

Abstract
Human XPF–ERCC1 is a DNA endonuclease that incises a damaged DNA strand on the 5′ side of a lesion during nucleotide excision repair and has additional role(s) in homologous recombination and DNA interstrand crosslink repair. We show that a truncated form of XPF lacking the N-terminal helicase-like domain in complex with ERCC1 exhibits a structure-specific endonuclease activity with similar specificity to that of full-length XPF–ERCC1. Two domains of ERCC1, a central domain and a C-terminal tandem helix–hairpin–helix (HhH2) dimerization domain, bind to ssDNA. The central domain of ERCC1 binds ssDNA/dsDNA junctions with a defined polarity, preferring a 5′ single-stranded overhang. The XPF–ERCC1 HhH2 domain heterodimer contains two independent ssDNA-binding surfaces, which are revealed by a crystal structure of the protein complex. A crystal structure of the central domain of ERCC1 shows its fold is strikingly similar to that of the nuclease domains of the archaeal Mus81/XPF homologs, despite very low sequence homology. A groove lined with basic and aromatic residues on the surface of ERCC1 has apparently been adapted to interact with ssDNA. On the basis of these crystallographic and biochemical studies, we propose a model in which XPF–ERCC1 recognizes a branched DNA substrate by binding the two ssDNA arms with the two HhH2 domains of XPF and ERCC1 and by binding the 5′-ssDNA arm with the central domain of ERCC1.
Branched DNA intermediates that arise during normal DNA metabolism are processed by structure-specific endonucleases, which cleave one DNA strand at a junction between double-stranded and single-stranded regions of DNA. These enzymes resolve Holliday junction intermediates during recombination, reconfigure stalled replication forks to enable replication restart, or remove segments of chemically modified DNA during damage repair (1). In vitro, structure-specific endonucleases of higher organisms cleave different types of DNA substrates, including four-way junctions, single-stranded flaps, or bubble-shaped regions of strand separation. Genetic inactivation of one of the nucleases can cause profound chromosomal instability and/or heightened susceptibility to cancer, highlighting their essential and nonredundant roles in normal cellular physiology.
The XPF/Mus81 family comprises a group of dimeric DNA endonucleases that cleave irregular DNA shapes arising during meiotic recombination, DNA replication, and nucleotide excision repair (NER) (2). The eukaryotic XPF and Mus81 proteins function as obligate heterodimers with ERCC1 and Eme1, respectively, but only XPF and Mus81 contain a functional nuclease active site (3, 4). Archaeal homologs of the XPF/Mus81 family function as homodimers and, hence, have nuclease domains in both subunits (5, 6). The biological function of the archaeal proteins, however, remains to be explored. Mus81-Eme1 nuclease functions in meiotic recombination and the restart of stalled replication forks (7) and preferentially cleaves 3′-flaps in dsDNA and nicked Holliday junctions resembling a stalled replication fork in vitro (8–10).
A well characterized member of this family is XPF–ERCC1. The main role of XPF-ERCC1 is in NER, where it is recruited to DNA lesions through interaction with NER damage-recognition proteins and makes the incision 5′ to the damaged site, leading to excision of a 24- to 32-mer oligonucleotide after concomitant incision 3′ to the damage by XPG (11). XPF-ERCC1 has additional, NER-independent roles in homologous recombination (12) and in the repair of interstrand DNA crosslinks (13–15). The profound growth retardation and very short life span of both ERCC1 and XPF knock-out mice (16, 17) contrasts with the classical phenotype of other NER gene knockouts (UV sensitivity and cancer predisposition), providing further evidence of XPF–ERCC1's pleiotropic biological functions.
Xeroderma pigmentosum patients belonging to the XP-F complementation group have reduced cellular levels of XPF and ERCC1 proteins, and, conversely, ERCC1-deficient cells show low levels of XPF, implying that formation of the heterodimeric complex stabilizes both proteins in vivo (18, 19). The interactions of XPF and ERCC1 subunits are mediated through C-terminal, tandem helix–hairpin–helix (HhH2) domains of both proteins (20) (Fig. 1), and small deletions at the C terminus of either protein result in a loss of activity (21). The XPF subunit harboring the nuclease active site (3) consists of three domains (22, 23) (Fig. 1). An N-terminal helicase-like domain of XPF (residues 4–457) is homologous to superfamily II helicases but lacks several key residues required for DNA unwinding activity. The helicase-like domain of XPF may contribute to DNA binding activity, although its function has not been examined in detail. The central nuclease domain of XPF (residues 656–813) harbors conserved metal-binding residues of a [V/I]ERKX3D motif as well as functionally important basic residues that presumably interact with DNA substrates (3). The C-terminal HhH2 domain of XPF (residues 837–905) specifically dimerizes with ERCC1 to form the functional nuclease and may additionally contribute to DNA binding (20, 24). The ERCC1 subunit consists of only two domains (Fig. 1), a conserved central domain (residues 96–214) predicted to be structurally similar to the XPF nuclease domain but devoid of residues characteristic of a nuclease domain (25). The C-terminal HhH2 domain of ERCC1 (residues 220–297) dimerizes with the equivalent domain in XPF and may bind to DNA.
Fig. 1.

Domain organization of XPF and ERCC1 and their homologs. The numbers designate homology-based domain boundaries for the human proteins. YRAH denotes the sequence in XPF that is similar to the DEAH sequence within conserved helicase motif II.
Crystal structures of the nuclease domain from Pyrococcus furiosus (5) and Aeropyrum pernix XPF homodimers complexed to dsDNA (26) show that the nuclease domains from both Mus81/XPF family members closely resemble the protein fold of type II restriction endonucleases (27). Based on this similarity and structures of DNA complexes with restriction enzymes, the interactions of XPF with a branched DNA substrate have been modeled (5, 26). The A. pernix XPF homodimer was crystallized with a short dsDNA lying outside of the active site and instead interacting with the HhH2 domain of one subunit (26). This interaction may position the 3′ flap of a DNA substrate in the active site of the nuclease domain at the junction between a 3′ flap and dsDNA. The other subunit of the A. pernix XPF dimer would then be oriented away from the site of cleavage (26).
The heterodimeric makeup and substrate specificity of mammalian XPF–ERCC1 distinguish this enzyme from the archaeal XPF nucleases. No archaeal homologs of ERCC1 have been identified, and ERCC1 has been proposed to evolve from the same gene as XPF as a result of gene duplication in lower eukaryotes (25). XPF–ERCC1 preferentially cleaves 3′ single-stranded tails at a junction with dsDNA (28, 29) and is poorly active toward the 3′ flap-type substrates preferred by Mus81-Eme1 and archaeal XPF proteins (6, 10). XPF–ERCC1 instead prefers to cleave either a DNA hairpin or the 3′ tail of a Y-shaped DNA known as a “splayed-arm” substrate (Fig. 7, which is published as supporting information on the PNAS web site) (29). In addition to stabilizing the XPF nuclease, ERCC1 probably contributes to DNA substrate selection by the nuclease. The central domain of ERCC1 (residues 91–118) also mediates an interaction with the XPA protein that is essential for NER (30–32).
We have determined crystal structures of both constituent domains of human ERCC1 and characterized their biochemical activities. Our experiments show that ERCC1's central domain, although resembling the nuclease fold of the archaeal XPF enzymes, binds to ssDNA and has basic and aromatic residues in place of a nuclease active site. The XPF–ERCC1 heterodimeric HhH2 domain also functions in ssDNA binding and contains two independent DNA-binding surfaces. The crystallographic models in combination with our enzymatic and DNA binding experiments suggest a model to explain the ssDNA/dsDNA junction-specific cleavage activity of XPF–ERCC1.
Materials and Methods
DNA Binding and Cleavage Activities. The oligonucleotide substrates used for DNA binding and nuclease cleavage experiments are shown in Table 1, which is published as supporting information on the PNAS web site, and Fig. 7. Oligos with a 5′ 6FAM fluorescent label were obtained from Integrated DNA Technologies (Coralville, IA) and unlabeled oligos were synthesized on an ABI394 instrument. All oligos were gel-purified by urea/PAGE then annealed by heating to 95°C and slowly cooling the mixture to room temperature in a 1:1.1 mixture of labeled and unlabeled oligonucleotides in an annealing buffer (10 mM Tris, pH 8.0/50 mM NaCl/0.05 mM EDTA). The DNA binding activity of XPFΔ655–ERCC1Δ95 and individual protein domains was measured by equilibrium fluorescence anisotropy and analyzed by following the methods described in detail in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. The DNA cleavage activity of full-length XPF–ERCC1 and XPFΔ655–ERCC1Δ95 was measured and analyzed as described in detail in Supporting Materials and Methods.
Protein Expression and Crystal Structure Determination. The subcloning of XPF and ERCC1 gene fragments, protein expression in Escherichia coli, and purification are described in Supporting Materials and Methods. Full-length XPF–ERCC1 was purified from overexpressing Sf9 insect cells as described in ref. 3. The central domain of ERCC1 (residues 96–230) and the XPF–ERCC1 HhH2 proteins were crystallized by hanging drop vapor diffusion, and the structures were determined by experimental phasing using mercury-derivatized crystals, as described in Supporting Materials and Methods. The crystallographic data statistics are shown in Table 2, which is published as supporting information on the PNAS web site, and the model coordinates for the central domain of ERCC1 [Protein Data Bank (PDB) ID code 2A1I) and the XPF–ERCC1 HhH2 heterodimer (PDB ID code 2A1J) have been deposited in the PDB.
Results and Discussion
A Minimal, Structure-Specific Nuclease Derived from XPF–ERCC1. The full-length human XPF–ERCC1 complex can only be purified in small quantities from HeLa cells or in recombinant form in baculovirus-infected Sf9 insect cells, whereas overexpression in E. coli leads to mostly aggregated protein (3). We therefore set out to identify minimal fragments of XPF and ERCC1 that retain the structure-specific DNA binding and cleavage activities of full-length XPF–ERCC1 that would also be suitable for crystallographic studies. A fragment of ERCC1 lacking 91 nonconserved, N-terminal residues is functional in both NER and interstrand DNA crosslink repair (25, 33), whereas deletion of just 5 residues at the C terminus of ERCC1 eliminates in vivo activity (21). Less is known about the function of the N-terminal helicase-like domain of XPF, or the 200 residue linker joining this domain to the nuclease domain (Fig. 1). Several crenarchaeal XPF proteins lack a helicase-like domain altogether (Fig. 1) (34), whereas euarchaeal XPF proteins have a functional helicase (35) appended to the nuclease and HhH2 domains. The helicase-like domain of mammalian XPF could support DNA binding activity.
We prepared N-terminally truncated human XPF lacking the helicase-like domain and adjoining linker, in combination with a fragment of ERCC1 spanning the conserved central and HhH2 domains (Fig. 1 and Fig. 8, which is published as supporting information on the PNAS web site) by coexpression of both subunits in E. coli. The purified ERCC1Δ95–XPFΔ655 protein is a structure-specific nuclease like full-length XPF–ERCC1, incising a hairpin DNA substrate at the junction between the 5′ end of the ssDNA and dsDNA (Figs. 2 and 7). More extensive deletions of the N terminus of ERCC1 coexpressed with XPFΔ655 caused protein aggregation (data not shown). We conclude that ERCC1Δ95–XPFΔ655 is the smallest functional mimic of XPF–ERCC1.
Fig. 2.
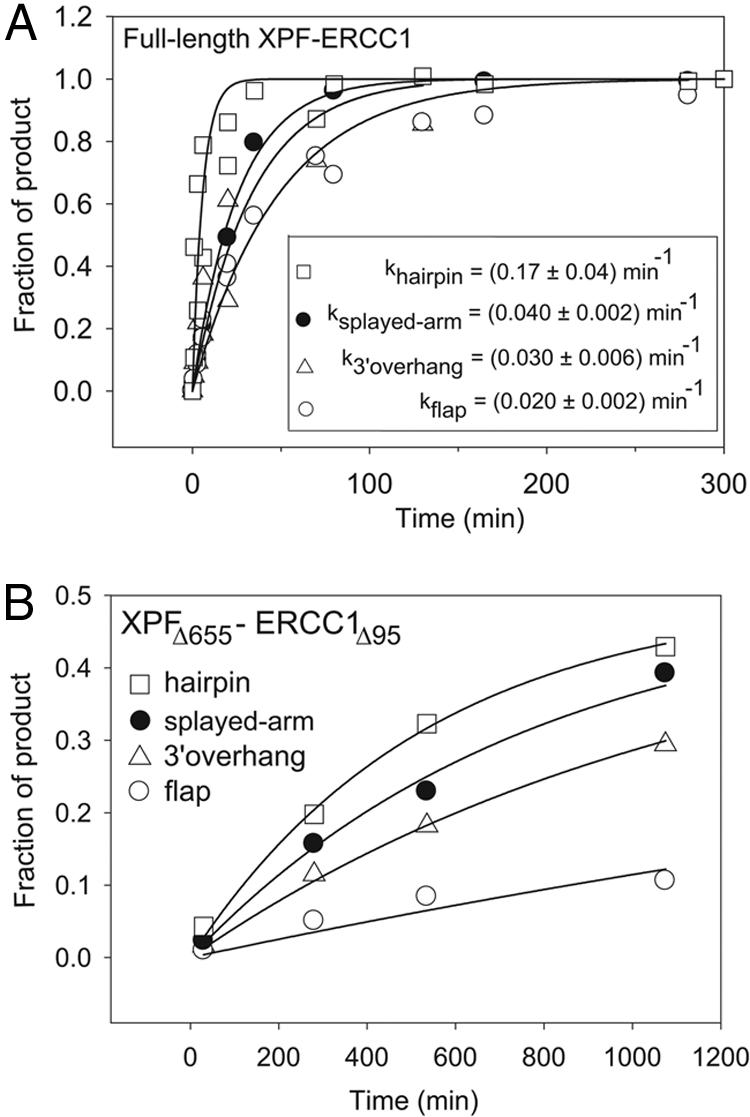
Kinetics of DNA cleavage by full-length XPF–ERCC1 (A) and XPFΔ655–ERCC1Δ95 (B). The fractions of major cleavage products in the overall DNA population are plotted as a function of reaction time. The DNA substrates are described in Fig 7. The data points represent the average of two independent experiments. (Inset) The best-fit kobs values for the full-length XPF–ERCC1 correspond to kcat. kobs values for XPFΔ655–ERCC1Δ95 should not be interpreted as kcat because of protein aggregation and are as follows: (0.30 ± 0.04) × 10–2 min–1 (hairpin), (0.14 ± 0.02) × 10–2 min–1 (splayed-arm), (0.08 ± 0.01) × 10–2 min–1 (3′ overhang), (0.04 ± 0.01) × 10–2 min–1 (3′ flap).
The DNA cleavage specificity of ERCC1Δ95–XPFΔ655 was compared to that of full-length human XPF–ERCC1 purified from Sf9 cells (Figs. 2 and 7). On a DNA hairpin substrate, ERCC1Δ95–XPFΔ655 cleaves specifically at the ssDNA/dsDNA junction (Fig. 7), albeit at a lower rate than full-length XPF–ERCC1. On a splayed-arm DNA, the truncated enzyme also generates a secondary cleavage product on the opposite DNA strand near the ssDNA/dsDNA junction (data not shown). Our results indicate that the helicase-like domain of XPF contributes strongly to the efficiency of DNA cleavage and modestly enhances cleavage specificity. The rates of DNA cleavage for various DNA substrates were determined with saturating concentrations of XPF–ERCC1 (Fig. 2A), whereas the truncated ERCC1Δ95–XPFΔ655 enzyme lost activity over the long time course of the experiment (Fig. 2B), and we were unable to achieve saturation. The loss of enzymatic activity may result from protein aggregation, as observed during purification in low-salt conditions, whereas higher salt concentrations decrease the DNA cleavage activity of full-length and truncated XPF–ERCC1 proteins.
ERCC1Δ95–XPFΔ655 exhibits the same rank order of cleavage rates for various DNA substrates as full-length XPF–ERCC1 (Figs. 2 A and B). The best substrate for both proteins is a DNA hairpin mimicking an NER bubble-type substrate (Figs. 2 and 7). A splayed-arm DNA with single-stranded 3′ and 5′ tails is cleaved faster than DNAs with a lone 3′ tail or a 3′ flap, emphasizing the importance of an unpaired 5′ tail for nuclease activity. Neither truncated nor full-length XPF–ERCC1 cleaved ssDNA under these conditions. This catalytic selectivity is different from that of eukaryotic Mus81-Eme1 and the archaeal XPF proteins, which preferentially cleave 3′ flaps embedded in dsDNA (10, 26). Our results indicate that ERCC1Δ95–XPFΔ655 retains all of the elements required for structure-specific DNA nuclease activity.
Crystal Structure of the Central Domain of ERCC1. A protein fragment spanning the central domain of ERCC1 (Fig. 1) was generated by proteolytic degradation during overexpression of ERCC1Δ95–XPFΔ655 in E. coli (Fig. 8). This ERCC1 fragment eluted separately from the complex during size exclusion chromatography, confirming that the central domain of ERCC1 does not stably interact with XPF (20). The amino acid sequence of ERCC1's central domain is weakly homologous to the nuclease domain of XPF (25), although ERCC1 lacks the active site residues of the nuclease (Fig. 9, which is published as supporting information on the PNAS web site). The function(s) of ERCC1's central domain had not been previously investigated.
We determined a crystal structure of the ERCC1 central domain (residues 96–214; Fig. 3) by using the single isomorphous replacement with anomalous scattering method at 1.9-Å resolution. The core structure consists of a six-stranded β-sheet flanked on both sides by α-helices, resembling the fold of type II restriction endonucleases (27, 36). Despite a sequence identity of only 11–14%, the structure of ERCC1's central domain superimposes very well on the nuclease domain of P. furiosus XPF [Hef (5)] (rms deviation = 1.34 Å for 81 Cα atoms) (Fig. 4) and A. pernix XPF (26) (rms deviation = 1.19 Å for 77 Cα atoms). A multiple sequence alignment of the ERCC1 proteins (Fig. 9) shows that the majority of universally conserved residues cluster in the hydrophobic core of ERCC1 or on the surface of a V-shaped groove (Fig. 3) analogous to the substrate binding site of the archaeal XPF endonucleases (5, 26) (Fig. 4). The basic and aromatic residues lining this groove (e.g., Arg-106, Arg-108, Arg-144, Arg-156, Phe-140, Tyr-145, and Tyr-152) (Figs. 4 and 9) could mediate the DNA-binding activity described below. In contrast, the corresponding surface of the P. furiosus and A. pernix XPF nucleases is populated by acidic, metal-binding active site residues (Fig. 4B) and the catalytic motif GDXnERKX3D (3).
Fig. 3.

Crystal structure and DNA binding of the central domain of ERCC1. (A) Cartoon/surface representation of the central domain of ERCC1. The solvent-exposed residues, absolutely conserved in the ERCC1 family (Fig. 8), are mapped as a yellow surface, and the less-conserved residues are displayed in pale green (see also Fig. 9). (B) The data for the equilibrium binding experiments with dsDNA (□), 3′ overhang (•), and 5′ overhang (○) are averages of two independent titrations. The theoretical curves were generated by using the KD values given in Table 1.
Fig. 4.

Structural (A and B) and electrostatic comparison (C and D) of the central domain of ERCC1 (A and C) with the nuclease domain of Hef nuclease (B and D) (5). (A and B) Basic and aromatic residues of ERCC1 proposed to interact with DNA and the residues required for cleavage activity of Hef nuclease (5) are shown in sticks and labeled in A and B, respectively (see also Fig. 9). (C and D) The electrostatic surface shows the difference between the positively charged (blue) DNA-binding groove of the central domain of ERCC1 (C) and the negatively charged (red) active site cleft of the Hef nuclease domain (D).
An extended linker (residues 215–219) (Fig. 1) connects the central domain of ERCC1 to the C-terminal HhH2 dimerization domain. Based on its exposed location and proteolytic sensitivity (Fig. 8), the interdomain linker is most likely unstructured and flexible, allowing the central and HhH2 domains to adopt different orientations. The conformation of the linker is stabilized by crystal packing interactions, as is one turn of an α-helix belonging to the C-terminal HhH2 domain described below.
Structure of the XPF–ERCC1 Heterodimeric HhH2 Complex. The C-terminal HhH2 domains of XPF and ERCC1, which are necessary and sufficient for dimerization (20), were crystallized, and a 2.7-Å structure of the complex was determined by using the multiwavelength anomalous dispersion method (Table 2). The heterodimer consists of two structurally similar, tandem HhH2 domains apposed in a pseudo twofold symmetrical arrangement (Fig. 5). The N-terminal α-helix of the ERCC1 HhH2 domain is also present in the structure of the ERCC1 central domain (Fig. 3), facilitating superposition of the two structures to reconstruct the entire conserved region of ERCC1 (minus nonconserved residues 1–95) functioning in DNA repair in vivo (21). The two HhH2 domains interact through an extensive network of hydrophobic interactions mediated by the N-terminal helices, a short helix in the middle of each subunit, and an extended segment at the C terminus of either subunit (Fig. 5). Several phenylalanine side chains interdigitate across the subunit interface, including Phe-293 of ERCC1, which inserts into a pocket formed by XPF residues Leu-841, Met-856, Val-859, and Ile-862 (Fig. 10, which is published as supporting information on the PNAS web site). ERCC1 lacking the last four residues still binds to XPF, whereas deletion of the next residue, Phe-293, eliminates XPF binding (20).
Fig. 5.

Crystal structure (A) and DNA binding (B) of XPF–ERCC1 heterodimeric HhH2 complex. (A) The labels indicate hairpin and adjacent basic residues structurally homologous to DNA binding residues in other HhH2 proteins (Fig. 9). The residues that are most likely to interact with DNA based on the conserved position as well as the nature of the side-chain are labeled in boldface type. Light brown indicates XPF, and green indicates ERCC1. (B) Binding titration of HhH2 heterodimer to a single-stranded 15-base oligomer ([DNA] = 4 μM) at 20 mM NaCl. In this regime [KD = 200 nM (Fig. 11) ≪ [DNA]], all added protein binds DNA until saturation is achieved. The saturation occurs at the molar ratio of two DNA molecules per one heterodimer.
The structure of the XPF–ERCC1 HhH2 heterodimer closely resembles the C-terminal HhH2 domains of A. pernix Mus81 (26) and E. coli UvrC (37) and RuvA (38, 39) proteins. The main-chain amides of hairpin residues within these HhH2 motifs donate hydrogen bonds to the DNA backbone, and nearby basic residues make electrostatic interactions with the DNA phosphates in DNA complexes with these proteins (26, 37–39). Structurally analogous residues of the XPF–ERCC1 HhH2 dimer are candidates for DNA binding (Fig. 5A), including ERCC1 residues Lys-243, Lys-247, Gly-276, and Gly-278 and XPF residues Gly-846, Lys-850, and Gly-878 (Fig. 9). The putative DNA binding residues cluster in two separate regions on the XPF–ERCC1 heterodimer, where they could function as independent DNA binding surfaces.
The Central Domain of ERCC1 Binds to ssDNA. Equilibrium binding experiments with structurally diverse DNAs show that the ERCC1 central domain and the XPF–ERCC1 HhH2 heterodimer are both single-stranded DNA binding proteins (Table 1). The DNA-binding activity of the ERCC1 central domain is markedly affected by the salt concentration of the buffer (Fig. 11, which is published as supporting information on the PNAS web site). In 15 mM NaCl buffer, the central domain binds with comparable affinity to ssDNA and dsDNA 40-mer oligomers, whereas binding to dsDNA is nearly undetectable at 80 mM NaCl. At this higher salt concentration, the central domain binds ssDNA in a saturable manner (KD = 10 μM). DNA binding affinity is practically independent of salt concentration above 50 mM NaCl (Fig. 11), with selective binding to ssDNA in the physiological range of salt concentration. These results highlight an electrostatic component of DNA binding interactions that is consistent with the positive electrostatic surface of the proposed DNA binding groove (Fig. 4C). Additional interactions with bases of single-stranded DNA may involve aromatic residues of the proposed binding groove.
The central domain of ERCC1 binds to ssDNA without regard to sequence but with regard to orientation. Its DNA binding affinity was measured for a series of dsDNAs containing 5′ and 3′ overhangs. The central domain binds with maximal affinity to single-stranded overhangs 15 nt or longer, whereas shorter overhangs bind less tightly (Table 1). The binding affinity was compared for duplex DNAs with a 5′ or 3′ overhang 15 nt in length (Fig. 3B) by using conditions (20 mM NaCl) that maximize binding affinity while maintaining strong discrimination against dsDNA binding (Fig. 11). The central domain of ERCC1 binds a 5′ overhang with 8-fold higher affinity than a 3′ overhang (Fig. 3B and Table 1, experiments 7 and 8). This difference in affinity shows that the central domain of ERCC1 binds ssDNA with a defined polarity. However, there is no indication of a favorable interaction with the ssDNA/dsDNA junction because the affinity for a long (40-mer) ssDNA is equal to the affinity for a 5′ tail abutting a duplex segment of DNA (Table 1). We conclude that the central domain of ERCC1 is a ssDNA-binding protein that is well suited for interactions with the 5′ tail of a branch-shaped DNA substrate (Fig. 6).
Fig. 6.

A model for XPFΔ655–ERCC1Δ95 binding to a splayed-arm DNA substrate. Light brown indicates XPF, and green indicates ERCC1. The cleavage site is shown by the orange sphere. The linker between the nuclease and the HhH2 domains of XPF is shown by the dashed line. The XPA binding region of ERCC1 (residues 99–118) (30) is shown in dark green.
DNA Binding by the XPF–ERCC1 HhH2 Complex. The HhH2 motif is a widely distributed DNA-binding motif that binds nonspecifically to dsDNA or ssDNA (24, 26, 37, 38). The XPF–ERCC1 HhH2 heterodimer binds to ssDNA with 6-fold higher affinity than does dsDNA in buffers of varying ionic strength (Fig. 11), and it binds to either DNA with higher affinity than does the central domain of ERCC1. Based on the crystal structure, two putative DNA binding sites were identified on the surface of the HhH2 heterodimer (Fig. 5A). The stoichiometry of binding to ssDNA was experimentally measured by titrating the HhH2 heterodimer at a fixed, high concentration (4 μM) of a 15-nt ssDNA under conditions in which KD = 0.2 μM for ssDNA (Fig. 5B). The data show that each HhH2 heterodimer binds two ssDNA oligomers (Fig. 5B), as is also suggested for the bacterial NER factor UvrC (37). We propose that the HhH2 dimerization domain of the XPF–ERCC1 heterodimer interacts with both unpaired strands of a bubble-type substrate for incision at the junction with dsDNA (Fig. 6).
A Model for DNA Substrate Recognition by XPF–ERCC1. As a structure-specific nuclease, XPF–ERCC1 cleaves DNA near a ssDNA/dsDNA junction at the 5′ end of the single-stranded region. The helicase-like domain of XPF is not required for DNA structure-specific cleavage activity (Figs. 2 and 7). The minimal ERCC1Δ95–XPFΔ655 nuclease cleaves a DNA hairpin and a splayed-arm DNA more readily than 3′ overhang and 3′ flap (Fig. 2), highlighting the importance of an unpaired DNA strand opposite the strand that is cleaved (Fig. 6). The eukaryotic XPF–ERCC1 heterodimeric nucleases have a different substrate preference than homodimeric archaeal XPF homologs (Fig. 4), which cleave 3′ flaps and are inactive on DNA bubble-like substrates (26).
Based on our structural and biochemical data, we have modeled ERCC1–XPF bound to a splayed-arm DNA substrate (Fig. 6). XPF nuclease domain, modeled from the structure of P. furiosus Hef (5), is bound at the dsDNA/ssDNA junction near the site of cleavage. Each single-stranded tail of the branched DNA substrate is bound to one HhH2 domain of the heterodimer, consistent with the stoichiometry of DNA binding by the HhH2 heterodimer (Fig. 5B). The DNA binding groove of the ERCC1 central domain engages the single-stranded 5′ tail, as suggested by the polarity of DNA binding (Fig. 3B). Although the isolated central domain of ERCC1 binds to ssDNA with modest affinity, it normally functions together with other DNA binding surfaces of the XPF–ERCC1 heterodimer that provide additional specificity. The yeast homolog of ERCC1, the Rad10 protein, also binds preferentially to ssDNA (40), and this conserved binding activity may position XPF–ERCC1-type nucleases on a branched DNA substrate (Fig. 6).
The central domain of ERCC1 does not stably interact with the nuclease domain of XPF (ref. 20 and this study), so we have modeled them separately on DNA. However, the archaeal XPF homodimers physically interact by means of their nuclease domains (5), and we cannot exclude a corresponding interaction between XPF and ERCC1 on DNA. Rad10, the functional homolog of ERCC1 in Saccharomyces cerevisiae, lacks a HhH2 domain altogether and dimerizes with its partner Rad1 through an interaction with the nuclease domain of Rad1 (41).
In conclusion, we demonstrate that the ERCC1 subunit of the XPF–ERCC1 heterodimer binds to ssDNA and propose that this activity is directed toward the uncleaved strand of a bubble-type substrate for NER. This interaction, together with the ssDNA binding activity of the tandem HhH2 domains, may explain the distinct substrate preference of XPF–ERCC1 in contrast to the homodimeric archaeal XPF homologs.
Supplementary Material
Acknowledgments
We thank Tapan Biswas, John Pascal, and Luis Brieba for helpful discussions and assistance. We are grateful to the staff of 19-ID synchrotron beamline at the Advanced Photon Source for help with x-ray data collection. This work was supported by National Institutes of Health Grant RO1GM52504, the Human Frontier Science Program, and the Swiss National Science Foundation. T.E. is the Hsien Wu and Daisy Yen Wu Professor of Biological Chemistry and Molecular Pharmacology at Harvard Medical School.
Author contributions: O.V.T. and T.E. designed research; O.V.T. performed research; O.V.T., J.H.E., and O.D.S. contributed new reagents/analytic tools; O.V.T., O.D.S., and T.E. analyzed data; and O.V.T., O.D.S., and T.E. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NER, nucleotide excision repair; HhH2, helix–hairpin–helix.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2A1I and 2A1J).
References
- 1.Nishino, T. & Morikawa, K. (2002) Oncogene 21, 9022–9032. [DOI] [PubMed] [Google Scholar]
- 2.Bardwell, A. J., Bardwell, L., Tomkinson, A. E. & Friedberg, E. C. (1994) Science 265, 2082–2085. [DOI] [PubMed] [Google Scholar]
- 3.Enzlin, J. H. & Scharer, O. D. (2002) EMBO J. 21, 2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heyer, W. D., Ehmsen, K. T. & Solinger, J. A. (2003) Trends Biochem. Sci. 28, 548–557. [DOI] [PubMed] [Google Scholar]
- 5.Nishino, T., Komori, K., Ishino, Y. & Morikawa, K. (2003) Structure (Cambridge, MA) 11, 445–457. [DOI] [PubMed] [Google Scholar]
- 6.Roberts, J. A. & White, M. F. (2005) J. Biol. Chem. 280, 5924–5928. [DOI] [PubMed] [Google Scholar]
- 7.Hollingsworth, N. M. & Brill, S. J. (2004) Genes Dev. 18, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, X. B., Melchionna, R., Denis, C. M., Gaillard, P. H., Blasina, A., Van de Weyer, I., Boddy, M. N., Russell, P., Vialard, J. & McGowan, C. H. (2001) Mol. Cell. 8, 1117–1127. [DOI] [PubMed] [Google Scholar]
- 9.Gaillard, P. H., Noguchi, E., Shanahan, P. & Russell, P. (2003) Mol. Cell. 12, 747–759. [DOI] [PubMed] [Google Scholar]
- 10.Bastin-Shanower, S. A., Fricke, W. M., Mullen, J. R. & Brill, S. J. (2003) Mol. Cell. Biol. 23, 3487–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Laat, W. L., Jaspers, N. G. & Hoeijmakers, J. H. (1999) Genes Dev. 13, 768–785. [DOI] [PubMed] [Google Scholar]
- 12.Niedernhofer, L. J., Essers, J., Weeda, G., Beverloo, B., de Wit, J., Muijtjens, M., Odijk, H., Hoeijmakers, J. H. & Kanaar, R. (2001) EMBO J. 20, 6540–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busch, D. B., van Vuuren, H., de Wit, J., Collins, A., Zdzienicka, M. Z., Mitchell, D. L., Brookman, K. W., Stefanini, M., Riboni, R., Thompson, L. H., et al. (1997) Mutat. Res. 383, 91–106. [DOI] [PubMed] [Google Scholar]
- 14.Kuraoka, I., Kobertz, W. R., Ariza, R. R., Biggerstaff, M., Essigmann, J. M. & Wood, R. D. (2000) J. Biol. Chem. 275, 26632–26636. [DOI] [PubMed] [Google Scholar]
- 15.Niedernhofer, L. J., Odijk, H., Budzowska, M., van Drunen, E., Maas, A., Theil, A. F., de Wit, J., Jaspers, N. G., Beverloo, H. B., Hoeijmakers, J. H. & Kanaar, R. (2004) Mol. Cell. Biol. 24, 5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McWhir, J., Selfridge, J., Harrison, D. J., Squires, S. & Melton, D. W. (1993) Nat. Genet. 5, 217–224. [DOI] [PubMed] [Google Scholar]
- 17.Weeda, G., Donker, I., de Wit, J., Morreau, H., Janssens, R., Vissers, C. J., Nigg, A., van Steeg, H., Bootsma, D. & Hoeijmakers, J. H. (1997) Curr. Biol. 7, 427–439. [DOI] [PubMed] [Google Scholar]
- 18.Biggerstaff, M., Szymkowski, D. E. & Wood, R. D. (1993) EMBO J. 12, 3685–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Vuuren, A. J., Appeldoorn, E., Odijk, H., Yasui, A., Jaspers, N. G., Bootsma, D. & Hoeijmakers, J. H. (1993) EMBO J. 12, 3693–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Laat, W. L., Sijbers, A. M., Odijk, H., Jaspers, N. G. & Hoeijmakers, J. H. (1998) Nucleic Acids Res. 26, 4146–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sijbers, A. M., van der Spek, P. J., Odijk, H., van den Berg, J., van Duin, M., Westerveld, A., Jaspers, N. G., Bootsma, D. & Hoeijmakers, J. H. (1996) Nucleic Acids Res. 24, 3370–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aravind, L., Walker, D. R. & Koonin, E. V. (1999) Nucleic Acids Res. 27, 1223–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sgouros, J., Gaillard, P. H. & Wood, R. D. (1999) Trends Biochem. Sci. 24, 95–97. [DOI] [PubMed] [Google Scholar]
- 24.Doherty, A. J., Serpell, L. C. & Ponting, C. P. (1996) Nucleic Acids Res. 24, 2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaillard, P. H. & Wood, R. D. (2001) Nucleic Acids Res. 29, 872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman, M., Murray-Rust, J., Lally, J., Rudolf, J., Fadden, A., Knowles, P. P., White, M. F. & McDonald, N. Q. (2005) EMBO J. 24, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aggarwal, A. K. (1995) Curr. Opin. Struct. Biol. 5, 11–19. [DOI] [PubMed] [Google Scholar]
- 28.Park, C. H., Bessho, T., Matsunaga, T. & Sancar, A. (1995) J. Biol. Chem. 270, 22657–22660. [DOI] [PubMed] [Google Scholar]
- 29.de Laat, W. L., Appeldoorn, E., Jaspers, N. G. & Hoeijmakers, J. H. (1998) J. Biol. Chem. 273, 7835–7842. [DOI] [PubMed] [Google Scholar]
- 30.Li, L., Peterson, C. A., Lu, X. & Legerski, R. J. (1995) Mol. Cell. Biol. 15, 1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, L., Peterson, C. A., Lu, X., Wei, P. & Legerski, R. J. (1999) Mol. Cell. Biol. 19, 5619–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saijo, M., Kuraoka, I., Masutani, C., Hanaoka, F. & Tanaka, K. (1996) Nucleic Acids Res. 24, 4719–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Duin, M., van den Tol, J., Warmerdam, P., Odijk, H., Meijer, D., Westerveld, A., Bootsma, D. & Hoeijmakers, J. H. (1988) Nucleic Acids Res. 16, 5305–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White, M. F. (2003) Biochem. Soc. Trans. 31, 690–693. [DOI] [PubMed] [Google Scholar]
- 35.Nishino, T., Komori, K., Tsuchiya, D., Ishino, Y. & Morikawa, K. (2005) Structure (Cambridge, MA) 13, 143–153. [DOI] [PubMed] [Google Scholar]
- 36.Holm, L. & Sander, C. (1998) Nucleic Acids Res. 26, 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh, S., Folkers, G. E., Bonvin, A. M., Boelens, R., Wechselberger, R., Niztayev, A. & Kaptein, R. (2002) EMBO J. 21, 6257–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hargreaves, D., Rice, D. W., Sedelnikova, S. E., Artymiuk, P. J., Lloyd, R. G. & Rafferty, J. B. (1998) Nat. Struct. Biol. 5, 441–446. [DOI] [PubMed] [Google Scholar]
- 39.Rafferty, J. B., Ingleston, S. M., Hargreaves, D., Artymiuk, P. J., Sharples, G. J., Lloyd, R. G. & Rice, D. W. (1998) J. Mol. Biol. 278, 105–116. [DOI] [PubMed] [Google Scholar]
- 40.Sung, P., Prakash, L. & Prakash, S. (1992) Nature 355, 743–745. [DOI] [PubMed] [Google Scholar]
- 41.Bardwell, A. J., Bardwell, L., Johnson, D. K. & Friedberg, E. C. (1993) Mol. Microbiol. 8, 1177–1188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.