

Abstract
Angiopoietins are a family of factors that play important roles in angiogenesis, and their receptor, Tie-2 receptor tyrosine kinase, is expressed primarily by endothelial cells. Three angiopoietins have been identified so far, angiopoietin-1 (Ang-1), angiopietin-2 (Ang-2), and angiopoietin-3 (Ang-3). It has been established that Ang-1 and Tie-2 play essential roles in embryonic angiogenesis. We have demonstrated recently that, unlike Ang-2, Ang-1 binds to the extracellular matrix, which regulates the availability and activity of Ang-1 (Xu, Y., and Yu, Q. (2001) J. Biol. Chem. 276, 34990–34998). However, the role and biochemical characteristics of Ang-3 are unknown. In our current study, we demonstrated that, unlike Ang-1 and Ang-2, Ang-3 is tethered on cell surface via heparan sulfate proteoglycans (HSPGs), especially perlecan. The cell surface-bound Ang-3 is capable of binding to its receptor, Tie-2; suggesting HSPGs concentrate Ang-3 on the cell surface and present Ang-3 to its receptor to elicit specific local reaction. Mutagenesis experiment revealed that the coiled-coil domain of Ang-3 is responsible for its binding to the cell surface. In addition, we demonstrated that the cell surface-bound Ang-3 but not soluble Ang-3 induces retraction and loss of integrity of endothelial monolayer, indicating the binding of Ang-3 to the cell surface via HSPGs is required for this bioactivity of Ang-3.
Angiogenesis plays important roles in embryonic organogenesis, postnatal tissue repair, female reproductive function, arthritis, diabetes, and tumor growth and metastasis (1–4). Numerous molecules, including pro- and anti-angiogenic factors and their receptors, proteases, adhesion receptors, and the ECM1 components, are involved in angiogenesis (5–7).
Angiopoietins are ligands of Tie-2 (tyrosine kinase with immunoglobulin and epidermal growth factor homology domains-2) receptor kinase, which is primarily expressed by endothelial cells and their precursors (8–11). Three Tie-2 ligands have been identified so far (12–14), angiopoietin-1 (Ang-1), angiopietin-2 (Ang-2), and angiopoietin-3 (Ang-3) in mouse or its ortholog in human, angiopoietin-4 (Ang-4). Angiopoietins all contain a signal peptide, an amino-terminal coiled-coil domain, a linker peptide region, and a carboxyl-terminal fibrinogen homology domain (FHD). Studies have shown that the coiled-coil region is responsible for dimerization/multimerization of angiopoietins, whereas the FHD binds to the Tie-2 receptor (13–15).
It has been shown that the angiopoietin-Tie-2 pathway plays an essential role during the late stages of vascular development. Knock out of Ang-1 or Tie-2, or overexpression of Ang-2, is embryonic lethal with similar vascular defects. These mice display normal vascular growth factor (VEGF)-dependent early vascular development, however, there are profound defects in remodeling, organization, and stabilization of the primitive vasculature in these mice (11, 13, 16, and 17). Studies have shown that, at least under some circumstances, Ang-2 and Ang-3 are naturally occurring antagonists of Tie-2 receptor (13, 14). Ang-2 and Ang-3 are believed to compete with Ang-1 for the binding of Tie-2 and block Tie-2 phosphorylation induced by Ang-1 (13, 14, 18, and 19). However, how Ang-3 affects endothelial cell behavior and how bioactivity of Ang-3 is regulated have not been established.
We have demonstrated previously that Ang-2 is primarily secreted and that Ang-1 is incorporated into the ECM and the ECM binding of Ang-1 regulates its availability and activity (20). In our current study, we demonstrated that, unlike Ang-1 and Ang-2, Ang-3 is tethered on cell surface via heparan sulfate proteoglycans (HSPGs), especially perlecan, through the coiled-coil domain of Ang-3. Perlecan is an HSPG that is present in the basement membrane and on the cell surface (21). Perlecan binds to the cell surface through integrin (22–24), and plays important roles in vasculogenesis, angiogenesis, and tumorigenesis (21, 31). In addition, we have shown that the cell surface-bound Ang-3 is capable of binding to Tie-2-Fc fusion protein. More importantly, the cell surface-bound Ang-3 but not soluble Ang-3 induces retraction and loss of integrity of the endothelial monolayer, indicating that the cell surface binding of Ang-3 is required for its function.
We have shown very recently that overexpression of Ang-3 inhibits pulmonary metastasis of Lewis lung carcinoma and TA3 mammary carcinoma cells by blocking tumor angiogenesis and promoting apoptosis of tumor cells (45). Furthermore, we have demonstrated that tethering Ang-3 on the cell surface is required for effective inhibition of Ang-3 on tumor angiogenesis and pulmonary metastasis (45). These results further confirmed that binding of Ang-3 to the cell surface via HSPGs enhances/facilitates the bioactivity of Ang-3.
The results reported in our previous study (20) and current study demonstrate that angiopoietins not only play different roles in regulating endothelial cell function, but also their activities are differentially regulated by the ECM and HSPGs, which likely provides the basis for modulating activities of angiopoietins in vivo therefore regulating angiogenesis in physiologic and pathologic situations.
EXPERIMENTAL PROCEDURES
Cells and Reagents
Lewis lung carcinoma (LLC) cells, A10 embryonic aorta smooth muscle (A10) cells, bovine pulmonary artery endothelial cells (CAPE), C2C12 myoblasts, and COS-7 cells were obtained from the Cell Center Services Facility of University of Pennsylvania. LLC transfectants were maintained in the condition as described previously (20, 25). Anti-v5 epitope (Invitrogen), anti-Tie2, and anti-Ang-1, -2, and -3, and anti-glypican-1 (Santa Cruz Biotechnology), anti-heparan sulfate (Calbiochem), anti-perlecan (NeoMarkers), and anti-syndecan-1 (BD Biosciences) antibodies, heparinases I and III (Sigma), Streptomyces hyaluronidase (ICN), and Tie-2-Fc (R&D Systems) were used in the experiments.
RT-PCR, Mutagenesis, and Expression Constructions
RT-PCR was performed as described previously (25). Briefly, full-length Ang-1, -2, and -3 cDNAs were amplified using mouse placenta cDNAs as templates together with Pfu DNA polymerase (Stratagene) and a pair of the primers corresponding to the 5′- and 3′-ends of 24 nucleotides of the coding sequence of each molecule derived from the GenBank™ under the accession numbers U83509, AF004326, and AF113707, respectively. The stop codons were omitted from these reverse primers to fuse Ang-1, -2, and -3 to the C-terminal v5 epitope tag, which is presented in the expression vector (pEF6/V5-His TOPO, Invitrogen). The resulting PCR fragments were inserted into pEF6/V5-His TOPO vectors. Authenticity and correct orientation of Ang-1, -2, and -3 inserts were verified by DNA sequencing.
The various deletion fragments of Ang-3 were generated as detailed in the Fig. 4 using the full-length Ang-3 in pEF6/v5-His vector as a template and the ExSite PCR-based site-directed mutagenesis kit (Stratagene). The accuracy of the deletions was confirmed by DNA sequencing.
Fig. 4.

The coiled-coil domain of Ang-3 mediates its binding to the cell surface. A, several deletional constructs of Ang-3 were made, which contain the full-length of Ang-3 (1), the coiled-coil domain plus the linker peptide region (2), the coiled-coil domain (3), and the linker peptide region plus FHD of Ang-3 (4). These expression constructs were used to transfect COS-7 cells. 72 h after the transfection, the cells were treated with (B, panel c) or without (B, panel b) trypsin, washed, and lysed. 50 μg of proteins derived from the cell culture supernatants (B, panel a) and the cell lysates (B, panels b and c) were analyzed by Western blotting with anti-v5 antibody. Arrows indicate the monomer, dimer, tetramer, and oligomer of the coiled-coiled fragments of Ang-3.
Transient and Stable Transfection
COS-7 cells and LLC cells were transfected using LipofectAMINE (Invitrogen) with the expression constructs containing cDNA inserts encoding mouse Ang-1, -2, or -3 or the deletional constructs of Ang-3. The transient transfected COS-7 cells were used 72 h after the transfection. The stably transfected LLC cells were selected for blasticidin resistance. The expression level of v5-epitope tagged Ang-1, Ang-2, and full-length and deletion fragments of Ang-3 in COS-7 and LLC transfectants was determined by Western blotting with anti-v5 antibody as described previously (20).
Protein Sample Preparation and Western Blot Analysis
To determine the distribution pattern of angiopoietins, Western blotting was performed using the proteins derived from the cell culture supernatants of LLC transfectants or transiently transfected COS-7 cells, lysates of the EDTA-lifted cells, and the ECM materials deposited on the cell culture dishes. The transfected cells were either treated with trypsin for 10 min at room temperature and washed with 10% FBS Dulbecco’s modified Eagle’s medium, or released from the culture dishes by phosphate-buffered saline (PBS) solution containing 5 mm EDTA and washed. The trypsin or EDTA-released cells were washed and lysed in 2× SDS Laemmli sample buffer. The ECM materials remaining on the cell culture dishes, after removal of the cells with EDTA solution, were washed and extracted with 2× SDS sample buffer.
To determine the binding between the cell surface heparan sulfate (HS) and Ang-3, the EDTA-lifted LLCAng-3 cells were treated with heparinase I (5 units/ml) plus heparinase III (0.5 unit/ml) or Streptomyces hyaluronidase (5 units/ml, as a control for heparinase) at 37 °C for 2 h, washed, lysed in 2× SDS sample buffer, and analyzed by Western blotting with anti-v5 antibody to determine whether heparinases have dissociated Ang-3 protein from LLCAng-3 cell surface.
To determine whether glycosylphosphatidylinositol-anchored glypicans bind to Ang-3, LLCAng-3 cells were treated with or without 1 unit/ml phosphatidylinositol-specific phospholipase C (PI-PLC) at 37 °C for 2 h. These LLC Ang-3 cells were washed and lysed with radioimmune precipitation assay buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1 μg/ml pepstatin A, 1 mm phenylmethyl-sulfonyl fluoride). 30 μl (50 μg) of lysed proteins was digested with or without heparinases I and III (1 unit/ml) at 37 °C for 6 h (26), and the reactions were terminated by adding 5 μl of 8× SDS sample buffer and analyzed by Western blot with anti-glypican-1 (Santa Cruz Biotechnology) or anti-v5 antibody. Immunoprecipitation was performed using the protein G bead-bound anti-glypican-1 antibody and the supernatants of the PI-PLC digestion reactions. The immunoprecipitated proteins were digested with or without heparinases as described above and analyzed by Western blot for the presence of glypican-1 and Ang-3 protein.
Production and Purification of Angiopoietins
Five liters of cell culture supernatants derived from LLC transfectants expressing mouse or human Ang-1, Ang-2, or Ang-3 protein were collected and purified through Ni+-Probond affinity columns (Invitrogen), and the affinity columns were conjugated with anti-v5 antibodies (Sigma). The purity of the purified proteins was determined by silver staining of SDS-10% PAGE gel loaded with the purified proteins under reducing conditions. The results showed that v5-epitope-tagged Ang-1, Ang-2, and Ang-3 reached more than 90% purity after two rounds of affinity purification, which was demonstrated by the presence of a single silver-stained band with the appropriate molecular weight. The concentration of the purified proteins was determined by Bio-Rad Bradford protein assay using a serial diluted bovine serum albumin (BSA, 2 mg/ml) as a standard.
Solid Phase and Cell-based Ligand Binding Assay
96-well ELISA plates were coated overnight at 4 °C with heparin, chondroitin sulfate, or hyaluronic acid (HA, 1 mg/ml, Sigma), or BSA (1 mg/ml, Sigma) in triplicate. The coated plates were washed and blocked with 0.5% BSA. Purified Ang-1v5 or Ang-3v5 proteins (500 ng/ml) were applied to the coated plates and incubated for overnight at 4 °C. After extensive washing with phosphate buffer containing 300 mm NaCl, the 96-well bound Ang-1v5 or Ang-3v5 proteins were detected and measured.
The cell-based ligand binding assays were performed by incubating purified Ang-3v5 (1 μg) or Ang-1v5 (1 μg, as a control of Ang-3v5) with the EDTA-lifted 1 × 106 of C2C12, A10, COS-7, and LLC cells; or by incubating purified Tie-2-Fc (2 μg, P&D System) or CD-8-Fc (2 μg, as a control of Tie-2-Fc) with the EDTA-lifted LLCAng-1, LLCAng-2, or LLCAng-3 cells at 4 °C for 2 h. The cells were then washed with PBS and lysed in 2× SDS sample buffer. The cell surface-bound v5-tagged angiopoietins or Tie-2-Fc fusion proteins were detected by Western blotting with anti-v5 mAb or anti-human IgG antibody, respectively.
Heparin Affinity Column
To determine the binding profile and relative affinity of Ang-3 to heparin, 20 μg of purified Ang-3v5 was applied into a heparin affinity column (heparin-conjugated agarose, Sigma). Ang-3 protein was eluted from the column using non-continuous gradient of NaCl (0.15, 0.3, 0.6, and 1.2 m). Three fractions (2 ml/each) were collected for each NaCl concentration, and a 20-μl sample from each fraction was analyzed by Western blotting with anti-v5 mAb.
Endothelial and Tumor Cell Co-culture Assay
Subconfluent LLCAng-3, LLCAng-2, and LLCAng-1 cells were labeled with CellTracker Green CMFDA fluorescein (Molecular Probes) as described previously (27) and lifted with the EDTA solution, which was known to preserve Ang-3 protein on the cell surface. These EDTA-lifted cells or soluble angiopoietin (200 ng/ml) were applied to the monolayers of bovine pulmonary artery endothelial cells (CAPE, ATCC) in triplicate for 4 h at 37 °C. The cells were then fixed and observed under microscope and photographed, and the endothelial retraction lesions were counted in ten randomly selected microscopic (40×) fields.
Immuocytochemistry
LLC transfectants expressing Ang-1, Ang-2, or Ang-3, or transfected with the empty expression vector were cultured in 35-mm dishes until confluence. Some cells were lifted with the EDTA solution as described (20), and the cell-free ECM and the confluence LLC transfectants were fixed with methanol at −20 °C for 15 min. The fixed cells and ECM were washed and blocked, and antibodies against v5 epitope, Ang-1, Ang-2, Ang-3, and perlecan were used to detected Ang-1, -2, -3, or perlecan.
To investigate the relative localization of Ang-3 protein and heparan sulfate (HS) on the LLCAng-3 cell surface, an immunocolocalization experiment was performed using anti-v5 antibody and TRITC-conjugated secondary antibody to detect Ang-3v5 protein and anti-heparan sulfate (Calbiochem) antibody and FITC-conjugated secondary antibody to detect HS, respectively. To determine the relative localization of Ang-3 and perlecan, glypican, or syndecan and determine whether HS side chains on proteoglycans are required for the binding of Ang-3 to cell surface, LLCAng-3 cells were cultured in the absence or presence of 100 mm sodium chlorate and fixed. Immunocolocalization experiments were performed using anti-v5 antibody with TRITC-conjugated secondary antibody to detect Ang-3v5, and anti-perlecan (NeoMarkers) or anti-glypican-1 antibody with FITC-conjugated secondary antibody to detect perlecan or glypican-1, respectively, or using FITC-conjugated anti-v5 antibody to detect Ang-3v5 protein and phycoerythrin-conjugated anti-syndecan-1 (BD Biosciences) antibody to detect syndecan-1. The inhibitory effect of sodium chlorate on the synthesis of HS in LLCAng-3 cells was revealed by anti-HS antibody (Calbiochem).
RESULTS
Unlike Ang-1 and Ang-2, Ang-3 Is Retained on the Surface of Various Cells
It was noted previously that, unlike producing Ang-2, it is difficult to produce and purify Ang-1 and Ang-3 proteins (13, 14). We have demonstrated recently that, unlike Ang-2, which is primarily secreted, Ang-1 is secreted and incorporated into the ECM and the binding of Ang-1 to the ECM regulates availability and activity of Ang-1 (20). To determine whether Ang-3 binds to the ECM as well, we investigated the distribution pattern of Ang-3 protein in the established LLC transfectants expressing the C-terminal v5 epitope-tagged Ang-3 (Ang-3v5). The v5 epitope is a 14-amino acid epitope derived from P and V proteins of the paramyxovirus, SV5 (28).
The proteins used in the Western blotting were derived from culture supernatants, from the ECM materials deposited on the cell culture dishes, and from the EDTA-lifted LLC transfectants expressing v5-tagged Ang-1 (LLCAng-1), Ang-2 (LLCAng-2), or Ang-3 (LLCAng-3). The results showed that Ang-2 is largely secreted, Ang-1 is incorporated into the ECM, and a large fraction of Ang-3 is associated with the transfected cells (Fig. 1). The similar result was obtained in several other cell lines tested, including COS-7, 293, and TA3 mammary carcinoma cells (data not shown). Like all the proteins containing the coiled-coil domain, angiopoietins are aggregated to form dimers and oligomers through their coiled-coil region, and treatment of the proteins with β-mercaptoethanol releases these aggregates to monomers (Refs. 12–15 and data not shown). Angiopoietins are glycosylated, which causes the multiple banding patterns in Western blot (12–14). The ECM-bound Ang-1 is aggregated to form oligomers, whereas the cell-associated Ang-3 exists in monomeric, dimeric, and some oligomeric forms (Figs. 1C and 2).
Fig. 1.
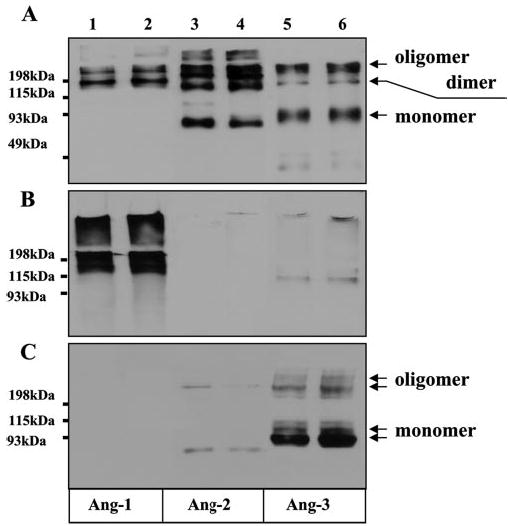
Ang-3 protein is associated with LLC cells. Western blotting was performed using anti-v5 antibody to determine the distribution patterns of the v5-epitope-tagged Ang-1 (Ang-1v5), Ang-2 (Ang-2v5), and Ang-3 (Ang-3v5) in the cell culture supernatants (A), the ECM materials (B), and the EDTA-lifted LLC transfectants (C) that express Ang-1 (lanes 1 and 2), Ang-2 (lanes 3 and 4), or Ang-3 (lanes 5 and 6). The Western blot analyses were performed under non-reducing conditions. Arrows indicate the monomer, dimmer, and oligomer of Ang-3.
To determine whether the cell surface-bound Ang-3 binds to Tie-2-Fc fusion protein, the methanol-fixed LLCAng-3 cells were washed, blocked, and incubated with Tie-2-Fc. The cell surface-bound Tie-2-Fc fusion protein was revealed by TRITC-conjugated anti-human Fc antibody, and Ang-3v5 protein in the same cells was detected with anti-v5 mAb with FITC-conjugated anti-mouse secondary antibody.
Fig. 2.
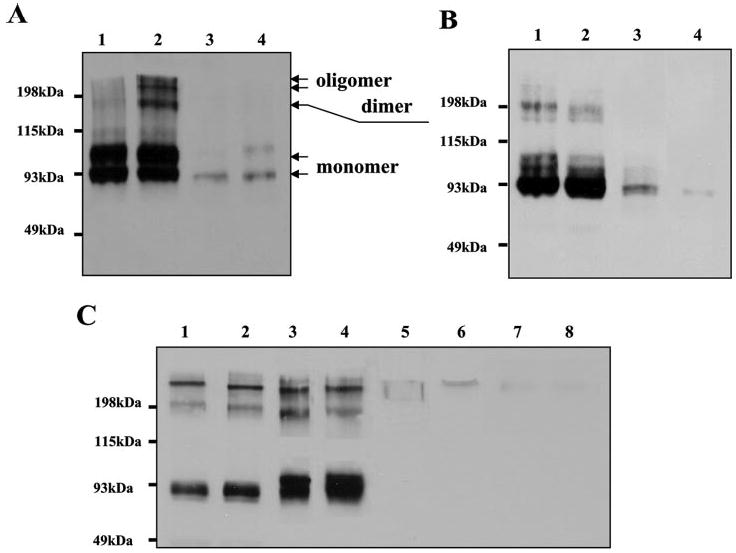
Ang-3 binds to the surface of various cells. A, the binding of Ang-3v5 to LLC cells is sensitive to the trypsin treatment (10 min in room temperature with 500 μg/ml trypsin). Two independent clonal LLCAng-3 transfectants were treated and lifted with trypsin (lanes 3 and 4) or EDTA solution (lanes 1 and 2), and these cells were washed and lysed with 2× SDS sample buffer and analyzed by Western blotting with anti-v5 mAb. B, Ang-3v5 protein is present in the cell membrane fraction. LLCAng3 transfectants were fractionated into the crude cell membrane (lanes 1 and 2) and soluble cytosolic fractions (lanes 3 and 4) as described previously (44). 30 μg of proteins from each fraction was analyzed by Western blotting with anti-v5 mAb. C, the cell-based binding assay was performed by incubating the purified Ang-3v5 (1 μg, lanes 1–4) or Ang-1v5 (1 μg, lanes 5–8) with 1 × 106 of C2C12 (lanes 1 and 5), A10 (lanes 2 and 6), COS-7 (lanes 3 and 7), and LLC (lanes 4 and 8) cells at 4 °C for 2 h. After washing with PBS, the cells were lysed in 2× SDS sample buffer. The cell-bound v5-tagged angiopoietins were detected by Western blotting with anti-v5 mAb. Arrows indicate the monomer, dimer, and oligomer of Ang-3.
To determine whether endogenous Ang-3 behaves in the same way as the transfected one, we performed Western blot analysis of the proteins derived from C2C12 myoblasts and A10 embryonic aortic smooth muscle cells, both of which express endogenous Ang-3 as assessed by RT-PCR (45). Proteins derived from lysates of these cells were analyzed by Western blotting with anti-Ang-3 antibody (Santa Cruz Biotechnology), and the results showed that Ang-3 protein is associated to A10 and C2C12 cells as well (data not shown).
To determine whether the association of Ang-3 with LLC cells merely reflects the presence of Ang-3 protein in the intra-cellular secretory pathway of the cells or represents binding of Ang-3 to the cell surface, we performed the following experiments. First, LLCAng-3 cells were treated with trypsin (0.5 mg/ml) or EDTA solution for 10 min at room temperature, lifted, washed, lysed, and analyzed by Western blotting with anti-v5 mAb. The result showed that the treatment of trypsin, but not EDTA, dislodges Ang-3 protein from the cells suggesting that Ang-3v5 is localized on the cell surface, where it is accessible to trypsin (Fig. 2A, lanes 3 and 4). To confirm this, we performed Western blot analysis of the proteins derived from the cell membrane and soluble cytosolic fractions of LLCAng-3 cells and found that Ang-3v5 is indeed present in the cell membrane fraction (Fig. 2B, lanes 1 and 2).
Finally, a cell-based binding assay was performed by incubating the EDTA-lifted C2C12, A10, COS-7, and LLC cells (1 × 106) with purified Ang-3v5 or Ang-1v5 (2 μg) at 4 °C for 2 h. The cells were washed with cold PBS, lysed, and analyzed by Western blotting with anti-v5 mAb. The result showed that Ang-3v5 but not Ang-1v5 binds to all the cells tested (Fig. 2C).
The Cell Surface-bound Ang-3 Is Capable of Binding to Tie-2-Fc Protein
RT-PCR results indicated that these non-endothelial cells that displayed the binding affinity to Ang-3 protein (Fig. 2C) do not express Tie-2 receptor (data not shown), suggesting that Ang-3 binds to these cell surface via binding partner(s) other than Tie-2 receptor. To determine whether the cell surface-bound Ang-3 is capable of binding to Tie-2-Fc fusion protein, 1 × 106 of the EDTA-lifted LLC transfectants (LLCAng-1, -2, and -3) were incubated with 2 μg of Tie-2-Fc or CD8-Fc (as a negative control) at 4 °C for 2 h. After extensive washing, the cells were lysed and Western blotting was performed using anti-human IgG antibody to detect cell surface-bound Tie-2-Fc or CD8-Fc. The result showed that Tie-2-Fc but not CD8-Fc binds to LLCAng-3 cells but not to LLCAng-1 or LLCAng-2 cells (Fig. 3A).
Fig. 3.

The cell surface-bound Ang-3 is capable of binding to Tie-2-Fc protein. A, Tie-2-Fc binds to LLCAng-3 cells. The binding assay was performed by incubating 2 μg of Tie-2-Fc (lanes 2, 4, and 6) or CD8-Fc (lanes 3, 5, and 7) fusion protein with 1 × 106 of the EDTA-lifted LLCAng-1 (lanes 2 and 3), LLCAng-2 (lanes 4 and 5), and LLCAng-3 (lanes 6 and 7) cells at 4 °C for 2 h. The cell surface-bound Tie-2-Fc proteins were visualized by Western blotting with HRP-conjugated anti-human IgG antibody. 100 ng of purified Tie-2-Fc fusion proteins were loaded in lane 1. B, the binding pattern of Tie-2-Fc to the cell surface-bound Ang-3 was revealed by applying 2 μg/ml Tie-2-Fc (B, panel b) or CD8-Fc (B, panel c) to the fixed LLCAng-3 cells with (B, panel d) or without (B, panel b) prior to incubation with 20 μg/ml purified soluble Ang-3. The cell surface-bound Tie-2-Fc or CD8-Fc was revealed by FITC-conjugated anti-human Fc antibody. The distribution of Ang-3v5 was revealed by anti-v5 antibody (B, panel a). C, Tie-2-Fc binds to LLC cell-tethered Ang-3v5. The fixed LLCAng-3 cells were reacted with Tie-2-Fc with TRITC-conjugated anti-human Fc antibody (C, panel b) and anti-v5 mAb with FITC-conjugated secondary antibody (C, panel a). Panels a and b were merged to show the colocalization of Tie-2-Fc and Ang-3 v5 in yellow color (C, panel c). Bar, 30 μm.
To confirm this result, confluent LLCAng-3 cells were fixed and blocked, and the binding assay was performed by applying 2 μg/ml Tie-2-Fc or CD8-Fc proteins (with or without prior incubation with 20 μg/ml soluble Ang-3 protein) to these fixed cells. The cell surface-bound Tie-2-Fc was revealed by FITC-conjugated anti-human Fc antibody. The result showed that Tie-2-Fc but not CD8-Fc binds to the surface of LLCAng-3 cells in a pattern that is similar to that of cell surface-tethered Ang-3 protein, and the binding is blocked by the prior incubation of T-e-2-Fc with an excess amount of soluble Ang-3 protein (Fig. 3B). To demonstrate the colocalization of the cell surface-tethered Ang-3 and Tie-2-Fc fusion protein, the distribution of Ang-3 and Tie-2-Fc was revealed by anti-v5 antibody with the FITC-conjugated secondary antibody, and TRITC-conjugated anti-human Fc antibody, respectively. The result showed that Tie-2-Fc is colocalized with the cell surface-tethered Ang-3 (Fig. 3C, panel c). Together, these results suggest that tethering Ang-3 on the cell surface likely provides a mechanism to localize, concentrate, and present Ang-3 to its receptor, rather than to sequester Ang-3, and that the domains of Ang-3 that bind to Tie-2 and the putative cell surface binding partner(s) are different.
The Coiled-coil Domain of Ang-3 Mediates the Cell Surface Binding of Ang-3
To determine which domain of Ang-3 mediates the binding of Ang-3 to the cell surface, we made several Ang-3 deletion constructs, which contain the coiled-coil domain plus the linker peptide region, the coiled-coil domain, or the linker peptide region plus the FHD (Fig. 4A). The cDNA fragment encoding the signal peptide (SP) of Ang-3 was constructed to the amino-terminal of all the deletion fragments, so that they can be expressed and secreted properly (20). In addition, v5-epitope was tagged at the carboxyl termini of these fragments for easy identification. The expression constructs containing full-length Ang-3 and various Ang-3 fragments were used to transfect COS-7 cells in triplicate.
72 h after the transfection, one set of the transfected COS-7 cells was treated with trypsin (0.5 mg/ml) at room temperature for 10 min, which was known to release the cell surface-bound Ang-3 (Fig. 2A). Equal amounts of the proteins, derived from culture supernatants (Fig. 4B, panel a) or cell lysates (Fig. 4B, panels b and c) of the transfected COS-7 cells that were treated with (panel c) or without (panel b) trypsin, were analyzed by Western blotting with anti-v5 mAb. The results showed that, like full-length Ang-3, the coiled-coil fragment of Ang-3 binds to the cells and the binding is sensitive to the trypsin treatment. On the contrary, the FHD fragment of Ang-3 is either secreted or resides inside the transfected cells and is insensitive to the trypsin treatment (Fig. 4B, panels b and c, lane 4). This result suggests that the coiled-coil domain of Ang-3 mediates the binding of Ang-3 to the cell surface. This conclusion was further supported by the immunocytochemistry analysis of these transfected COS-7 cells. The results showed that the coiled-coil fragment of Ang-3 displayed a cell surface distribution pattern that is similar to that of full-length Ang-3, whereas the FHD fragment is indeed localized in the intracellular compartment (data not shown).
Perlecan Is a Major HSPG That Mediates the Cell Surface Binding of Ang-3
Our RT-PCR results indicated that the cells that displayed binding affinity to Ang-3 (Fig. 2C) do not express Tie-2 (data not shown), suggesting that Ang-3 binds to the cell surface via protein(s) other than Tie-2. To determine which of the one or more cell surface proteins binds to Ang-3, we first investigated localization of Ang-3 in LLCAng-3 cells by immunocytochemistry. We found that the localization of Ang-3 resembles that of HS glycosaminoglycans (GAGs) observed previously (not shown). To determine whether Ang-3 is tethered on the cell surface via HS, we performed a solid phase binding assay to assess the binding affinity of purified Ang-3v5 protein to heparin (an analog of HS), chondroitin sulfate, and hyaluronan (HA); the latter two of which are both negatively charged GAGs and used as controls of heparin. Purified Ang-1v5 was used as a control of Ang-3v5 in the assay. The result showed that Ang-3v5 but not Ang-1v5 binds to heparin but not to chondroitin sulfate or HA (Fig. 5A), suggesting that Ang-3 likely binds to the cell surface via HSPGs.
Fig. 5.

Ang-3 binds to and colocalizes with HS. A, Ang-3 specifically binds to heparin in a solid phase binding assay. Heparin, chondroitin sulfate (CS), hyaluronan (HA), and BSA (1 mg/ml) were coated onto 96-well ELISA plates in triplicate. Ang-3v5 or Ang-1v5 (500 ng/ml) was incubated with the plates at 4 °C overnight. After washing, the ELISA plate-bound Ang-3v5 or Ang-1v5 was detected and measured. B, binding of Ang-3 to a heparin affinity column. 20 μg of Ang-3v5 was applied onto a heparin column. The flow through (FT) was collected (lane 2), and the column was washed with a non-continuous gradient of NaCl from 0.15 m (lanes 3–5), 0.3 m (lanes 6–8), 0.6 m (lanes 9–11), to 1.2 m (lanes 12–14), and collected for Western blot analysis using anti-v5 mAb. 100 ng of Ang-3v5 was loaded into lane 1. C, Ang-3 colocalizes with HS on the LLCAng-3 cell surface. Confluent LLCAng-3 (C, panels a–c and f), LLCAng-1 (C, panel d), and LLCAng-2 (C, panel e) cells were fixed, and the localization of Ang-3v5 and HS were detected with anti-v5 mAb and TRITC-conjugated secondary antibody (C, panel a, red fluorescence) or anti-HS antibody and FITC-conjugated secondary antibody (C, panel b, green fluorescence), respectively. In C, panels a and b were merged to show the colocalization of Ang-3v5 and HS in yellow color (C, panel c). Ang-1v5 (C, panel d) and Ang-2v5 (C, panel e) were detected by anti-v5 mAb. In C, panel f, only secondary antibody was used. Bar, 32 μm. D, binding of Ang-3 to the cell surface is sensitive to heparinase treatment. 1 × 106 of LLCAng-3 cells were incubated with 50 mm Tris-HCl buffer alone (150 mm NaCl, 0.1% BSA, and 3.5 mm CaCl2, lane 1), or containing heparinase I (5 units/ml) plus heparinase III (0.5 unit/ml, lane 2), or Streptomyces hyaluronidase (lane 3, 5 units/ml) at 37 °C for 2 h, washed, and lysed. The remaining cell-bound Ang-3v5 was detected by Western blotting with anti-v5 mAb.
To determine the binding profile and relative affinity of Ang-3 to heparin, we applied purified Ang-3 to a heparin affinity column, and Ang-3 was eluted using non-continuous gradient of NaCl (0.15, 0.3, 0.6, and 1.2 m). The result showed that Ang-3 protein was eluted at two different salt concentrations, 0.3 and 0.6 m NaCl, which implies the presence two subsets of Ang-3 proteins that bind to heparin with different affinities (low and high, Fig. 5B).
To determine whether Ang-3 colocalizes with HS on the cell surface, we performed immunocolocalization experiment using anti-v5 mAb and TRITC-conjugated secondary antibody to detect Ang-3v5 and anti-heparan sulfate antibody and FITC-conjugated secondary antibody to detect HS, respectively. Our results showed that Ang-3v5 is colocalized with HS on the surface of LLCAng-3 cells (Fig. 5C, panels a–c). It is well established that HS can exist as side chains of protein core structures in the forms of HSPGs, which are present in the ECM and on the cell surface (29–31). To confirm that Ang-3 binds to the cell surface via HSPGs, the EDTA-lifted LLCAng-3 cells were treated with heparinase or hyaluronidase (as a control of heparinase) at 37 °C for 2 h, washed, lysed, and analyzed by Western blotting with anti-v5 mAb. The results showed that the treatment of LLCAng-3 cells with heparinase but not hyaluronidase releases Ang-3v5 from the cell surface (Fig. 5D), suggesting that Ang-3 is bound to the cell surface through HSPGs.
The major cell surface HSPGs are syndecans, glypicans, CD44 variants containing v3 exon (CD44v3) (32, 33), and perlecan (21, 30, 31). Our RT-PCR results demonstrated that LLC cells express syndecan-1, -2, and -4, glypican-1, -2, -3, and -6, and perlecan, and LLC cells express little CD44 variant isoforms containing v3 exon (data not shown). Thus, CD44 is not likely involved in the binding of Ang-3 to LLC cell surface. To investigate whether syndecans bind to Ang-3, immunoprecipitation was performed by incubating Ang-3v5 with the protein-A beads that were bound with syndecan-1-Fc, syndecan-2-Fc, syndecan-4-Fc fusion proteins, or human IgG (as a control of syn-Fc). The results showed that syndecan-1-Fc and to a lesser extent syndecan-4-Fc and syndecan-2-Fc, but not human-IgG, bind to Ang-3v5 (data not shown). This result indicates that syndecans binds to Ang-3 in vitro.
To determine whether syndecan, perlecan, and/or glypican are responsible for tethering Ang-3 on surface of the cells, we performed immunocolocalization of Ang-3v5 and perlecan, syndecan-1, or glypican-1. The results showed that Ang-3v5 is colocalized with perlecan in both LLC and TA3 cells (Figs. 6 and 7 (G–I)), however, Ang-3 did not display significant colocalization with syndecan-1 (Fig. 7, A–C) or glypican-1 (Fig. 7, D–F). To determine whether HS side chains on perlecan are required for the binding of Ang-3 to the cell surface, LLCAng-3 cells were cultured in the absence or presence of 100 mm sodium chlorate. The inhibitory effect of sodium chlorate on synthesis of HS was demonstrated by the absence of HS on the chlorate-treated LLCAng-3 cells using anti-HS antibody (Fig. 6H). Immunocolocalization experiments showed that perlecan core protein, lacking HS side chains, is incapable of tethering Ang-3 to LLC cells. We only detected Ang-3 protein located inside of the chlorate-treated LLCAng-3 cells (Fig. 6F). Together, these data suggest that perlecan tethers Ang-3 to the cell surface through HS side chains.
Fig. 6.
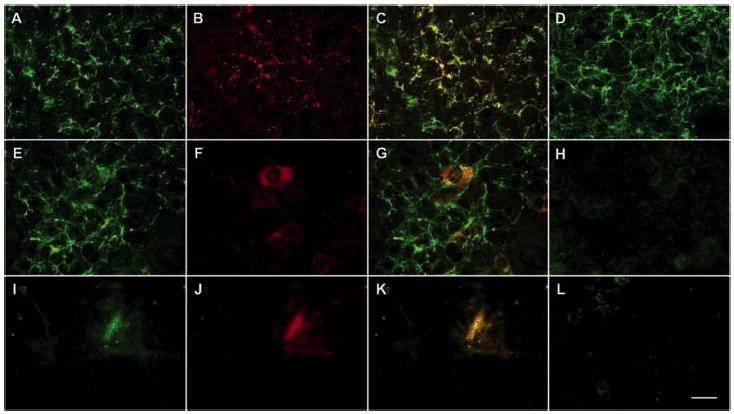
Ang-3 is colocalized with perlecan on the surface of LLCAng-3 cells, and HS side chains on perlecan are required for the colocalization. LLCAng-3 cells were cultured in the presence (E–H) or absence (A–D and I–L) of 100 mm sodium chlorate. The confluence cells (A–H, and L) or the cell-free ECM (I–K) were fixed. The localizations of perlecan and Ang-3v5 on the surface of LLCAng-3 cells (A–H) or in the ECM materials (I–K) deposited by LLCAng-3 cells were detected with anti-perlecan antibody and FITC-conjugated secondary antibody (A, E, and I; green fluorescence) and anti-v5 mAb and TRITC-conjugated secondary antibody (B, F, and J; red fluorescence), respectively. Panels A and B, E and F, and I and J were merged to show the colocalization of Ang-3v5 and perlecan HSPG on the LLCAng-3 cell surface (C) in the ECM (K) in yellow color and non-colocalization of Ang-3v3 and perlecan core protein on the LLCAng-3 cell surface (G). HS synthesized by LLCAng-3 cells was revealed by anti-HS antibody (D and H). In panel L, only secondary antibody was used. Bar, 35 μm.
Fig. 7.

Ang-3 is colocalized with perlecan but not with syndecan-1 or glypican-1. LLCAng-3 (A–F) and TA3Ang-3 (G–I) cells were fixed, and the relative localization of Ang-3v5 and syndecan-1 were detected with FITC-conjugated anti-v5 mAb (A, green fluorescence) or TRITC-conjugated anti-syndecan-1 antibody (B, red fluorescence), respectively. Panels A and B were merged to show that there is little colocalization between Ang-3v5 and syndecan-1 (C). The relative localizations of glypican-1 (D, green fluorescence) and Ang-3v5 (E, red fluorescence) were revealed by anti-glypican-1 antibody with FITC-conjugated secondary antibody and anti-v5 mAb with TRITC-conjugated secondary antibody, respectively. Panels D and E were merged to show that there is little colocalization between Ang-3v5 and glypican-1 (F). The relative localizations of Ang-3v5 (G, green fluorescence) and perlecan (H, red fluorescence) in the fixed TA3Ang-3 cells were revealed by anti-v5 mAb with FITC-conjugated secondary antibody and anti-perlecan antibody with TRITC-conjugated secondary antibody, respectively. Panels G and H were merged to show that Ang-3v5 and perlecan are colocalized on surface of these cells (I). Bar, 35 μm.
Western blot result demonstrated that some Ang-3 is deposited into the ECM (Fig. 1). To investigate whether the ECM-bound Ang-3 is colocalized with perlecan, confluent Ang-3 cells were lifted with the EDTA solution, the cell-free ECM on the cell culture dishes were fixed, and immunocytochemistry was performed to detect the ECM-bound Ang-3 or perlecan. The result showed that the EDTA treatment lifted the LLC cells and the pericellular perlecan; however, there were patches of perlecan remaining in the cell-free ECM (Fig. 6I), and Ang-3 was found to be colocalized with these perlecan patches in the ECM (Fig. 6J and K).
To further confirm that syndecan and glypican are not the major HSPGs that are responsible for the cell surface binding of Ang-3, we performed the following biochemical experiments. We treated LLCAng-3 cells without or with PI-PLC, which releases glycosylphosphatidylinositol-anchored proteins, including glypicans from the cell surface. The cells were washed and lysed. The protein lysates were digested with or without heparinases and analyzed for the presence of glypican-1 or Ang-3, respectively, by Western blotting. The reaction supernatants were immunoprecipitated to recover cleaved glypican-1 and potential glypican-1-bound Ang-3. The results showed that the PI-PLC treatment did release a large fraction of glypican-1 from the cell surface; however, it had little effect on the cell surface binding of Ang-3. In addition, the released glypican-1 did not form a complex with Ang-3 protein in the reaction supernatant, suggesting that glypicans are not the major cell surface HSPGs that bind to Ang-3 in LLC cells (Fig. 8C).
Fig. 8.
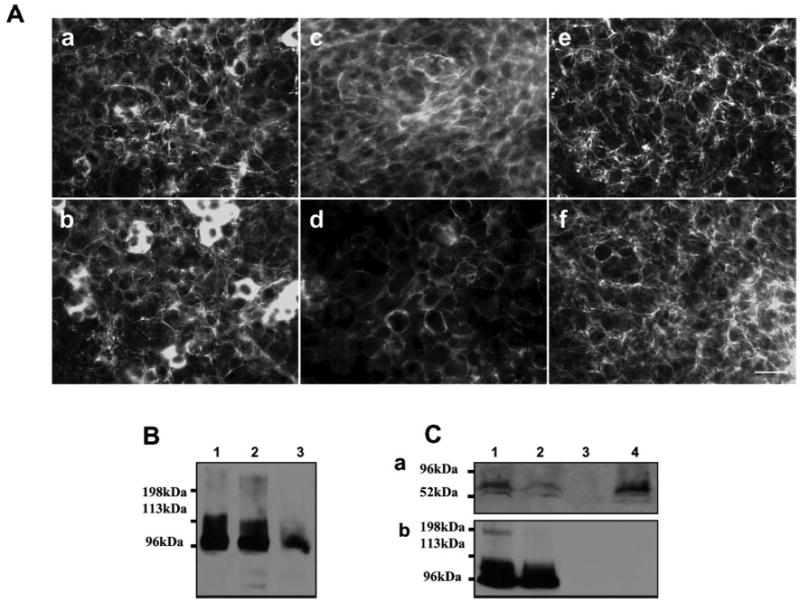
Syndecan-1 and glypican-1 do not play major roles in tethering Ang-3 on LLC cell surface. LLCAng-3 cells were treated without (A, panels a, c, and e; B, lane 1) or with (A, panels b, d, and f; B, lane 2) 10 μg/ml trypsin on ice for 15 min, or with 100 μg/ml trypsin in room temperature for 15 min (B, lane 3). These cells were either lysed and analyzed by Western blotting with anti-v5 antibody (B) to detect the cell-bound Ang-3v5 or fixed and analyzed by immunocytochemistry (A) to detect the cell-bound Ang-3v5, syndecan-1, or perlecan using anti-v5 (A, panels a and b), anti-syndecan-1 (A, panels c and d), or anti-perlecan (A, panels e and f) antibody. Bar, 35 μm. C, LLCAng-3 cells were treated without (C, panels a and b, lane 1) or with 1 unit/ml phosphatidylinositol-specific phospholipase C (PI-PLC, C, panels a and b, lane 2) at 37 °C for 2 h, washed, and lysed. The lysed proteins were analyzed directly for the cell surface-tethered Ang-3 (C, panel b, lanes 1 and 2) or digested with heparinases I and III (1 unit/ml) at 37 °C for 6 h and analyzed by Western blot with anti-glypican-1 antibody to detect cell surface-bound glypican-1 (C, panel a, lanes 1 and 2). Immunoprecipitation was performed using the protein G bead-bound anti-gnypican-1 antibody and the supernatants derived from the PBS treated (lane 3) or PI-PLC digestion (lane 4) LLCAng-3 cells. The immunoprecipitated proteins were analyzed by Western blot for the presence of glypican-1 (C, panels a, lanes 3 and 4) or Ang-3 protein (C, panels b, lanes 3 and 4).
To confirm that syndecan-1 play no major role in tethering Ang-3 on LLC cells, we treated LLCAng-3 cells without or with 10 μg/ml trypsin on ice for 15 min, which is known to be sufficient to cleave syndecans from the epithelial cell surface (34), or with 100 μg/ml trypsin in room temperature for 15 min. The cells were then washed and lysed for Western blotting analysis. The mild trypsin treatment (10 μg/ml) did not lift LLCAng-3 cells from the culture dishes, which allowed us to perform immunocytochemistry to determine whether the treatment released syndecan-1 from the cell surface. Our results showed that there is a significant reduction of syndecan-1 but not perlecan-1 level on the surface of these LLCAng-3 cells, which were treated with 10 μg/ml trypsin (Fig. 8A and B); however, there was a small change in the level of the cell surface-bound Ang-3 (Fig. 8A and B), suggesting that syndecan-1 does not play a major role in tethering Ang-3 on the surface of LLC cells. Together, these results (Fig. 6 –8) indicate that, at least in LLC and TA3 cells, perlecan is the major HSPG that tethers Ang-3 to the cell surface.
The Cell Surface-bound Ang-3 Induces Retraction and Loss of Integrity of the Endothelial Monolayer
It is well established that the interaction between endothelial-endothelial cells and endothelial-periendothelial cells is important for maintaining the vascular integrity and formation of functional blood vessels. Ang-1 has been shown to play an important role in maintaining integrity of blood vessels (35–40). To assess function of the cell surface-tethered Ang-3, we performed tumor and endothelial cell co-culture assay by seeding green fluorescein-labeled LLCAng-3, LLCAng-2, and LLCAng-1 cells onto the monolayers of bovine pulmonary artery endothelial (CAPE) cells. As controls, purified soluble angiopoietins (200 ng/ml) alone were applied to the monolayers. The cells were cultured for 4 h at 37 °C, fixed, and observed under a microscope. The results demonstrated that the cell surface-bound Ang-3, but not soluble Ang-3, LLCAng-1, or LLCAng-2 cells, induces retraction and loss of integrity of the CAPE monolayers (Fig. 9).
Fig. 9.

The cell surface-bound Ang-3 induces retraction and loss of integrity of the endothelial monolayers. 5 × 105 of green fluorescein-labeled LLCAng-3 (G–I), LLCAng-1 (A–C), or LLCAng-2 (D–F) cells in 2% FBS medium, 2% FBS medium alone (J), or containing soluble Ang-1 (K) or Ang-3 (L, 200 ng/ml) were applied to the monolayers of CAPE (bovine pulmonary artery endothelial) cells for 4 h at 37 °C. The cells were fixed and observed under light (A, D, G, J, K, and L) or fluorescence microscope (B, E, and H). The images under the same microscopic fields of light or fluorescence were overlaid on top of each other to show the exact location of the green fluorescein-labeled tumor cells and the unlabeled CAPE cells (C, F, and I). The results showed that the cell surface-bound Ang-3, but not the soluble Ang-3, LLCAng-1, or LLCAng-2 cells, induces retraction and loss of integrity of CAPE monolayers. Bar, 150 μm.
The extent of endothelial monolayer retraction was determined by counting the retraction lesions in ten randomly selected 40× microscopic fields. There are 17 retraction lesions/microscopic field that were induced by LLCAng-3 cells, whereas only 0.2, 0.33, or 0 retraction lesions/microscopic field were induced by LLCAng-1, LLCAng-2 cells, or by soluble Ang-3 alone. Thus, we have demonstrated that inducing endothelial monolayer retraction is a specific function of the cell surface-tethered Ang-3. This result suggests that binding of Ang-3 to the cell surface via HSPG regulates Ang-3 activity.
DISCUSSION
Ang-3 Is Tethered on the Cell Surface via HSPGs, Especially Perlecan, and the Cell Surface-tethered Ang-3 Binds to Tie-2-Fc Fusion Protein
In our current study, we demonstrated that, unlike Ang-1 and Ang-2, Ang-3 binds to the cell surface via the interaction between its coiled-coil domain and HSPGs. The cell surface binding of Ang-3 is saturable, and the access amount of Ang-3 is present in soluble form and secreted (Fig. 1 and data not shown), suggesting the local concentration of HSPGs can be used to establish the Ang-3 gradient, which may be important during morphogenesis of the blood vessels. At least in LLC and TA3 cells, perlecan is a major HSPG that tethers Ang-3 to the cell surface, which does not exclude the possibility that other HSPGs may mediate cell surface binding of Ang-3 in other cell types.
HSPGs often serve as binding proteins for growth factors and play important roles in modulating functions of these factors (29–31, 41). In the current study, we have shown that the cell surface-bound Ang-3 is capable of binding to Tie-2-Fc fusion protein (Fig. 3). This result suggests that, instead of sequestering Ang-3 from its receptor, tethering of Ang-3 on the cell surface such as that of perivascular cells localizes and presents Ang-3 to Tie-2 receptor on adjacent endothelial cells to elicit specific local reaction. Furthermore, binding of Ang-3 to the cell surface via HSPGs and sharing the common binding partners with other HSPG-binding growth factors such as basic fibroblast growth factor and VEGF imply that Ang-3 may cross-talk with these factors, and vice versa.
It has been established that the coiled-coil region of angiopoietins is responsible for dimerization/multimerization of the proteins, whereas the FHD binds to Tie-2 receptor (13–15). We have shown that soluble Ang-3 protein tends to form oligomers, whereas the cell surface-bound Ang-3 tends to form monomers, dimers, and some oligomers (Figs. 1, 2, and 4). Because the HSPG binding domain of Ang-3 is in the coiled-coil region, binding of Ang-3 to HSPG is likely responsible for the altered aggregation pattern. Thus, tethering Ang-3 on the cell surface by HSPGs likely concentrates Ang-3 protein in the particular aggregated forms, which may generate the distinct local signal that is different from the one derived from soluble Ang-3 protein.
The Function of Ang-3 Is Regulated by Its Binding to Cell Surface
It is well established that the interaction between endothelial-endothelial cells and endothelial-periendothelial cells is important for maintaining vascular health and integrity. Ang-1 has been shown to enhance the interactions between endothelial-endothelial and endothelial-periendothelial mural cells (17, 35, 38, 39, 40, 42). However, previous work has shed little light on the function of Ang-3. In our present study, we have shown that the cell surface-bound Ang-3, but not soluble Ang-3, induces retraction and loss of integrity of the endothelial monolayers (Fig. 9). This result implied that the cell surface-bound Ang-3 plays a distinct important role in regulating endothelial cell behavior, and mechanisms that regulate the cell-surface binding of Ang-3 and release Ang-3 from the cell surface (including shedding of HSPGs from the cell surface by matrix metalloproteinases (30, 43)) likely regulate the function of Ang-3.
Very recently, we have shown that overexpression of Ang-3 inhibits pulmonary metastasis of LLC and TA3 mammary carcinoma cells by inhibiting tumor angiogenesis and promoting apoptosis of the tumor cells, and that the cell surface binding of Ang-3 is required for its effective inhibition on tumor angiogenesis and metastasis. Furthermore, we have demonstrated that Ang-3 inhibits endothelial cell proliferation and survival and blocks Ang-1- and VEGF-induced activation of extracellular signal-regulated kinase 1/2 and Akt kinase, which likely underlie the Ang-3-mediated inhibition on tumor angiogenesis and metastasis (45). Together with the results obtained in the current study, these results suggest that Ang-1 and Ang-3 play opposite roles in maintaining endothelial layer integrity and in inducing endothelial cell sprouting/retraction, proliferation, and survival. Ang-1 and Ang-3 likely represent two important factors that are produced by periendothelial cells and play antagonistic roles in maintaining health and integrity of blood vessels in adult tissues. Furthermore, these data imply that the balanced activity of Ang-1 and Ang-3 is important for angiogenesis during embryogenesis and tissue repairing; the imbalanced up-regulation of Ang-3 activity and/or down-regulation of Ang-1 activity may contribute to blood vessel regression and other vascular diseases such as atherosclerosis and restenosis.
HSPGs may enhance/regulate Ang-3 bioactivity by the following mechanisms. We found that Ang-3 protein is cleaved (data not shown). Thus, tethering Ang-3 to the cell surface via HSPGs likely protects Ang-3 from proteolytic cleavage and, thereby, extends its half-life. In addition, binding of Ang-3 to the cell surface via HSPGs concentrates Ang-3 protein in a small area in a certain configuration to evoke specific local reaction. Finally, Ang-3 and HSPG, together in a complex, may generate signals that are different from the ones derived from soluble Ang-3 and HSPGs separately.
Acknowledgments
We are grateful to Dr. Renato Iozzo for his excellent suggestions on the work related to perlecan. We also thank Dr. Ivan Stamenkovic and Dr. Wilfried Weber for their generous support and acknowledge the generous support of the School from the Commonwealth of Pennsylvania.
Footnotes
This work was supported by funds from the University of Pennsylvania, School of Veterinary Medicine and by National Institutes of Health Grant 5RO1HL074117.
The abbreviations used are: ECM, extracellular matrix: Ang-1, angiopoietin-1; Ang-2, angiopoietin-2; Ang-3, angiopoietin-3; HS, heparan sulfate; HSPG, heparan sulfate proteoglycan; HA, hyaluronan; CS, chondroitin sulfate; GAG, glycosaminoglycan; PBS, phosphate-buffed saline; mAb, monoclonal antibody; LLC, Lewis lung carcinoma; FHD, fibrinogen homology domain; BSA, bovine serum albumin; VEGF, vascular endothelial growth factor; RT, reverse transcriptase; CAPE, bovine pulmonary artery endothelial cells; FBS, fetal bovine serum; PI-PLC, phosphatidylinositol-specific phospholipase C; TRITC, tetramethyl rhodamine isothiocyanate; FITC, fluorescein isothiocyanate; ELISA, enzyme-linked immunosorbent assay.
References
- 1.Folkman J. N. Engl. J. Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Nat. Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D. Science. 1997;277:48–50. doi: 10.1126/science.277.5322.48. [DOI] [PubMed] [Google Scholar]
- 5.Ingber DE, Folkman J. Cell. 1989;58:803–805. doi: 10.1016/0092-8674(89)90928-8. [DOI] [PubMed] [Google Scholar]
- 6.Folkman J, D’Amore P. Cell. 1996;87:1153–1155. doi: 10.1016/s0092-8674(00)81810-3. [DOI] [PubMed] [Google Scholar]
- 7.Risau W. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 8.Sato TN, Quin Y, Kozak CA, Audus KL. Proc. Natl. Acad. Sci. U. S. A. 1993;90:9355–9358. doi: 10.1073/pnas.90.20.9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnurch H, Risau W. Development. 1993;119:957–968. doi: 10.1242/dev.119.3.957. [DOI] [PubMed] [Google Scholar]
- 10.Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, Breitman ML. Genes Dev. 1994;8:1897–1909. doi: 10.1101/gad.8.16.1897. [DOI] [PubMed] [Google Scholar]
- 11.Dumont DJ, Gradwohl GJ, Fong GH, Auerbach R, Breitman ML. Oncogene. 1993;8:1293–1301. [PubMed] [Google Scholar]
- 12.Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Cell. 1996;87:1161–1169. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- 13.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 14.Valenzuela DM, Griffiths JA, Rojas J, Aldrich TH, Jones PF, Zhou H, McClain J, Copeland NG, Gilbert DJ, Jenkins NA, Huang T, Papadopoulos N, Maisonpierre PC, Davis S, Yancopoulos GD. Proc. Natl. Acad. Sci. U. S. A. 1999;96:1904–1909. doi: 10.1073/pnas.96.5.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Procopio WN, Pelavin PI, Lee WMF, Yeilding NM. J. Biol. Chem. 1999;274:30196–30201. doi: 10.1074/jbc.274.42.30196. [DOI] [PubMed] [Google Scholar]
- 16.Sato TN, Tozawa Y, Deutsch U, Wolburg-Burcholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 17.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 18.Holash J, Maisonpierre D, Compton D, Boland P, Alexander CR, Zagzag D, Alexander CR, Zagzag D, Yancopoulos GD, Wiegland SJ. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 19.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 20.Xu Y, Yu Q. J. Biol. Chem. 2001;276:34990–34998. doi: 10.1074/jbc.M103661200. [DOI] [PubMed] [Google Scholar]
- 21.Iozzo RV. J. Clin. Invest. 2001;108:165–167. doi: 10.1172/JCI13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battaglia C, Aumailley M, Mann K, Mayer U, Timpl R. Eur. J. Cell Biol. 1993;61:92–99. [PubMed] [Google Scholar]
- 23.Chakravarti S, Horchar T, Jefferson B, Laurie GW, Hassell JR. J. Biol. Chem. 1995;270:404–409. doi: 10.1074/jbc.270.1.404. [DOI] [PubMed] [Google Scholar]
- 24.Brown JC, Sasaki T, Gohring W, Yamada Y, Timpl R. Eur. J. Biochem. 1997;250:39–46. doi: 10.1111/j.1432-1033.1997.t01-1-00039.x. [DOI] [PubMed] [Google Scholar]
- 25.Yu Q, Stamenkovic I. Am. J. Pathol. 2001;158:563–570. doi: 10.1016/S0002-9440(10)63998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleeff J, shiwata T, Kumbasar A, Friess H, Buchler MW, Lander AD, Korc M. J. Clin. Invest. 1998;102:1662–1673. doi: 10.1172/JCI4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Q, Toole BP, Stamenkovic I. J. Exp. Med. 1997;186:1985–1996. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Southern JA, Young DF, Heaney F, Gaumgartner W, Randall RE. J. Gen. Virol. 1991;72:1551–1557. doi: 10.1099/0022-1317-72-7-1551. [DOI] [PubMed] [Google Scholar]
- 29.Sanderson RD. Cell Dev. Biol. 2001;12:89–98. doi: 10.1006/scdb.2000.0241. [DOI] [PubMed] [Google Scholar]
- 30.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Annu. Rev. Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 31.Iozzo RV, San Antonio JD. J. Clin. Invest. 2001;108:349–355. doi: 10.1172/JCI13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett KL, Jackson DG, Simon JC, Tanczos E, Peach R, Modrell B, Stamenkovic I, Plowman G, Aruffo A. J. Cell Biol. 1995;128:687–698. doi: 10.1083/jcb.128.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson DG, Bell JI, Dickinson R, Timans J, Shields J, Whittle N. J. Cell Biol. 1995;128:673–685. doi: 10.1083/jcb.128.4.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rapraeger A, Bernfield M. J. Biol. Chem. 1985;260:4103–4109. [PubMed] [Google Scholar]
- 35.Koblizek TI, Weiss C, Yancopoulos GD, Deutsch U, Risau W. Curr. Biol. 1998;8:529–532. doi: 10.1016/s0960-9822(98)70205-2. [DOI] [PubMed] [Google Scholar]
- 36.Kim I, Moon S-O, Koh KN, Kim H, Uhm C-S, Kwak HJ, Kim N-G, Koh GY. J. Biol. Chem. 1999;274:26523–26528. doi: 10.1074/jbc.274.37.26523. [DOI] [PubMed] [Google Scholar]
- 37.Kim I, Kim HG, So J-N, Kim JH, Kwak HJ, Koh GY. Circ. Res. 2000;86:24–29. doi: 10.1161/01.res.86.1.24. [DOI] [PubMed] [Google Scholar]
- 38.Hayes AJ, Huang W-Q, Mallah J, Yang D, Lippman ME, Li L-Y. Microvasc. Res. 1999;58:224–237. doi: 10.1006/mvre.1999.2179. [DOI] [PubMed] [Google Scholar]
- 39.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 40.Thurston G, Rudge JS, offe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD. Nat. Med. 2000;6:460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 41.Rapraeger AC, Krufka A, Olwin BB. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 42.Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas L, Rudge J, Yancopoulos G, Vadas MA. Circ. Res. 2000;87:603–607. doi: 10.1161/01.res.87.7.603. [DOI] [PubMed] [Google Scholar]
- 43.Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M. Nat. Med. 1998;4:691–697. doi: 10.1038/nm0698-691. [DOI] [PubMed] [Google Scholar]
- 44.Yu Q, Stamenkovic I. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu, Y., Liu, Y.-j., and Yu, Q. (2004) Cancer Res, in press [DOI] [PubMed]