic fragment cycle. Htt is cleaved by caspases, calpains, and aspartyl proteases (30), yielding different N-terminal htt fragments that may be differentially toxic. In 2003, Yu et al. (31) demonstrated that an N-terminal fragment of 208 aa produced the greatest caspase activation and toxicity in a cell culture model, significantly greater than that caused by a shorter exon 1 fragment or longer htt fragments. The hypothesis that different fragments of a disease-causing protein can be differentially toxic is well recognized in the Alzheimer's disease field, where the Aβ1-42 fragment is more toxic in vivo than the Aβ1-40 fragment, despite only a 2-aa difference (32). Nonsteroidal antiinflammatory drugs, which act to reduce the proportion of Aβ1-42 relative to shorter fragments, have been associated with a decreased rate of developing Alzheimer's disease, suggesting differential toxicity of fragments (33).
The inability of the N-terminal fragment expressed by the shortstop mouse to initiate the toxic fragment cycle, which ultimately leads to neuronal degeneration in the full-length YAC128 mouse, indicates that there is an important role for the full-length mutant protein or a longer fragment in the pathogenesis of the disorder. It appears that not only the fragment but from where the fragment is derived is important. The necessity of the correct protein context is illustrated by comparison of the polyQ disorders, which shows that despite the identical underlying genetic mutation, the site of pathology is different for each disorder. Phosphorylation of ataxin-1 is required for neurodegeneration in spinocerebellar ataxia type 1 (34), and testosterone is required for neurodegeneration in spinobulbar muscular atrophy (35), revealing the importance of protein domains other than the polyQ stretch for neurodegeneration in these related disorders.
The lack of neuronal degeneration in the shortstop mouse suggests that other portions or longer fragments of htt are required for the initiation of the specific toxic cycle that leads to the specific sequence of behavioral, cognitive, and neuropathological abnormalities in HD. Htt is phosphorylated by Akt (36) on serine 421, a residue not present in the shortstop mouse, and further research is required to determine the effect that phosphorylation or other posttranslational modifications have on the development of neurodegeneration. Full-length WT htt has recently been shown to promote transport of BDNF, an important neurotrophic factor implicated in HD (37), whereas a htt exon 1 fragment was incapable of similarly effecting BDNF transport (38).
The importance of other portions or domains of htt in HD-related neurodegeneration was recently demonstrated in a study of susceptibility to excitotoxicity (16, 39). The YAC128 mouse demonstrates increased susceptibility to excitotoxicity compared with the shortstop mouse both by intrastriatal injections of QA in adult mice and treatment of primary striatal neuronal cultures with NMDA. These results demonstrate a correlation between enhanced susceptibility to excitotoxicity and neuronal degeneration in the full-length YAC128 model and the absence of excitotoxicity with no neuropathology in the shortstop model. This correlation further reveals the importance of excitotoxicity as an initiating mechanism in HD pathogenesis.
The ability to differentiate phenotypic changes that relate to neurodegeneration (e.g., excitotoxicity) from those changes that are simply a benign product of polyQ expansion (e.g., inclusions) reveals the importance of further investigations using the shortstop mice. Delineation of the morphological and functional differences between shortstop and YAC128 mice may be a fertile area of research for identification of critical pathways for the pathogenesis of HD.
Supplementary Material
Acknowledgments
We thank Declan Bradley and Zoe Murphy for technical assistance, Dr. Xiao-Jiang Li (Emory University, Atlanta) for the EM48 antibody, and Stefanie Butland (University of British Columbia Bioinformatics Centre) for sequence advice. This work was supported by the Canadian Institutes of Health Research (M.R.H., B.R.L., E.J.S., and R.K.G.), the Huntington's Disease Society of America (M.R.H. and B.R.L.), the High Q Foundation (M.R.H. and B.R.L.), the Michael Smith Foundation for Health Research (E.J.S. and R.K.G.), and the Hereditary Disease Foundation (R.W. and A.P.O.). M.R.H. holds a Canada Research Chair in Human Genetics.
Author contributions: E.J.S., R.K.G., A.P.O., R.S.D., and M.R.H. designed research; E.J.S., R.K.G., A.P.O., R.S.D., G.L., Y.D., J.P., K.V., and N.B. performed research; R.W. contributed new reagents/analytic tools; E.J.S., R.K.G., A.P.O., and R.S.D. analyzed data; and E.J.S. and M.R.H. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HD, Huntington disease; htt, huntingtin; polyQ, polyglutamine; AF, aggregation foci; YAC, yeast artificial chromosome; QA, quinolinic acid; NI, neuronal inclusion.
References
- 1.Gutekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S., Jones, R., Rye, D., Ferrante, R. J., Hersch, S. M. & Li, X. J. (1999) J. Neurosci. 19, 2522-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies, S. W., Turmaine, M., Cozens, B. A., DiFiglia, M., Sharp, A. H., Ross, C. A., Scherzinger, E., Wanker, E. E., Mangiarini, L. & Bates, G. P. (1997) Cell 90, 537-548. [DOI] [PubMed] [Google Scholar]
- 3.Slow, E. J., van Raamsdonk, J., Rogers, D., Coleman, S. H., Graham, R. K., Deng, Y., Oh, R., Bissada, N., Hossain, S. M., Yang, Y. Z., et al. (2003) Hum. Mol. Genet. 12, 1555-1567. [DOI] [PubMed] [Google Scholar]
- 4.Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R. & Finkbeiner, S. (2004) Nature 431, 805-810. [DOI] [PubMed] [Google Scholar]
- 5.Kim, M., Lee, H. S., Laforet, G., McIntyre, C., Martin, E. J., Chang, P., Kim, T. W., Williams, M., Reddy, P. H., Tagle, D., et al. (1999) J. Neurosci. 19, 964-973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., Scaravilli, F., Easton, D. F., Duden, R., O'Kane, C. J., et al. (2004) Nat. Genet. 36, 585-595. [DOI] [PubMed] [Google Scholar]
- 7.Smith, D. L., Portier, R., Woodman, B., Hockly, E., Mahal, A., Klunk, W. E., Li, X. J., Wanker, E., Murray, K. D. & Bates, G. P. (2001) Neurobiol. Dis. 8, 1017-1026. [DOI] [PubMed] [Google Scholar]
- 8.Apostol, B. L., Kazantsev, A., Raffioni, S., Illes, K., Pallos, J., Bodai, L., Slepko, N., Bear, J. E., Gertler, F. B., Hersch, S., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 5950-5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrante, R. J., Andreassen, O. A., Dedeoglu, A., Ferrante, K. L., Jenkins, B. G., Hersch, S. M. & Beal, M. F. (2002) J. Neurosci. 22, 1592-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N. R., Doi, H., Kurosawa, M., Nekooki, M. & Nukina, N. (2004) Nat. Med. 10, 148-154. [DOI] [PubMed] [Google Scholar]
- 11.Saudou, F., Finkbeiner, S., Devys, D. & Greenberg, M. E. (1998) Cell 95, 55-66. [DOI] [PubMed] [Google Scholar]
- 12.Hodgson, J. G., Agopyan, N., Gutekunst, C. A., Leavitt, B. R., LePiane, F., Singaraja, R., Smith, D. J., Bissada, N., McCutcheon, K., Nasir, J., et al. (1999) Neuron 23, 181-192. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg, Y. P., Andrew, S. E., Clarke, L. A. & Hayden, M. R. (1993) Hum. Mol. Genet. 2, 635-636. [DOI] [PubMed] [Google Scholar]
- 14.Chen, S. & Wetzel, R. (2001) Protein Sci. 10, 887-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berghorn, K. A., Bonnett, J. H. & Hoffman, G. E. (1994) J. Histochem. Cytochem. 42, 1635-1642. [DOI] [PubMed] [Google Scholar]
- 16.Zeron, M. M., Hansson, O., Chen, N., Wellington, C. L., Leavitt, B. R., Brundin, P., Hayden, M. R. & Raymond, L. A. (2002) Neuron 33, 849-860. [DOI] [PubMed] [Google Scholar]
- 17.Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S. W., et al. (1996) Cell 87, 493-506. [DOI] [PubMed] [Google Scholar]
- 18.Hackam, A. S., Singaraja, R., Wellington, C. L., Metzler, M., McCutcheon, K., Zhang, T., Kalchman, M. & Hayden, M. R. (1998) J. Cell Biol. 141, 1097-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., McCutcheon, K., Singaraja, R., Kazemi-Esfarjani, P., Devon, R., Kim, S. U., et al. (1998) Nat. Genet. 18, 150-154. [DOI] [PubMed] [Google Scholar]
- 20.Van Raamsdonk, J. M., Pearson, J., Slow, E. J., Hossain, S. M., Leavitt, B. R. & Hayden, M. R. (2005) J. Neurosci. 25, 4169-4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klement, I. A., Skinner, P. J., Kaytor, M. D., Yi, H., Hersch, S. M., Clark, H. B., Zoghbi, H. Y. & Orr, H. T. (1998) Cell 95, 41-53. [DOI] [PubMed] [Google Scholar]
- 22.Cummings, C. J., Reinstein, E., Sun, Y., Antalffy, B., Jiang, Y., Ciechanover, A., Orr, H. T., Beaudet, A. L. & Zoghbi, H. Y. (1999) Neuron 24, 879-892. [DOI] [PubMed] [Google Scholar]
- 23.Bowman, A. B., Yoo, S. Y., Dantuma, N. P. & Zoghbi, H. Y. (2005) Hum. Mol. Genet. 14, 679-691. [DOI] [PubMed] [Google Scholar]
- 24.Zhang, X., Smith, D. L., Meriin, A. B., Engemann, S., Russel, D. E., Roark, M., Washington, S. L., Maxwell, M. M., Marsh, J. L., Thompson, L. M., et al. (2005) Proc. Natl. Acad. Sci. USA 102, 892-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klivenyi, P., Ferrante, R. J., Gardian, G., Browne, S., Chabrier, P. E. & Beal, M. F. (2003) J. Neurochem. 86, 267-272. [DOI] [PubMed] [Google Scholar]
- 26.Bates, G. (2003) Lancet 361, 1642-1644. [DOI] [PubMed] [Google Scholar]
- 27.Hockly, E., Cordery, P. M., Woodman, B., Mahal, A., van Dellen, A., Blakemore, C., Lewis, C. M., Hannan, A. J. & Bates, G. P. (2002) Ann. Neurol. 51, 235-242. [DOI] [PubMed] [Google Scholar]
- 28.Carter, R. J., Lione, L. A., Humby, T., Mangiarini, L., Mahal, A., Bates, G. P., Dunnett, S. B. & Morton, A. J. (1999) J. Neurosci. 19, 3248-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wellington, C. L. & Hayden, M. R. (1997) Curr. Opin. Neurol. 10, 291-298. [DOI] [PubMed] [Google Scholar]
- 30.Wellington, C., Ellerby, L., Leavitt, B., Roy, S., Nicholson, D. W. & Hayden, M. R. (2003) Clin. Neurosci. Res. 3, 129-139. [Google Scholar]
- 31.Yu, Z. X., Li, S. H., Evans, J., Pillarisetti, A., Li, H. & Li, X. J. (2003) J. Neurosci. 23, 2193-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iijima, K., Liu, H. P., Chiang, A. S., Hearn, S. A., Konsolaki, M. & Zhong, Y. (2004) Proc. Natl. Acad. Sci. USA 101, 6623-6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eriksen, J. L., Sagi, S. A., Smith, T. E., Weggen, S., Das, P., McLendon, D. C., Ozols, V. V., Jessing, K. W., Zavitz, K. H., Koo, E. H., et al. (2003) J. Clin. Invest. 112, 440-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emamian, E. S., Kaytor, M. D., Duvick, L. A., Zu, T., Tousey, S. K., Zoghbi, H. Y., Clark, H. B. & Orr, H. T. (2003) Neuron 38, 375-387. [DOI] [PubMed] [Google Scholar]
- 35.Katsuno, M., Adachi, H., Doyu, M., Minamiyama, M., Sang, C., Kobayashi, Y., Inukai, A. & Sobue, G. (2003) Nat. Med. 9, 768-773. [DOI] [PubMed] [Google Scholar]
- 36.Humbert, S., Bryson, E. A., Cordelieres, F. P., Connors, N. C., Datta, S. R., Finkbeiner, S., Greenberg, M. E. & Saudou, F. (2002) Dev. Cell 2, 831-837. [DOI] [PubMed] [Google Scholar]
- 37.Zuccato, C., Ciammola, A., Rigamonti, D., Leavitt, B. R., Goffredo, D., Conti, L., MacDonald, M. E., Friedlander, R. M., Silani, V., Hayden, M. R., et al. (2001) Science 293, 493-498. [DOI] [PubMed] [Google Scholar]
- 38.Gauthier, L. R., Charrin, B. C., Borrell-Pages, M., Dompierre, J. P., Rangone, H., Cordelieres, F. P., De Mey, J., MacDonald, M. E., Lessmann, V., Humbert, S., et al. (2004) Cell 118, 127-138. [DOI] [PubMed] [Google Scholar]
- 39.Zeron, M. M., Chen, N., Moshaver, A., Lee, A. T., Wellington, C. L., Hayden, M. R. & Raymond, L. A. (2001) Mol. Cell. Neurosci. 17, 41-53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}