

Abstract
Rho GTPases integrate control of cell structure and adhesion with downstream signaling events. In keratinocytes, RhoA is activated at early times of differentiation and plays an essential function in establishment of cell–cell adhesion. We report here that, surprisingly, Rho signaling suppresses downstream gene expression events associated with differentiation. Similar inhibitory effects are exerted by a specific Rho effector, CRIK (Citron kinase), which is selectively down-modulated with differentiation, thereby allowing the normal process to occur. The suppressing function of Rho/CRIK on differentiation is associated with induction of KyoT1/2, a LIM domain protein gene implicated in integrin-mediated processes and/or Notch signaling. Like activated Rho and CRIK, elevated KyoT1/2 expression suppresses differentiation. Thus, Rho signaling exerts an unexpectedly complex role in keratinocyte differentiation, which is coupled with induction of KyoT1/2, a LIM domain protein gene with a potentially important role in control of cell self renewal.
Keywords: epithelial cell differentiation, gene expression, Rho signaling, stem cell potential
The switch between epithelial cell growth and differentiation involves a close coordination between changes in cell structure and adhesion and downstream gene expression events. Calcium-induced differentiation of primary mouse keratinocytes in culture provides a well established model for the complex program of differentiation that occurs in vivo in the transition from the basal to upper epidermal layers (1). Essential structural aspects of this program include the establishment of E-cadherin-mediated cell–cell adhesion with associated reorganization of the actin and keratin cytoskeleton, followed by upwards migration of cells and loss of attachment to the substrate. Early biochemical events associated with the differentiation process trigger a signaling cascade culminating with several cell cycle and transcription regulatory changes leading to growth arrest and terminal differentiation. In addition to calcium-induced differentiation, loss of keratinocyte attachment to the substrate, either spontaneous or experimentally induced, provides a powerful stimulus for expression of the various biochemical markers of terminal differentiation, bypassing the ordered structural changes that accompany this process (1).
Small GTPases of the Rho family function as molecular switches for the integrated control of cell structure and adhesion with downstream signaling pathways and gene expression (2). Regulation of transcription by Rho can affect diverse biological functions in different cellular systems, such as cell cycle progression, differentiation, and cell fate determination during development (3, 4). Several direct Rho effectors have been identified, including kinases such as Rho kinase I and II (ROCK I/II or ROK β/α, p160ROCK), Citron kinase (CRIK), and PKN/PRK2, as well as scaffold proteins such as mDia 1/2 and rhotekin (5). ROCK and CRIK are structurally homologous kinases with a catalytic domain at their N terminus that is homologous to that of myotonic dystrophy protein kinase (6–8), whereas the catalytic domains of PKN and PRK2 are related to those of PKC family members (9). By contrast, mDia1/2 proteins are devoid of enzymatic activity and they function by a still poorly understood mechanism, although various proteins have been shown to interact with mDia1/2 that could account for effects on the cytoskeleton as well as transcription (10). Unlike other Rho effectors, CRIK has been implicated in cytokinesis (7, 11) and specific neuronal arborization processes (12), but not in regulation of gene expression.
Rho signaling is activated at early stages of the keratinocyte differentiation process and plays an essential function in the establishment of calcium-dependent cell–cell adhesion (13, 14). The function of Rho in this context is mediated, at least in part, by the PKN/PRK2-dependent activation of the Fyn tyrosine kinase with consequent tyrosine phosphorylation and strengthening of adherens junction complexes (14). Here, we have explored the possibility that Rho signaling plays a more general role in keratinocyte growth/differentiation control beyond the establishment of cell–cell adhesion. Unexpectedly, we have found that increased Rho activity suppresses expression of biochemical markers of terminal differentiation, particularly the “early” ones characteristic of the spinous layer (keratins 1 and 10), whereas inhibition of endogenous Rho signaling is by itself sufficient to induce expression of these markers even in cells that maintain active DNA replication. Such negative control of differentiation is likely mediated by the CRIK kinase, which is the only Rho effector to be selectively down-modulated during the keratinocyte differentiation process. At the transcription level, we have uncovered a link between Rho/CRIK signaling and control of the KyoT1/2 (FHL1) gene, which is highly expressed in stem cell populations of various tissues, including skin (15, 16), and codes for overlapping LIM domain proteins with a role in modulation of integrin-mediated processes (17) and/or Notch signaling (18).
Materials and Methods
Cells. Primary mouse keratinocytes were cultured and induced to differentiate by addition of CaCl2 to the medium (2 mM) or suspension culture as described (19). Primary human keratinocytes were cultured in serum-free keratinocyte medium (Gibco) supplemented with bovine pituitary extract (25 μg/ml) and epidermal growth factor (0.2 ng/ml). Bacterially expressed C3 transferase toxin or GST control proteins were purified and used as described (14). Y27632 was from Calbiochem.
Antibodies. Monoclonal antibodies against PRK2, PKN, p140mDia, and ROCK II were from BD Bioscience. Polyclonal antibodies against Citron (C-20) and ROCK I and monoclonal antibodies against p21WAF1/Cip1 and RasGap were from Santa Cruz Biotechnology. γ-Tubulin antibody was from Sigma. Antibodies against the various differentiation markers were from Covance Research Products (Denver, CO), except those against human involucrin (SY5), which were from Abcam. Rabbit antibodies against amino acids 168–194 of KyoT2 (FHL1C) (20) were a generous gift of Mary Waye (Chinese University of Hong Kong, Hong Kong). imagej (http://rsb.info.nih.gov/ij) was used for densitometry.
Plasmids and Recombinant Adenoviruses. The cDNA of the hemagglutinin-tagged ROCK II fragment from amino acid 1 to 543 was cloned into the pXJ40 vector (21) and was a kind gift of Andy McClatchey (Massachusetts General Hospital, Charlestown). Recombinant adenoviruses for activated RhoA (RhoV14) and PRK2 have been described (14). cDNAs of KyoT1 and KyoT2 were obtained in a Myc-tagged form by RT-PCR using the primers 5′-CCA CCA TGG AGC AGA AGC TGA TCT CCG AGG AGG ACC TGA ACT CGG AGA AGT TCG ACT GTC-3′ (forward) and 5′-GCC ATT TTA CAG GAC AGG AGC C-3′ (reverse). The Myc-tagged KyoT1 and KyoT2 cDNAs, as well as a cDNA for Flag-tagged CRIK-SK (8), were subcloned into the adenoviral shuttle vector pAdTrack-CMV (22). Recombinant adenoviruses were produced and used at a multiplicity of infection of 100, unless otherwise specified, as described (14). Expression of CRIK-SK and KyoT1 and KyoT2 proteins in adenovirally infected keratinocytes was confirmed by immunoblotting with anti-Flag and anti-Myc antibodies, respectively.
Real-Time RT-PCR and Microarray Analysis. One microgram of total RNA was used for cDNA preparation and real-time PCR analysis as described (23). Oligonucleotide primers are listed in Supporting Text, which is published as supporting information on the PNAS web site.
Total RNA from primary mouse keratinocytes infected with adeno-GFP, adeno-RhoV14, or adeno-CRIK-SK for 48 h was used for biotin-labeled cRNA probe preparation and hybridization to Affymetrix U74A gene chips as described (23). cRNA probes from each condition were tested in duplicate.
Results
Rho Signaling Down-Regulates Expression of Keratinocyte Terminal Differentiation Markers, with No Effects on the Cell Cycle. Calcium-induced differentiation of mouse primary keratinocytes provides a well established model for the complex program of differentiation that occurs in vivo in the transition from the basal to upper epidermal layers (1). Rho activity is induced at early times of calcium-induced keratinocyte differentiation and is required for the establishment of cell–cell adhesion associated with this process (13, 14). To assess whether Rho activity is involved in other aspects of keratinocyte differentiation besides these early structural events, we adopted two complementary approaches. In the first, to suppress endogenous Rho activity, cells were treated with the exoenzyme C3, a toxin produced by Clostridium botulinum that selectively inhibits RhoA, -B, and -C, but not other Rho/Rac family members. Treatment of growing keratinocytes with C3 did not affect the proliferative activity of these cells as assessed by BrdUrd labeling assays (data not shown). However, immunoblot as well as real-time RT-PCR analysis revealed that C3 treatment was, by itself, sufficient to induce strong expression of keratins 1 and 10 (K1 and K10), two “early” differentiation markers of the spinous epidermal layer (Fig. 1 A and B). By contrast, levels of involucrin and filaggrin, which are expressed at later stages of differentiation (early and late granular layers, respectively), were little or not induced (Fig. 1 A and B).
Fig. 1.

Rho signaling negatively regulates keratinocyte differentiation marker expression. (A) Mouse primary keratinocytes under growing conditions were treated with the C3 toxin as a GST-fusion product or GST control as described (14), and further incubated in growth medium or induced to differentiate by the addition of CaCl2 (2 mM) for 7 h. Total protein extracts were analyzed by immunoblotting with antibodies against the indicated proteins. Filaggrin is produced as a high molecular precursor, profilaggrin, which is converted into multiple products migrating as a diffuse set of bands on a gel. Keratin 5 is constitutively expressed in cultured keratinocytes irrespective of their growing or differentiating conditions and was used as equal loading control. (B) Mouse primary keratinocytes were treated with the C3 toxin or GST control proteins as in A, followed by total RNA preparation and real-time RT-PCR analysis for the indicated markers. Values are expressed as relative mRNA levels after normalization for GAPDH mRNA expression. (C) Mouse primary keratinocytes were infected with an adenovirus expressing an activated form of RhoA (RhoV14) or GFP control (GFP) as described (14), and either kept under growing conditions or induced to differentiate by high calcium concentrations for 9 h, followed by immunoblot analysis for the indicated markers. (D) Keratinocytes were treated with the C3 and GST proteins, or infected with the adenoviruses indicated above in parallel with an adenovirus expressing CRIK-SK (8), plus/minus calcium treatment for the last 9 h of the experiment. Levels of p21WAF1/Cip1 expression were determined at the RNA level, by real-time PCR analysis with p21WAF1/Cip1 specific primers and normalization for GAPDH expression (Upper). Alternatively, total protein extracts were analyzed by immunoblotting with antibodies against p21 and RasGAP as an equal loading control (Lower).
As a second approach, to evaluate the effects of increased Rho activity on keratinocyte differentiation, we expressed a constitutively active form of RhoA (RhoV14) by adenoviral infection. Opposite to the C3 effects, activated RhoA suppressed expression of the K1 marker in keratinocytes induced to differentiate by elevated extracellular calcium concentrations, exerting a similar although lesser inhibition on filaggrin (Fig. 1C). Suppression of K1 and filaggrin expression was also exerted by activated RhoA in keratinocytes induced to differentiate by suspension culture, as shown further below (see Fig. 5F). In the case of involucrin, significant down-modulation was observed in most but not all cases (four of six independent experiments) (Fig. 1C and data not shown); for this reason, further analysis was focused mostly on the other markers.
Fig. 5.

KyoT1/2 gene expression is up-regulated by the Rho/CRIK pathway and suppresses differentiation. (A) Keratinocytes were treated with the GST-C3 fusion protein or GST control for 9 h, and total RNA was analyzed by real-time RT-PCR with primers that recognize a common region of the KyoT1/2 transcript, as well as with primers specific for either one of these forms. The same RNAs were also analyzed by real-time RT-PCR for levels of K10 and K1 expression. (B) Keratinocytes were infected with the RhoV14, CRIK-SK, and GFP adenoviruses for 48 h, and total RNA was analyzed for levels of KyoT1 and KyoT2 as in the previous panel. To evaluate effects on K1 and K10 expression, a similar parallel analysis was performed on keratinocytes induced to differentiate by 9 h of calcium treatment. (C) Keratinocytes were infected with the RhoV14, CRIK-SK, and GFP adenoviruses, and total cell extracts were analyzed, in parallel with extracts from C2C12 myoblasts, by immunoblotting with antibodies against the KyoT2 protein. Immunoblotting for RasGAP was used as equal loading control. The larger size protein recognized by the anti-KyoT2 antibodies in the CRIK-SK expressing cells is indicated by an asterisk. (D and E) Mouse primary keratinocytes were infected with recombinant adenoviruses expressing the KyoT1 or KyoT2 proteins or GFP control for 48 h, and either kept under growing conditions or induced to differentiate by calcium for the last 10 h of the experiment. Total cell extracts were analyzed by immunoblotting with antibodies against the indicated differentiation markers (D) or against p21WAF1/Cip1 and RasGAP as equal loading control (E). (F) Primary keratinocytes were infected with the same adenoviruses as in the previous experiments in parallel with Ad-RhoV14 and Ad-CRIK-SK. Twenty-four hours after infection, cells were trypsinized and induced to differentiate by culture in suspension for another 12 h. Total cell extracts were analyzed by immunoblotting.
It has been shown that in primary rodent fibroblasts Rho activation suppresses the induction of the cyclin/CDK inhibitor p21WAF1/Cip1 by oncogenic Ras, thereby promoting transformation (24). Induction of p21WAF1/Cip1 expression is one of the early cell cycle regulatory events that contribute to cell cycle withdrawal of differentiating keratinocytes (25). Although C3 treatment had little or no effect on levels of p21WAF1/Cip1 expression, expression of activated Rho suppressed induction of this CKI in differentiating cells, as assessed by both immunoblot and quantitative RT-PCR analysis (Fig. 1D). However, activated Rho expression was not sufficient to prevent differentiation-associated growth arrest (data not shown), consistent with this process being accompanied by other cell cycle inhibitory events, in addition to increased p21 expression (25).
Expression of CRIK Is Selectively Down-Modulated in Keratinocyte Differentiation, Whereas Sustained CRIK Activity Counteracts this Process. Although endogenous Rho is activated with differentiation (14), the above results indicate that sustained Rho activity can play a suppressive rather than enhancing function in this process. A possible explanation for this apparent paradox is that one or more effectors that mediate the inhibitory function of Rho on differentiation are selectively down-modulated during this process. Immunoblot analysis of keratinocytes under growing versus differentiating conditions revealed that, among all Rho effectors that were examined, the CRIK kinase was selectively down-modulated a few hours after induction of differentiation (Fig. 2A). Expression of the other effectors did not change or, in the case of PKN and p140mDia, appeared to increase (Fig. 2A). Down-modulation of CRIK expression was confirmed at the mRNA level by Northern blotting (Fig. 2B). In parallel with the in vitro results, immunofluorescence analysis showed that, even in vivo, the CRIK kinase is preferentially expressed in growing keratinocytes of the basal layer, whereas its expression decreases progressively in the outer layers (Fig. 2C).
Fig. 2.

CRIK expression is down-modulated with differentiation. (A) Expression of different Rho effectors was assessed in keratinocytes under growing conditions and at various times (hours) of calcium-induced differentiation by immunoblot analysis with antibodies against the indicated proteins. Immunoblotting for RasGAP was used as equal loading control. (B) mRNA levels of CRIK were evaluated in keratinocytes under growing versus differentiating conditions by Northern blot analysis with a CRIK specific probe as previously described (8). The same membrane was stripped and rehybridized with a GAPDH probe as equal loading control. (C) Frozen skin sections from newborn mice were analyzed by immunofluorescence with anti-CRIK rabbit antibodies (Upper) or affinity-purified Rabbit serum as nonimmune control (Lower).
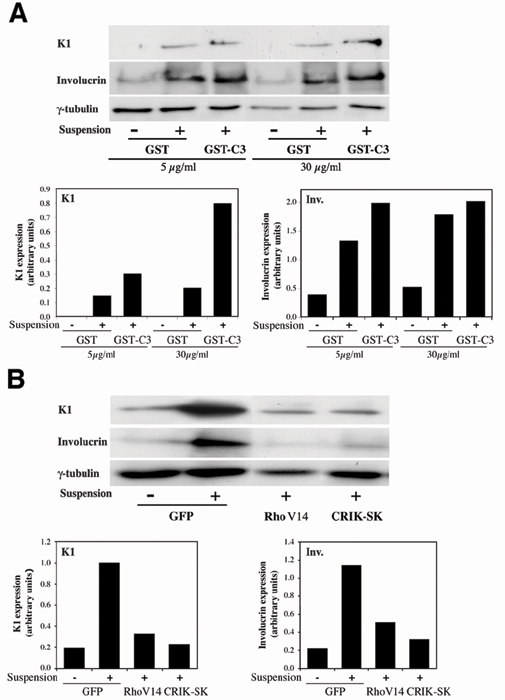
To test whether down-modulation of CRIK activity is required for differentiation, we constructed a recombinant adenovirus expressing the “shorter” cDNA form of CRIK, which is expressed in keratinocytes alongside the full-length form and codes for the constitutively active kinase domain (CRIK-SK) (8). Expression of CRIK-SK inhibited substantially K1 and filaggrin expression in keratinocytes induced to differentiate by either calcium (Fig. 3A Left) or suspension culture (as shown in Fig. 5F). On the other hand, differentiation maker expression was not affected by overexpression of the PRK2 kinase, the Rho effector involved in cell–cell junction formation (14) (Fig. 3A Right). As with Rho, activated CRIK expression prevented the increase of p21WAF1/Cip1 expression associated with differentiation (Fig. 1D).
Fig. 3.

CRIK activity negatively regulates keratinocyte differentiation marker expression. (A) Keratinocytes were infected with recombinant adenoviruses expressing the CRIK-SK or PRK2 kinases (14) or GFP control for 48 h and either kept under growing conditions or induced to differentiate by calcium for the indicated times (hours). Total cell extracts were analyzed by immunoblotting with antibodies against the indicated proteins. (B) Primary keratinocytes derived from CRIK–/– mice and wild-type littermate controls were infected with the RhoV14 or GFP adenoviruses (multiplicity of infection, 10) for 48 h and either kept under growing conditions or induced to differentiate by calcium for the last 12 h of the experiment. Differentiation marker expression was monitored by immunoblot analysis as in the A.
To assess the extent to which endogenous CRIK mediates the inhibitory effects of Rho on differentiation, primary keratinocytes were prepared from mice with a disruption of the CRIK gene and littermate wild-type controls. CRIK-deficient mice die before adulthood and show defective neurogenesis due to massive apoptosis as a consequence of impaired cytokinesis, but form an apparently normal skin (11). Even when cultured under growing conditions in medium at low calcium concentrations, a fraction of keratinocytes spontaneously differentiates, eventually detaching from the dish. The basal levels of K1 expression under low calcium conditions were found to be higher in CRIK–/– keratinocytes than in the controls, and a more pronounced increase of this marker, as well as filaggrin, was observed in the knockout cells upon induction of differentiation (Fig. 3B). Importantly, induction of these markers was suppressed by activated Rho expression to a lesser extent in the CRIK–/– than wild-type keratinocytes (Fig. 3B).
The CRIK and ROCK Kinases Exert Overlapping Compensatory Effects as Negative Regulators of Differentiation. The inhibitory effects of activated Rho on differentiation were reduced, but not completely suppressed, in CRIK–/– keratinocytes, suggesting that other Rho effectors could compensate for lack of CRIK function upon Rho activation. The ROCK kinases share a significant degree of homology with CRIK that extends beyond the N terminus catalytic domain (8) and, although not down-modulated with differentiation, they could still exert negative effects on this process. To test this possibility, keratinocytes were transfected with a vector expressing the constitutively active catalytic domain of ROCK II together with an expression vector for GFP, and were induced to differentiate by culture in suspension. Immunoblot analysis of transfected cells, purified by sorting for GFP expression, showed that activated ROCK II can significantly suppress K1 and filaggrin expression (Fig. 4A). To assess the function of endogenous ROCK, keratinocytes were treated with Y27632, a widely used inhibitor with much greater specificity for the ROCK than CRIK kinases, that allows to discriminate between the two (26). Y27632 treatment caused an increase of K1 and filaggrin expression even in keratinocytes under basal growing conditions, and a higher expression of these markers than in the controls upon induction of differentiation by calcium or suspension culture (Fig. 4 B and C).
Fig. 4.

ROCK and CRIK kinases exert overlapping compensatory effects as negative regulators of differentiation. (A) Primary keratinocytes were transfected with an expression vector for a constitutively active form of ROCK II (amino acids 1–543) or empty vector control alongside an expression vector for GFP and induced to differentiate by culture in suspension for 24 h. Transfected GFP-positive cells were purified by sorting, and total cell extracts were analyzed by immunoblotting with antibodies against the indicated proteins (Fil, Filaggrin). (B) Keratinocytes were treated with the ROCK inhibitor Y27632 (5 μM) for 24 h and either kept under growing conditions or induced to differentiate by calcium for the indicated times (hours) before termination of the experiment. Total cell extracts were analyzed by immunoblotting as before. (C) Keratinocytes were treated with Y27632 at the indicated concentrations for 12 h followed by induction of differentiation by suspension culture for additional 12 h. Total cell extracts were analyzed by immunoblotting. (D) Keratinocytes were infected with recombinant adenoviruses expressing RhoV14 or GFP control for 48 h, with induction of differentiation by calcium for the last 12 h of the experiment. Cells were either untreated or treated with Y27632 (5 μM) for the indicated times (hours) before termination of the experiment. Samples were analyzed by immunoblotting with antibodies against the keratin 1 and 5 markers. (E) Keratinocytes from CRIK–/– mice (KO) and wild-type littermate controls (WT) were infected with the GFP and RhoV14 adenoviruses (multiplicity of infection, 10) for 48 h, and either kept under growing conditions or induced to differentiate by calcium for the last 12 h of the experiment. Some cultures were also concomitantly treated with Y27632 (5 μM) for the last 24 h of the experiment, as indicated. Expression of the K1 and K5 proteins was monitored by immunoblotting (Left). Data were quantified by densitometry and expressed as arbitrary units after normalization for levels of K5 expression (Right).
To assess the extent to which the inhibitory effects of Rho on differentiation are mediated by endogenous ROCK, activated Rho was expressed in keratinocytes that were concomitantly treated with the Y27632 compound. As shown in Fig. 4D, suppression of K1 expression by activated Rho was counteracted by the ROCK inhibitor, but only to a limited extent. To assess the possible complementary effects of the ROCK and CRIK kinases, the same experiment was repeated with primary keratinocytes derived from CRIK knockout mice in parallel with littermate controls. Suppression of K1 expression by activated Rho was prevented to a large extent in Y27632-treated CRIK–/– keratinocytes, whereas, as before, a more limited reversal of Rho inhibition was observed in Y27632-treated wild-type keratinocytes or CRIK–/– cells that were not treated with this compound (Fig. 4E).
Negative Regulation of Differentiation Marker Expression by Rho/CRIK Signaling Extends to Human Keratinocytes. A recent report indicated that, in human primary keratinocytes induced to differentiate in suspension, inhibition of Rho activity by C3 treatment can suppress differentiation, as assessed by involucrin expression, whereas enhancement of RhoA activity exerts opposite inducing effects (27). The apparently conflicting observations with our present results may be due to the use of keratinocytes of human versus mouse origin, different culture and differentiation-inducing conditions, or the analysis of a marker, involucrin, which may be controlled differently from the others. To assess these possibilities, primary human keratinocytes were induced to differentiate by suspension culture as in the previous studies, and the effects of inhibited or increased RhoA activity were evaluated. Induction of the K1 marker in human keratinocytes in suspension was enhanced by treatment with C3 toxin at the concentration used in all of our experiments, and it was also slightly increased at the lower concentration used in the other study (27) (Fig. 7A, which is published as supporting information on the PNAS web site). Induction of involucrin in these cells was either unaffected or slightly increased by the C3 toxin at either concentration (Fig. 7A). Conversely, expression of K1 in differentiating human keratinocytes was suppressed by activated RhoA or CRIK expression, and similar inhibition was also seen for involucrin (Fig. 7B).
Thus, overall, our data are consistent with differentiation being negatively regulated by RhoA/CRIK signaling in both mouse and human keratinocytes, with some possible differences existing at the level of individual markers and/or treatment conditions.
Rho/CRIK Signaling Modulates a Global Program of Gene Expression Related to Keratinocyte Growth and Differentiation. One endpoint of Rho activation is control of gene expression, which can explain its cell type-specific effects on differentiation. To gain further insights into the role of this pathway in keratinocytes, we first analyzed the global pattern of gene expression in keratinocytes in which Rho activity was suppressed by treatment with the C3 toxin. By microarray hybridization, we found 166 up-regulated genes (Z score >+3) and 109 genes down-regulated (Z score < –3) (Table 1, which is published as supporting information on the PNAS web site). Among the up-regulated genes were those for several terminal differentiation markers, whereas integrin β1, whose expression is suppressed with differentiation (1), was among the down-modulated genes. A number of genes with a positive role in the cell cycle appeared also to be suppressed in the C3-treated cells, including cyclin D1, whose regulation by the Rho pathway in other cell types has been reported (28). c-Jun, a key transcription factor with a positive role in keratinocyte differentiation (1), was also induced as a consequence of Rho inhibition.
We next assessed which genes are modulated in keratinocytes by increased Rho and/or CRIK activity even before induction of differentiation, in a way that may explain their inhibitory effects on this process. Genes that are oppositely regulated by Rho/CRIK activation and Rho suppression would be the most interesting, because they might explain the opposite effects on differentiation. A relatively small number of genes were found that meet this criterion (Table 2, which is published as supporting information on the PNAS web site), one of which with a known cell regulatory function that, as discussed further below, could be of relevance in our context: KyoT1/2.
Rho/CRIK Signaling Controls KyoT Expression, a LIM Domain Protein Gene Involved in Growth/Differentiation. KyoT1/2 is the murine counterpart of the human FHL1 gene, and codes for overlapping LIM domain proteins that have been previously implicated in early stages of myoblast differentiation (17). Real-time RT-PCR confirmed that, in contrast to the induction of K1 and K10, transcripts for both KyoT1 and -2 are suppressed by C3 treatment and are induced in keratinocytes by expression of activated Rho and CRIK (Fig. 5 A and B). Immunoblotting of total cell extracts with antibodies against KyoT2 showed detectable expression of this protein in control keratinocytes as well as in C2C12 myoblasts, as reported for skeletal muscle and heart (Fig. 5C and ref. 20). KyoT2 protein levels were induced by activated Rho expression, whereas CRIK caused the appearance of a higher molecular weight form that may correspond to an additional product of differential splicing retaining the KyoT2-specific domain, as reported for human cells (20), and/or result from further posttranslational modification (Fig. 5C).
To assess whether increased KyoT1 and/or KyoT2 expression can have a direct impact on keratinocyte differentiation, we constructed recombinant adenoviruses expressing the individual proteins in comparable amounts. Infection of cells with either virus suppressed K1 and filaggrin expression in keratinocytes induced to differentiate by increased extracellular calcium, and similar inhibitory effects were observed on p21WAF1/Cip1 expression (Fig. 5 D and E). An even more drastic suppression of terminal differentiation markers, similar to that exerted by activated RhoA and CRIK expression, was found in keratinocytes induced to differentiate in suspension (Fig. 5F), whereas p21WAF1/Cip1 is already spontaneously down-modulated under these conditions (19).
Discussion
The keratinocyte differentiation program is a complex process with cell cycle regulatory events as well as sequential induction of terminal differentiation markers of overlying epidermal layers. Our present findings are overall consistent with a dual mode of Rho function in this process (Fig. 6). On one hand, activation of this pathway plays a positive role at very early times, promoting establishment of cell–cell adhesion through activation of a specific effector, the PKN/PRK2 kinase (14). On the other hand, Rho signaling can unexpectedly suppress downstream gene expression events associated with differentiation. Such inhibitory effects are exerted by a specific Rho effector, CRIK (Citron kinase), which is selectively down-modulated with differentiation, thereby allowing the normal process to occur. Sustained activation of the Rho/CRIK pathway may be of significance in vivo under conditions such as wound healing, where the normal differentiation process is arrested and/or reversed.
Fig. 6.

Diagrammatic illustration of the dual mode of Rho function in keratinocyte differentiation. As discussed in the text, available data are consistent with a model whereby activation of the RhoA pathway plays a positive role at early times of keratinocyte differentiation, promoting establishment of cell–cell adhesion through activation of a specific effector, the PKN/PRK2 kinase. Rho signaling can have a second suppressive function on downstream gene expression events associated with differentiation. Such inhibitory function is mediated by a specific Rho effector, CRIK (Citron kinase), which is selectively down-modulated with differentiation, through an as yet uncharacterized mechanism, thereby allowing the normal process to occur.
We have shown that activated Rho and CRIK expression inhibit p21 as well as K1, K10, and filaggrin expression, whereas these differentiation markers, but not p21, are induced by suppression of Rho, CRIK, and/or ROCK signaling. Involucrin expression, which is less consistently induced in differentiating keratinocytes in culture, was less affected by modulation of Rho-CRIK signaling. An apparently conflicting report that increased Rho signaling promotes keratinocyte differentiation was based mostly on evaluation of involucrin expression (27). Additionally, the previous studies examined only primary keratinocytes of human origin, where, unlike in calcium-treated mouse keratinocytes (14), activation of Rho signaling does not take place (27). However, given the positive role of Rho in promoting keratinocyte cell–cell adhesion, it remains possible that, under certain conditions, activation of this pathway can also serve to promote rather than suppress differentiation marker expression. This dual function of Rho signaling will have to be ultimately dissected in vivo, under conditions of normal versus altered homeostasis.
Although functionally overlapping with ROCK, the CRIK kinase is the only Rho effector that is down-modulated with differentiation, suggesting that it plays the most significant physiological role in negative control of this process. Within this context, a downstream transcriptional target under positive control of Rho/CRIK signaling is KyoT1/2. KyoT1/2 is the murine counterpart of the human FHL1 gene, and codes for partially overlapping LIM domain proteins. KyoT1 contains “four and a half” LIM domains, prevalently localizes to the actin cytoskeleton and focal adhesions, and interferes with integrin-mediated processes (29), whereas KyoT2 is mostly nuclear and contains the first “two and a half” LIM domains of KyoT1, followed by a short stretch of amino acids that bind and inactivate the RBP-Jκ DNA binding protein, the main mediator of Notch effects on transcription (18). Both mechanisms may impinge on keratinocyte differentiation, as indicated by the similar inhibitory effects exerted by the separate expression of the KyoT1 and -2 proteins. Alternatively, suppression of differentiation may be the result of a common function of the first “two and a half” LIM domains shared by the two proteins.
In addition to being more highly expressed in keratinocyte stem cell populations of the bulge region (16), KyoT1/2 expression is selectively elevated in squamous carcinoma cells as opposed to other cancer cell lines (30). A tempting speculation for future studies is that the complex role of Rho signaling in cell fate determination and tumorigenesis, in keratinocytes and possibly other cell types, involves modulation of KyoT1/2 expression and function.
Supplementary Material
Acknowledgments
We thank Dr. Mary Waye for her generous gift of anti-KyoT2 antibodies, and Drs. Anna Lyubimova and Sara Cabodi for their help in the initial part of these studies. This work was supported by National Institutes of Health Grants CA16038, CA73796, and AR39190 and a grant of the Swiss National Foundation (to G.P.D.).
Author contributions: M.G., A.H.-F., and G.P.D. designed research; M.G., A.H.-F., A.T.D.V., and E.C. performed research; P.O., S.L., G.C., and G.P.D. analyzed data; and G.P.D. wrote the paper.
References
- 1.Dotto, G. P. (1999) Crit. Rev. Oral Biol. Med. 10, 442–457. [DOI] [PubMed] [Google Scholar]
- 2.Ridley, A. J. (2001) Trends Cell Biol. 11, 471–477. [DOI] [PubMed] [Google Scholar]
- 3.Etienne-Manneville, S. & Hall, A. (2002) Nature 420, 629–635. [DOI] [PubMed] [Google Scholar]
- 4.Sordella, R., Jiang, W., Chen, G. C., Curto, M. & Settleman, J. (2003) Cell 113, 147–158. [DOI] [PubMed] [Google Scholar]
- 5.Bishop, A. L. & Hall, A. (2000) Biochem. J. 348, 241–255. [PMC free article] [PubMed] [Google Scholar]
- 6.Fujisawa, K., Fujita, A., Ishizaki, T., Saito, Y. & Narumiya, S. (1996) J. Biol. Chem. 271, 23022–23028. [DOI] [PubMed] [Google Scholar]
- 7.Madaule, P., Eda, M., Watanabe, N., Fujisawa, K., Matsuoka, T., Bito, H., Ishizaki, T. & Narumiya, S. (1998) Nature 394, 491–494. [DOI] [PubMed] [Google Scholar]
- 8.Di Cunto, F., Calautti, E., Hsiao, J., Ong, L., Topley, G., Turco, E. & Dotto, G. P. (1998) J. Biol. Chem. 273, 29706–29711. [DOI] [PubMed] [Google Scholar]
- 9.Mukai, H. (2003) J. Biochem. (Tokyo) 133, 17–27. [DOI] [PubMed] [Google Scholar]
- 10.Wallar, B. J. & Alberts, A. S. (2003) Trends Cell Biol. 13, 435–446. [DOI] [PubMed] [Google Scholar]
- 11.Di Cunto, F., Imarisio, S., Hirsch, E., Broccoli, V., Bulfone, A., Migheli, A., Atzori, C., Turco, E., Triolo, R., Dotto, G. P., et al. (2000) Neuron 28, 115–127. [DOI] [PubMed] [Google Scholar]
- 12.Di Cunto, F., Ferrara, L., Curtetti, R., Imarisio, S., Guazzone, S., Broccoli, V., Bulfone, A., Altruda, F., Vercelli, A. & Silengo, L. (2003) Brain Res. Bull. 60, 319–327. [DOI] [PubMed] [Google Scholar]
- 13.Braga, V. M., Machesky, L. M., Hall, A. & Hotchin, N. A. (1997) J. Cell Biol. 137, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calautti, E., Grossi, M., Mammucari, C., Aoyama, Y., Pirro, M., Ono, Y., Li, J. & Dotto, G. P. (2002) J. Cell Biol. 156, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramalho-Santos, M., Yoon, S., Matsuzaki, Y., Mulligan, R. C. & Melton, D. A. (2002) Science 298, 597–600. [DOI] [PubMed] [Google Scholar]
- 16.Tumbar, T., Guasch, G., Greco, V., Blanpain, C., Lowry, W. E., Rendl, M. & Fuchs, E. (2004) Science 303, 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGrath, M. J., Mitchell, C. A., Coghill, I. D., Robinson, P. A. & Brown, S. (2003) Am. J. Physiol. 285, C1513–C1526. [DOI] [PubMed] [Google Scholar]
- 18.Taniguchi, Y., Furukawa, T., Tun, T., Han, H. & Honjo, T. (1998) Mol. Cell. Biol. 18, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Cunto, F., Topley, G., Calautti, E., Hsiao, J., Ong, L., Seth, P. K. & Dotto, G. P. (1998) Science 280, 1069–1072. [DOI] [PubMed] [Google Scholar]
- 20.Ng, E. K., Lee, S. M., Li, H. Y., Ngai, S. M., Tsui, S. K., Waye, M. M., Lee, C. Y. & Fung, K. P. (2001) J. Cell Biochem. 82, 1–10. [DOI] [PubMed] [Google Scholar]
- 21.Leung, T., Chen, X. Q., Manser, E. & Lim, L. (1996) Mol. Cell. Biol. 16, 5313–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He, T. C., Zhou, S., da Costa, L. T., Yu, J., Kinzler, K. W. & Vogelstein, B. (1998) Proc. Natl. Acad. Sci. USA 95, 2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okuyama, R., Nguyen, B. C., Talora, C., Ogawa, E., di Vignano, A. T., Lioumi, M., Chiorino, G., Tagami, H., Woo, M. & Dotto, G. P. (2004) Dev. Cell 6, 551–562. [DOI] [PubMed] [Google Scholar]
- 24.Olson, M. F., Paterson, H. F. & Marshall, C. J. (1998) Nature 394, 295–299. [DOI] [PubMed] [Google Scholar]
- 25.Missero, C., Di Cunto, F., Kiyokawa, H., Koff, A. & Dotto, G. P. (1996) Genes Dev. 10, 3065–3075. [DOI] [PubMed] [Google Scholar]
- 26.Yamashiro, S., Totsukawa, G., Yamakita, Y., Sasaki, Y., Madaule, P., Ishizaki, T., Narumiya, S. & Matsumura, F. (2003) Mol. Biol. Cell 14, 1745–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McMullan, R., Lax, S., Robertson, V. H., Radford, D. J., Broad, S., Watt, F. M., Rowles, A., Croft, D. R., Olson, M. F. & Hotchin, N. A. (2003) Curr. Biol. 13, 2185–2189. [DOI] [PubMed] [Google Scholar]
- 28.Welsh, C. F., Roovers, K., Villanueva, J., Liu, Y., Schwartz, M. A. & Assoian, R. K. (2001) Nat. Cell Biol. 3, 950–957. [DOI] [PubMed] [Google Scholar]
- 29.Robinson, P. A., Brown, S., McGrath, M. J., Coghill, I. D., Gurung, R. & Mitchell, C. A. (2003) Am. J. Physiol. 284, C681–C695. [DOI] [PubMed] [Google Scholar]
- 30.Morgan, M. J. & Whawell, S. A. (2000) Biochem. Biophys. Res. Commun. 273, 776–783. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}