

Abstract
The intraflagellar transport (IFT) proteins Ift172/Wimple and Polaris/Ift88 and the anterograde IFT motor kinesin-II are required for the production and maintenance of cilia. These proteins are also required for the activation of targets of the mouse Hedgehog (Hh) pathway by Gli transcription factors. The phenotypes of the IFT mutants, however, are not identical to mutants that lack Smoothened (Smo), an essential activator of the Hh pathway. We show here that mouse embryos that lack both Ift172 and Smo are identical to Ift172 single mutants, which indicates that Ift172 acts downstream of Smo. Ift172 mutants have a weaker neural patterning phenotype than Smo mutants, because Ift172, but not Smo, is required for proteolytic processing of Gli3 to its repressor form. Dnchc2 and Kif3a, essential subunits of the retrograde and anterograde IFT motors, are also required for both formation of Gli activator and proteolytic processing of Gli3. As a result, IFT mutants display a loss of Hh signaling phenotype in the neural tube, where Gli activators play the major role in pattern formation, and a gain of Hh signaling phenotype in the limb, where Gli3 repressor plays the major role. Because both anterograde and retrograde IFT are essential for positive and negative responses to Hh, and because cilia are present on Hh responsive cells, it is likely that cilia act as organelles that are required for all activity of the mouse Hh pathway.
Keywords: Dnchc2, Gli proteins, IFT172, intraflagellar transport, node
Specification of ventral cell types in the mammalian neural tube depends on Sonic Hedgehog (Shh) released from the notochord (1, 2). As in the Drosophila Hedgehog (Hh) signaling pathway, binding of Shh to its receptor, Patched homolog 1 (Ptch1), allows activation of the membrane protein Smoothened (Smo), which regulates the Ci/Gli transcription factors that control Hh target gene expression. However, there are significant differences between Drosophila and mammals in the mechanisms that regulate Gli proteins in response to Hh ligands. For example, we discovered that intraflagellar transport (IFT) is required in the mouse Hh pathway downstream of Smo (3), whereas there is apparently no role for IFT in Drosophila Hh signaling.
IFT is required for the assembly and maintenance of all cilia in plants, protists, and animals (4). Cilia are microtubule-based cell surface protrusions that extend from the basal body (centriole), and nonmotile primary cilia are present on most vertebrate cells. IFT involves bidirectional movement of IFT particles beneath the plasma membrane of cilia along the axonemal microtubules, which allows material to be transported to the distal tip of the cilium, where new components are incorporated into the axoneme. IFT particles are composed of 16 known IFT proteins that form two multiprotein IFT complexes. The movement of IFT particles from the base to the tip of cilia is powered by the anterograde kinesin-II motor complex, and the movement back to the base is powered by the retrograde dynein motor complex.
We found that mutations in two IFT complex B proteins (Ift172 and Polaris) and an anterograde IFT motor subunit (Kif3a) prevent the specification of Shh-dependent ventral cell types in the mouse neural tube (3). Phenotypic and double-mutant analysis showed that Ift172, Polaris, and Kif3a are required for normal activation of Hh target genes and act at a step downstream of Ptch1. However, those experiments did not distinguish whether IFT proteins act as essential components of the signaling pathway or modulate the activity of classical Hh pathway components. Nor was it clear whether IFT proteins are required for a cytoplasmic transport process or in cilia for neural patterning. Both the lack of a requirement for IFT proteins in Drosophila Hh signaling and the fact that the IFT mutant phenotypes in the neural tube are weaker than those of Shh or Smo mutants were consistent with an indirect role of IFT proteins in Hh signaling.
Here we show that IFT proteins play an integral role in the pathway and are essential for all activity of the mouse Hh pathway downstream of Smo. Our experiments show that, in the absence of IFT proteins, cells can neither promote formation of Gli activators nor regulate production of Gli3 repressor in response to Hh ligands or upstream signaling components. We go on to show that loss of either the retrograde IFT motor subunit Dnchc2 or the anterograde IFT motor subunit Kif3a causes both loss of ventral neural cell types and reduction of Gli3 repressor protein. The identical phenotypes caused by loss of anterograde and retrograde IFT argue that cilia provide a structure that is essential for Hh signal transduction.
Materials and Methods
Mouse Mutations and Strains. Ift172wim and Smobnb are null ENU-induced alleles (3, 5). Other mutant strains used were Gli3XT-J, an intragenic deletion allele (6); Ptch1tm1Mps, a lacZ gene insertion allele (7); nodaltm1Rob, a lacZ gene insertion allele (8); and Kif3atm1Maz, a targeted null allele (9). Mutant alleles were genotyped as described (3, 5–9).
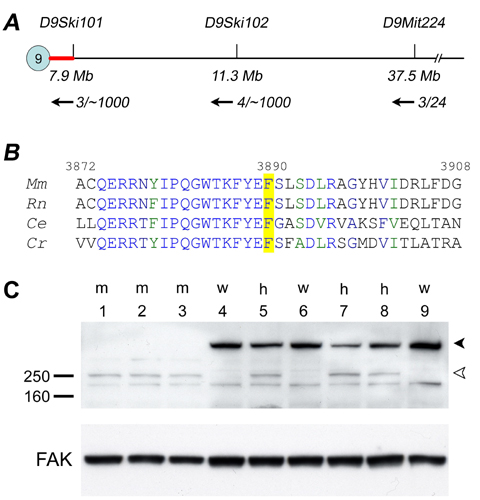
Dnchc2GT ES cells (RRM278) were obtained from the Bay Area Functional Genomics Consortium (http://baygenomics.ucsf.edu). The gene trap allele was predicted to produce a truncated protein (the N-terminal 734 amino acids) fused with β-geo (Fig. 3A). The Dnchc2GT allele was genotyped by using the generic neo primers (The Jackson Laboratory); the wild-type allele was distinguished from the Dnchc2GT allele by using primers (AACTGGCTTAGCGACTGTGG, GCCTCCAGACCCACTTGATA) that amplify the intron of the gene trap insertion. Phenotypic analysis was carried out as described (3).
Fig. 3.

Dnchc2 phenotypes. (A) The motor domain of Dnchc2 protein has six AAA (ATPase associated with cellular activities) motifs (A1–A6) (35). The Dnchc2lln mutation in A6 is likely to disrupt the motor activity of the protein. The Dnchc2GT mutation produced a truncated protein lacking all six of the AAA motifs. (B) Exencephaly (arrows) and reversal of heart looping (arrowhead) in Dnchc2 mutants at e10.5. (C) Occasional Dnchc2GT mutants that survived to e13.5 showed polydactyly in both hindlimbs (asterisk) and forelimbs (FL).
Mapping and Cloning. We identified ling-ling (lln, renamed Dnchc2lln after identification of the gene) in an ongoing screen for recessive ENU-induced mutations that cause morphological abnormalities at embryonic day (e)9.5 (10). Genetic mapping was carried out as described (3, 11). Dnchc2lln was mapped to an estimated 7.9-Mb interval in the proximal end of chromosome 9 (Fig. 7A, which is published as supporting information on the PNAS web site), which contained a Celera predicted transcript mCT178658, homologous to rat Dnchc2 (GenBank accession no. NM_023024). The coding region of Dnchc2 contained a T-to-C mutation in Dnchc2lln mutants. This mutation created a HinfI restriction fragment length polymorphism, which was assayed in genomic DNA by using the marker Dnchc2-G1 (TCATGTTTTTGTGGTCAGTGC, CACAGGCATAGCAGTCAGGA) to confirm the linkage of the mutation with the Dnchc2lln phenotype. Dnchc2lln/Dnchc2GT embryos had phenotypes similar to Dnchc2lln or Dnchc2GT homozygotes, confirming that Dnchc2lln and Dnchc2GT are allelic.
Western Blot Analysis of Gli3 Processing. Gli3 proteins were immunoprecipitated from e9.5 whole embryo extracts by a rabbit polyclonal Gli3 antibody, resolved on a 4–12% gradient PAGE gel and detected by a chicken polyclonal Gli3 antibody. Each lane was loaded with protein from a single embryo, with the exception of Smo mutants, where two mutants were used for each lane because of their smaller size. nih image software was used to quantify the results. Ratios of Gli3 repressor to full length protein were compared in littermates analyzed in the same experiment.
Results
IFT Components Are Required for All Activity of the Hh Pathway. We showed previously that two IFT proteins and the anterograde IFT motor are required for normal activity of the Hh pathway (3). However, the phenotypes of the IFT mutants appeared to be milder than mutants that lack Shh or Smo (1, 5, 12, 13). For example, V2 interneurons (a ventrolateral neural cell type marked by Chx10 expression) are absent in Shh mutants and Smo mutant clones but are present in Ift172 and polaris mutants. Similarly, Shh and Smo are required to repress Pax7 and restrict its expression to the dorsal half of the neural tube, but Pax7 is expressed in its normal dorsal position in Ift172 and polaris mutants. Ptch1 transcription, a direct target of Hh signaling in the neural tube (7), is not detected in Shh mutants (14) but is expressed at low levels in the ventral neural tube of the IFT mutants (3). Because all these mutations are null alleles, the differences in the neural tube phenotypes cannot be explained by residual gene function.
To investigate the relationship between Smo and Ift172, we examined embryos that lacked both genes. Smo is required for both Shh and Indian Hedgehog signaling during development (5, 12, 13). Smo mutants fail to complete embryonic turning and arrest at around e9.0 with a linear heart tube, a closed but narrow neural tube, and a small body size. Ift172 mutants, in contrast, survive to e10.5–11.5, have an open rostral neural tube, and show randomized heart looping (3). Ift172 Smo double mutants survived to e10.5–11.5, longer than Smo single mutants, and had the same morphology as Ift172 single mutants (Fig. 1A). Neural patterning in Ift172 Smo double mutants was identical to that in Ift172 single mutants (Fig. 1B). In particular, the V2 interneurons marked by Chx10, which are absent in Shh and Smo mutants (1, 5, 12, 13), were present in large numbers in a ventrally expanded domain in the double mutant neural tube. A few motor neurons were present in the Ift172 Smo double mutants but were never detected in Shh or Smo mutants. Pax7 was expressed in only the dorsal half of the spinal cord in both Ift172 Smo double and Ift172 single mutants, rather than throughout the spinal cord as in Shh or Smo mutants. Combined with our previous analysis showing Ift172 is epistatic to Ptch1 (3), the data indicate that, in the absence of Ift172, either full activation of the pathway, caused by loss of Ptch1, or complete blocking of upstream Hh signaling caused by loss of Smo has no effect on downstream signaling. These findings demonstrate that IFT components are absolutely required for all activity of the Hh signaling pathway downstream of Ptch1 and Smo.
Fig. 1.

Ift172 Smo double mutant analysis. (A) Phenotypes of wild-type (WT), Ift172, and Smo single mutants and Ift172 Smo double mutants at e10.5. Both the small body size and early lethality of Smo mutants were rescued in Ift172 Smo double mutants. Heart-looping reversal was observed in about one-half (five of nine) Ift172 Smo double mutants, similar to Ift172 single mutants (3). (B) Immunofluorescent staining of neural tube markers (Shh, Nkx2.2, Lhx3, Chx10, and Pax7) in e10.5 wild-type and Ift172 Smo double mutants.
Ift172 Is Required for Gli3 Processing. The Gli transcription factors mediate all Hh signaling in the mouse (15, 16). Gli2 is the primary transcriptional activator that mediates Hh signaling and proteolytically processed Gli3 is the principal transcriptional repressor that keeps targets off in the absence of ligand. Although Ift172 single mutants have a milder spinal cord phenotype than Shh or Smo mutants, neural patterning in Ift172 Gli3 double mutants is similar to that in Shh Gli3 or Smo Gli3 double mutants (1, 3, 5, 12, 13, 17). This suggested that the difference between the Ift172 and Smo phenotypes could be due to different amounts of Gli3 repressor. Proteolytic processing of full-length Gli3 protein to the repressor form is regulated by Hh signaling and can be monitored in extracts from embryonic tissues (18). We therefore tested the effect of Ift172 mutations on Gli3 processing in vivo.
Both full length and processed (repressor) Gli3 proteins were detected in wild-type e9.5 embryos (Fig. 2A). In Ptch1 mutant embryos, full length Gli3 was present, but very little Gli3 repressor was detected (Fig. 2 A) as expected, because full activation of the pathway caused by loss of Ptch1 should prevent formation of the repressor. In contrast, in Smo mutants, the pathway is completely inactive, and Gli3 repressor levels were elevated as expected: the Gli3 repressor/full length (R/FL) ratio was 3.5 times that in wild-type littermates (Fig. 2 B and D). The balance between the full length and processed Gli3 was also affected in Ift172 mutants but in the opposite direction seen in Smo mutants: the Gli3 R/FL ratio in Ift172 mutants was less than one-eighth that in wild-type embryos (Fig. 2 B and D). Thus, wild-type Ift172 protein is required for the production of normal levels of Gli3 repressor.
Fig. 2.

Gli3 processing. (A) Both full length (FL) and repressor (R) forms of Gli3 were detected in wild-type embryos but absent in Gli3 mutants, confirming these bands correspond to Gli3 proteins. The repressor form was undetectable in Ptch1 mutants. (B) Gli3 processing was affected in both Smo and Ift172 mutants but in opposite directions: Smo mutants showed a relative increase in Gli3 repressor, whereas Ift172 mutants showed a relative decrease in Gli3 repressor. (C) Mutants lacking the anterograde and retrograde motors, Kif3a and Dnchc2, both showed decreased Gli3 repressor, as did Ift172 mutants. (D and E) Quantification of results in B and C.(D) The average repressor to full length Gli3 protein ratios (R/FL) in wild-type, Smo, and Ift172 mutants were 0.74 ± 0.10 (mean ± SD), 2.60 ± 0.07, and 0.089 ± 0.044, respectively. (E) In an independent experiment (C), the average R/FL ratios in wild-type, Kif3a, and Dnchc2GT mutants were 1.11 ± 0.13, 0.34 ± 0.27, and 0.26 ± 0.05.
The reduction of Gli3 repressor in IFT mutants relative to the increased Gli3 repressor in Smo mutants correlated well with the specification of ventrolateral Shh-dependent neural cell types, such as V2 interneurons, in the IFT mutants and not in Smo mutants (3, 5, 12, 13). In addition, because Gli3 repressor is required for normal anterior–posterior patterning in the limb, the reduction of Gli3 repressor can account for the polydactyly phenotype seen in partial loss-of-function polaris mutants (3, 19), similar to that seen in Gli3 mutants (6). Thus the IFT mutants displayed a loss of Hh signaling phenotype in the neural tube, where Gli activators play the major role in pattern formation, and showed a gain of Hh signaling phenotype in the limb, where Gli3 repressor plays the major role in digit specification.
Dnchc2 Is Required for Retrograde IFT in Nodal Cilia. The essential requirement for Ift172, Polaris, and Kif3a in the Hh pathway could be explained in two ways. One possibility is that cilia are organelles that are essential for Hh signaling, which is consistent with the fact that all known functions of IFT proteins are related to cilia. Alternatively, it is possible that these anterograde IFT proteins have two independent roles, one in cilia formation and a second in localizing Hh pathway components to a specific subcellular compartment. To help distinguish between these possibilities, we examined the phenotypes of mouse mutants lacking the retrograde IFT motor.
Dnchc2 is the mammalian homologue of Dhc1b (20), which is required for retrograde IFT in both Chlamydomonas reinhardtii and Caenorhabditis elegans (21–23). We identified two mutant alleles of mouse Dnchc2 (Fig. 3A, Materials and Methods). Dnchc2lln, an ENU-induced allele, carried a phenylalanine-to-serine (F3890S) mutation in a conserved residue of the Dnchc2 motor domain (Fig. 7B). The second allele, Dnchc2GT, was generated from mouse ES cells carrying a gene trap insertion (24) that was predicted to produce a truncated protein lacking the entire dynein motor domain fused to β-geo. Western blot analysis confirmed that no full length Dnchc2 protein was made in Dnchc2GT homozygotes (Fig. 7C), so Dnchc2GT is a null allele. Homozygous Dnchc2lln, Dnchc2GT, and transheterozygous Dnchc2lln/Dnchc2GT mutants died at approximately e12.5 with abnormal brain morphology and frequent heart-looping reversal, indicating the two alleles of Dnchc2 were of similar strength.
Approximately one-half (44/97) of Dnchc2lln embryos were exencephalic and lacked the groove at the ventral midline of the brain; these embryos were indistinguishable from Ift172 mutants (Fig. 3B) (3). The remainder of the Dnchc2lln embryos were not exencephalic but exhibited abnormal brain morphology similar to that seen in partial loss-of-function polaris mutants (3). The morphology of Dnchc2 mutants was similar to that of embryos that lack dynein 2 light intermediate chain (D2LIC), a nonmotor subunit of the retrograde IFT motor complex, although we did not observe the class of early arrest mutants described for D2LIC (25). Occasional Dnchc2 mutants that survived to e13.5 were polydactylous in both forelimbs and hindlimbs (Fig. 3C), as seen in partial loss-of-function polaris mutants (3, 19).
The randomization of heart-looping polarity in Dnchc2 mutants suggested a defect in specification of left–right asymmetry, and an early marker of the left side, nodal (8), was expressed bilaterally in the lateral plate mesoderm in most Dnchc2lln embryos (Fig. 4A). Dnchc2 was expressed in the node at e7.5, as visualized by β-galactosidase activity of the gene-trap allele (Fig. 4B). Cilia were present on the surface of the node in Dnchc2lln embryos, but they all had abnormal morphology: some cilia were short, and others were of approximately normal length but had a bulge along their length (Fig. 4C). These cilia were similar in morphology to flagella in Chlamydomonas mutants that lack retrograde IFT (22, 26), so we conclude that Dnchc2 encodes a bona fide subunit of the retrograde IFT motor.
Fig. 4.

Dnchc2 is required for ciliogenesis. (A) Bilateral expression of nodal-lacZ in the lateral plate mesoderm of Dnchc2lln mutants (8 of 10). (B) Lateral view of Dnchc2-lacZ staining in the node of head-fold-stage embryos (Dnchc2GT/+) in whole-mount and section (rostral to the right). (C) Nodal cilia visualized by scanning electron microscopy (SEM). The average length of cilia in Dnchc2lln mutants was ≈70% of those in wild types (n = 51 in wild types, n = 37 in Dnchc2lln mutants). Cilia in Dnchc2lln mutants showed an unusual bulging morphology (arrowheads). (Bar, 1 μm.)
Similar Phenotypes Caused by Loss of Anterograde and Retrograde IFT Components Support a Requirement for Cilia in the Hh Pathway. The similar external morphology of Dnchc2 and other IFT mutants suggested that the retrograde IFT motor might also act in the Hh pathway. We therefore examined neural tube patterning in e10.5 Dnchc2 mutants. As in Ift172 and polaris mutants (3), Shh was expressed in the notochord, but ventral Shh-dependent neural cell types failed to be specified at all axial levels: floor-plate cells (marked by expression of Shh and Foxa2) and V3 interneuron precursors (Nkx2.2) were absent in Dnchc2 mutants (Fig. 5 and data not shown). As in the other IFT mutants, Pax7, a marker of dorsal neural progenitors, was expressed in the normal dorsal domain, and some ventrolateral cell types (Lhx3 and Chx10) were present (Fig. 5). Motor neurons, marked by expression of HB9, were absent in the rostral half of the spinal cord (data not shown), as in the other IFT mutants (3). However, in contrast to Ift172 and polaris mutants, motor neurons were present in a domain that spanned the ventral midline of the caudal Dnchc2 spinal cord. Similarly, Olig2, a marker of motor neuron and oligodendrocyte precursors, was expressed in the caudal but not the rostral Dnchc2 spinal cord (Fig. 8A, which is published as supporting information on the PNAS web site). The greater requirement for Dnchc2 in rostral regions is consistent with its expression pattern: at e9.5 and e10.5, the expression of Dnchc2-β-geo was higher in the rostral neural tube than in the caudal neural tube (Fig. 8B).
Fig. 5.

Expression of neural tube markers (Shh, Nkx2.2, Lhx3, Chx10, and Pax7) in e10.5 wild-type and Dnchc2lln mutant embryos.
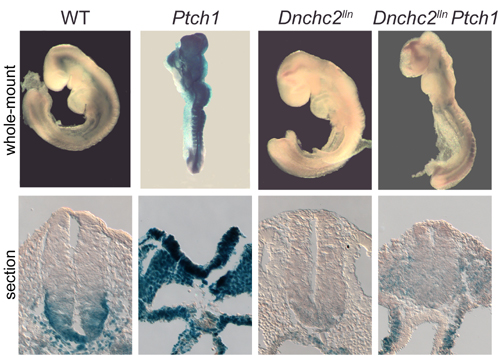
As in Ift172 and polaris mutants (3), Ptch1-lacZ expression was reduced in the Dnchc2lln neural tube (Fig. 9, which is published as supporting information on the PNAS web site), indicating that Hh signaling was impaired in these mutants. To define the step in the Hh pathway that requires Dnchc2, we analyzed the phenotypes of double mutants that lacked both Ptch1 and Dnchc2lln. In the absence of Ptch1, the Hh pathway is fully activated; Ptch1 mutants arrest at e9.0–9.5 and express high levels of Hh target genes, including Ptch1 itself (7). Dnchc2lln Ptch1 double mutants, in contrast, survived to ≈e12.5, and resembled Dnchc2lln single mutants. The ectopic expression of Ptch1-lacZ was suppressed in the double mutants (Fig. 9), and expression of neural markers in e10.5 Dnchc2lln Ptch1 double mutants was similar to Dnchc2lln single mutants (data not shown). Thus, as in the case of Ift172, polaris, and Kif3a (3), loss of Dnchc2 blocked the activity of the Hh pathway at a step downstream of Ptch1.
As in Ift172 mutants, Gli3 processing was affected by loss of Dnchc2. Extracts from mutants lacking either the anterograde motor subunit, Kif3a, or the retrograde motor subunit, Dnchc2 (Fig. 2 C and E), had a decreased amount of repressor Gli3 compared to the unprocessed protein. Thus both the anterograde and the retrograde IFT motors promote formation of Gli3 repressor. The similarity of the phenotypes caused by loss of either anterograde or retrograde IFT argues against a role for IFT in trafficking Hh pathway components to one of two alternative cytoplasmic locations and is more consistent with a role for cilia as organelles that are required for responses to Hh ligands.
In support of a role for cilia in Hh responsiveness, we found that cilia were present on neural progenitors and other cell types that respond to Hh signaling. γ-tubulin, a marker of basal bodies of cilia, was expressed in a punctate pattern along the ventricle of the neural tube (Fig. 6A). Acetylated α-tubulin, which marks the stable microtubules of cilia, was present in cilia-like protrusions into the ventricle of the neural tube (Fig. 6 B and C). Cilia-like expression of acetylated α-tubulin was also observed in Hh-responsive mesenchymal cells adjacent to the neural tube and in the limb bud (Fig. 6B and data not shown).
Fig. 6.

Presence of cilia in the neural tube at e10.5. Punctate expression of γ-tubulin (A) and cilia-like expression of acetylated α-tubulin (B and C) in the neural tube using confocal microscopy. A and B are ventral neural tubes; arrowhead points to the notochord. (C) A high-magnification view of the boxed area in B, with the arrow pointing to cilia-like protrusions into the lumen of the neural tube. Acetylated α-tubulin is also expressed in the notochord and the surrounding mesenchymal cells.
Discussion
The Roles of IFT Proteins in the Mouse Hh Pathway. The data presented here demonstrate that IFT proteins are required for Hh signaling because they are essential for both Gli activator and repressor functions. In the neural tube, where Gli activators are required to specify ventral cell types, both Smo and IFT mutants display a loss of Hh signaling phenotype. In contrast, Smo and IFT proteins have opposite effects on the formation of Gli3 repressor. Active Smo blocks formation of Gli3 repressor, whereas IFT172, Kif3a, and Dnchc2 proteins are required for formation of Gli3 repressor. Similar results were also obtained recently for polaris mutants (19). As a result, in the limb, where Gli3 repressor plays a major role in digit specification, the IFT mutants show a gain of Hh signaling phenotype. Because the Ift172 Smo double-mutant phenotype is identical to that of the Ift172 single mutant, we infer that regulation of both Gli activation and Gli3 processing by Hh signaling depends on IFT.
Although our biochemical analysis shows that IFT is required for the production of Gli3 repressor, our previous genetic studies showed that enough Gli3 repressor is present in Ift172 mutants to prevent the specification of motor neurons (3). Thus, although IFT proteins are required for normal levels of processing of Gli3 repressor, a small amount of functional Gli3 repressor can be made in the absence of IFT.
A Vertebrate-Specific Function for Cilia in Hh Signaling. The nearly ubiquitous presence of primary cilia on vertebrate cells (27) and their antenna-like structure have led to speculation that primary cilia may be used to relay extracellular signals to the cell body. Some specialized primary cilia monitor fluid flow: bending of cilia in cultured kidney epithelial cells or in the embryonic node causes an influx of extracellular calcium through the calcium channel protein Polycystin-2 (28–30). The function of other primary cilia is less clear, although the somatostatin receptor 3 and serotonin 5-HT6 receptor are both localized to cilia on specific classes of brain neurons (31, 32), suggesting these cilia have chemosensory functions.
Our results suggest that IFT proteins are required for mammalian Hh signaling, because normally structured cilia are required for cells to respond to Hh signals. Both the anterograde and retrograde motors have the same effect on Hh signaling: both are required for activation of Hh targets in the neural tube and for processing of Gli3. A dedicated role of IFT proteins for cilia formation and function is supported by the existence of a ciliary proteome, which includes the IFT proteins, that is found specifically in organisms with cilia or flagella (33, 34). The absence of IFT proteins in organisms that lack cilia argues that IFT proteins are required only for the production and maintenance of cilia and not for other intracellular transport processes.
Because IFT proteins are required for both Gli-activator functions and Gli3 processing, we predict that more than one component of the Hh signaling pathway may be localized to cilia. The subcellular locations of mammalian proteins that transduce the Hh signal, including Ptch1, Smo, PKA, Su(fu), and the Glis, are not known. Development of reagents to test whether Hh pathway components are localized to cilia would be a first step toward understanding how cilia integrate positive and negative inputs on Gli protein activity.
Supplementary Material
Acknowledgments
We thank N. Lampen for assistance with SEM and E. Robertson (Harvard University, Cambridge, MA), M. Scott (Stanford University, Palo Alto, CA), C. Cepko (Harvard Medical School, Boston), T. Li (Harvard Medical School, Boston), and R. B. Vallee (Columbia University College of Physicians and Surgeons, New York). We thank T. Bestor, T. Caspary, M. García-García, D. Laird, J. Lee, K. Liem, A. Rakeman, and S. Weatherbee for helpful comments on the manuscript. Monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank, which was developed under the auspices of the National Institute of Child Health and Human Development and is maintained by the Department of Biological Sciences, University of Iowa. Genome sequence analysis used Ensembl and the Celera Discovery System and associated databases, made possible in part by the AMDeC Foundation. This work was supported by the Lita Annenberg Hazen Foundation and National Institutes of Health Grant NS44385 (to K.A.).
Author contributions: D.H. and K.V.A. designed research, performed research, analyzed data, and wrote the paper.
Abbreviations: Shh, Sonic Hedgehog; Hh, Hedgehog; IFT, intraflagellar transport; Ptch1, Patched homolog 1; Smo, Smoothened; en, embryonic day n.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. DQ104402).
References
- 1.Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L., Westphal, H. & Beachy, P. A. (1996) Nature 383, 407–413. [DOI] [PubMed] [Google Scholar]
- 2.Ingham, P. W. & McMahon, A. P. (2001) Genes Dev. 15, 3059–3087. [DOI] [PubMed] [Google Scholar]
- 3.Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L. & Anderson, K. V. (2003) Nature 426, 83–87. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbaum, J. L. & Witman, G. B. (2002) Nat. Rev. Mol. Cell. Biol. 3, 813–825. [DOI] [PubMed] [Google Scholar]
- 5.Caspary, T., García-García, M. J., Huangfu, D., Eggenschwiler, J. T., Wyler, M. R., Rakeman, A. S., Alcorn, H. L. & Anderson, K. V. (2002) Curr. Biol. 12, 1628–1632. [DOI] [PubMed] [Google Scholar]
- 6.Hui, C. C. & Joyner, A. L. (1993) Nat. Genet. 3, 241–246. [DOI] [PubMed] [Google Scholar]
- 7.Goodrich, L. V., Milenkovic, L., Higgins, K. M. & Scott, M. P. (1997) Science 277, 1109–1113. [DOI] [PubMed] [Google Scholar]
- 8.Collignon, J., Varlet, I. & Robertson, E. J. (1996) Nature 381, 155–158. [DOI] [PubMed] [Google Scholar]
- 9.Marszalek, J. R., Ruiz-Lozano, P., Roberts, E., Chien, K. R. & Goldstein, L. S. (1999) Proc. Natl. Acad. Sci. USA 96, 5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-García, M. J., Eggenschwiler, J. T., Caspary, T., Alcorn, H. L., Wyler, M. R., Huangfu, D., Rakeman, A. S., Lee, J. D., Feinberg, E. H., Timmer, J. R., et al. (2005) Proc. Natl. Acad. Sci. USA 102, 5913–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasarskis, A., Manova, K. & Anderson, K. V. (1998) Proc. Natl. Acad. Sci. USA 95, 7485–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang, X. M., Ramalho-Santos, M. & McMahon, A. P. (2001) Cell 106, 781–792. [PubMed] [Google Scholar]
- 13.Wijgerde, M., McMahon, J. A., Rule, M. & McMahon, A. P. (2002) Genes Dev. 16, 2849–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eggenschwiler, J. T., Espinoza, E. & Anderson, K. V. (2001) Nature 412, 194–198. [DOI] [PubMed] [Google Scholar]
- 15.Bai, C. B., Stephen, D. & Joyner, A. L. (2004) Dev. Cell 6, 103–115. [DOI] [PubMed] [Google Scholar]
- 16.Motoyama, J., Milenkovic, L., Iwama, M., Shikata, Y., Scott, M. P. & Hui, C. C. (2003) Dev. Biol. 259, 150–161. [DOI] [PubMed] [Google Scholar]
- 17.Litingtung, Y. & Chiang, C. (2000) Nat. Neurosci. 3, 979–985. [DOI] [PubMed] [Google Scholar]
- 18.Wang, B., Fallon, J. F. & Beachy, P. A. (2000) Cell 100, 423–434. [DOI] [PubMed] [Google Scholar]
- 19.Liu, A., Wang, B. & Niswander, L. A. (2005) Development (Cambridge, U.K.) 132, 3103–3111. [DOI] [PubMed] [Google Scholar]
- 20.Criswell, P. S., Ostrowski, L. E. & Asai, D. J. (1996) J. Cell Sci. 109, 1891–1898. [DOI] [PubMed] [Google Scholar]
- 21.Signor, D., Wedaman, K. P., Orozco, J. T., Dwyer, N. D., Bargmann, C. I., Rose, L. S. & Scholey, J. M. (1999) J. Cell Biol. 147, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pazour, G. J., Dickert, B. L. & Witman, G. B. (1999) J. Cell Biol. 144, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter, M. E., Bower, R., Knott, J. A., Byrd, P. & Dentler, W. (1999) Mol. Biol. Cell 10, 693–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stryke, D., Kawamoto, M., Huang, C. C., Johns, S. J., King, L. A., Harper, C. A., Meng, E. C., Lee, R. E., Yee, A., L'Italien, L., et al. (2003) Nucleic Acids Res. 31, 278–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rana, A. A., Barbera, J. P. M., Rodriguez, T. A., Lynch, D., Hirst, E., Smith, J. C. & Beddington, R. S. P. (2004) Development (Cambridge, U.K.) 131, 4999–5007. [DOI] [PubMed] [Google Scholar]
- 26.Pazour, G. J., Wilkerson, C. G. & Witman, G. B. (1998) J. Cell Biol. 141, 979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pazour, G. J. & Witman, G. B. (2003) Curr. Opin. Cell Biol. 15, 105–110. [DOI] [PubMed] [Google Scholar]
- 28.Praetorius, H. A., Frokiaer, J., Nielsen, S. & Spring, K. R. (2003) J. Membr. Biol. 191, 193–200. [DOI] [PubMed] [Google Scholar]
- 29.Nauli, S. M., Alenghat, F. J., Luo, Y., Williams, E., Vassilev, P., Li, X., Elia, A. E., Lu, W., Brown, E. M., Quinn, S. J., et al. (2003) Nat. Genet. 33, 129–137. [DOI] [PubMed] [Google Scholar]
- 30.McGrath, J., Somlo, S., Makova, S., Tian, X. & Brueckner, M. (2003) Cell 114, 61–73. [DOI] [PubMed] [Google Scholar]
- 31.Händel, M., Schulz, S., Stanarius, A., Schreff, M., Erdtmann-Vourliotis, M., Schmidt, H., Wolf, G. & Höllt, V. (1999) Neuroscience 89, 909–926. [DOI] [PubMed] [Google Scholar]
- 32.Brailov, I., Bancila, M., Brisorgueil, M. J., Miquel, M. C., Hamon, M. & Vergé, D. (2000) Brain Res. 872, 271–275. [DOI] [PubMed] [Google Scholar]
- 33.Li, J. B., Gerdes, J. M., Haycraft, C. J., Fan, Y., Teslovich, T. M., May-Simera, H., Li, H., Blacque, O. E., Li, L., Leitch, C. C., et al. (2004) Cell 117, 541–552. [DOI] [PubMed] [Google Scholar]
- 34.Avidor-Reiss, T., Maer, A. M., Koundakjian, E., Polyanovsky, A., Keil, T., Subramaniam, S. & Zuker, C. S. (2004) Cell 117, 527–539. [DOI] [PubMed] [Google Scholar]
- 35.Mikami, A., Tynan, S. H., Hama, T., Luby-Phelps, K., Saito, T., Crandall, J. E., Besharse, J. C. & Vallee, R. B. (2002) J. Cell Sci. 115, 4801–4808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}