

Abstract
𝜋‐Conjugated polymers, including those based on acetylenic repeating units, are an exciting class of materials that offer narrow optical band gaps and tunable frontier orbital energies that lead to their use in organic electronics. This work expands the knowledge of structure‐property relationships of acetylenic polymers through the synthesis and characterization of a series of Glaser‐Hay‐coupled model compounds and random copolymers comprised of BF2 formazanate, fluorene, and/or bis(alkoxy)benzene units. The model compounds and copolymers synthesized exhibit redox activity associated with the reversible reduction of the BF2 formazanate units and the irreversible reduction of the fluorene and bis(alkoxy)benzene units. The copolymers exhibit absorption profiles characteristic or intermediate of their respective models and homopolymers, leading to broad absorption of UV–vis light. The alkyne linkages of the model compounds and copolymers are reacted with [Co2(CO)8] to convert the alkyne functional groups into cobalt carbonyl clusters. This transformation leads to blue‐shifted absorption profiles due to a decrease in π‐conjugation, demonstrating the ability to tune the properties of these materials through post‐polymerization functionalization. The redox activity and broad absorption bands of the polymers reported make them excellent candidates for use in photovoltaics and other light‐harvesting applications.
Keywords: alkynes, BF2 formazanate dyes, cyclic voltammetry, Glaser‐Hay Coupling, UV–vis spectroscopy
A series of Glaser‐Hay‐coupled model compounds and random copolymers comprised of combinations of BF2 formazanate (BF2 ), fluorene (FL), and/or bis(alkoxy)benzene (Bn) units exhibit redox activity and broad absorption of UV–vis light. Reactions of model compounds and copolymers with [Co2(CO)8] demonstrate the ability to tune optical properties through post‐polymerization structural variation.
![]()
1. Introduction
The scientific community is continuously exploring the development of new 𝜋‐conjugated polymers with unique charge‐transport, light absorption, and light emission properties for use in organic electronics and the health sciences.[ 1 , 2 ] 𝜋‐Conjugated polymers can be defined as polymers composed of alternating single and double/triple bonds in their backbone, where the 𝜋‐electrons are delocalized over the repeating monomeric units due to overlapping p orbitals of neighboring sp2 atoms.[ 1 , 3 ] These polymers are characterized by narrow optical band‐gaps, tunable frontier orbital energies, and semiconducting capabilities,[ 2 , 3 ] which afford their utility in organic electronics such as field effect transistors,[ 4 ] photovoltaics,[ 5 , 6 ] and light‐emitting diodes.[ 7 ] In comparison to traditional, purely inorganic materials, 𝜋‐conjugated polymers represent an exciting class of materials for the development of organic electronics due to their advantageous low cost, lightweight, efficiency, stability, flexibility, and solution processability.[ 3 ]
Acetylenic polymers are a class of 𝜋‐conjugated polymers that have gained interest in recent decades due to their interesting optical and electronic properties.[ 8 ] They contain repeat units that are linked together by unsaturated triple bonds which lead to electronic delocalization across the polymer backbone.[ 9 ] Alkyne linkages offer several beneficial traits to polymers such as increased 𝜋‐conjugation,[ 10 , 11 ] improved intermolecular packing due to minimized steric and conformational hindrances,[ 10 , 12 ] and low energy lowest unoccupied molecular orbitals (LUMOs) due to the electron‐accepting behavior of carbon‐carbon triple bonds.[ 12 , 13 ] Glaser coupling, which is an efficient oxidative coupling method for linking terminal alkynes together under mild conditions (Cu catalyst, supporting ligand, and air), has allowed for a large variety of conjugated acetylenic polymers (Figure 1 ) to be synthesized with diverse applications.[ 9 , 14 ] For example, Xie and colleagues used Glaser coupling to synthesize conjugated microporous polymer 1, which was found to have a large specific surface area of 1008 m2 g−1 allowing for high CO2 and CH4 uptake capacities of 3.78 mmol g−1 and 0.95 mmol g−1, leading to potential applications in gas storage.[ 15 ] Glaser coupling has also been used to polymerize hexaethynylbenzene monomers (2) leading to the formation of well‐defined graphdiyne nanowalls with remarkably stable field‐emission properties.[ 16 ]
Figure 1.

Examples of Glaser‐ and Glaser‐Hay‐coupled polymers and the monomers used for copolymerization in this work.
While Glaser coupling can be used to produce polymers with interesting structures, other polymers with intriguing optical properties can also be achieved (Figure 1). Anderson et. al. designed conjugated porphyrin‐based polymer 3 via Glaser‐Hay coupling, which specifically employs CuCl and TMEDA to facilitate carbon–carbon bond formation,[ 14 ] of meso‐diethynyl zinc porphyrin monomers.[ 17 ] The porphyrin‐containing polymers were found to possess near‐infrared absorption as well as third‐order nonlinear optical properties, leading to potential applications in telecommunication optical switches.[ 17 ] Hu and coworkers explored the synthesis of conjugated polyelectrolytes with high molecular weights of up to 38900 g mol−1 using Glaser‐Hay coupling followed by quaternization reactions with amines.[ 18 ] Polymer 4 was found to exhibit aggregation‐induced emission (AIE) and the related polyelectrolyte 5 was shown to act as a fluorescent sensor for biomolecules such as heparin, RNA, and human serum albumin (HSA).[ 18 ] Sun et al. designed conjugated acetylenic polymers 6 and 7 with tunable band gaps using a Glaser polycondensation reaction on Cu foam.[ 19 ] Polymer 6 (Eg = 2.17 eV) was found to have a lower optical band gap compared to polymer 7 (Eg = 2.36 eV) due to greater conjugation within the polymer backbone.[ 19 ] Furthermore, it was shown that the alkyne bonds of 6 were intrinsically activated for water‐splitting and hydrogen evolution upon irradiation with solar light.[ 19 ]
Fluorene and bis(alkoxy) benzene units are often incorporated into 𝜋‐conjugated polymers due to their attractive properties. Fluorene can be described by its planar, rigid, and conjugated structure that governs its unique properties, such as high photoluminescent quantum yields as well as high thermal and chemical stability.[ 20 ] Fluorene can be combined with other aromatic units to form copolymers with increased stability, solubility, and tunable electronic properties.[ 21 ] 𝜋‐Conjugated polymers containing fluorene units have garnered attention in the realm of optoelectronics due to their solution processability, blue emissivity, and structurally tunable electronic properties.[ 22 ] Bis(alkoxy) benzene units have been integrated into 𝜋‐conjugated polymers due to their bis(alkoxy) substituents which offer several beneficial traits.[ 23 , 24 ] The oxygen atoms increase electron density along the polymer backbone, leading to smaller optical band gaps, while also participating in intramolecular interactions, affecting molecular aggregation.[ 23 , 24 ] Furthermore, the alkyl chains allow for increased solubility and solution processability.[ 24 ] Baier and coworkers employed Glaser coupling under aqueous miniemulsion conditions to produce highly fluorescent nanoparticles of fluorene and bis(2‐ethylhexyloxy)benzene based polymers (Figure 1).[ 25 ] These 𝜋‐conjugated units were combined with small amounts of a perylenediimide dye (8) resulting in a shift in the wavelength of emission, demonstrating efficient energy transfer from the conjugated backbone to the dye.[ 25 ]
Boron difluoride (BF2) formazanate dyes are a class of conjugated compounds that are comprised of an [NNCNN]− backbone bound to a [BF2]+ unit, which implements structural rigidity and stability.[ 26 ] BF2 formazanate dyes are known for their redox activity and unique optical properties, which can be fine‐tuned by varying the N‐aryl substituents[ 27 − 30 ] or by extending the degree of 𝜋‐conjugation (Figure 2 ).[ 31 − 35 ] These tunable properties have allowed BF2 formazanates to find applications in organic photovoltaics,[ 32 ] unusual BN heterocycles,[ 36 , 37 ] near‐infrared theranostics,[ 38 ] fluorescence cell‐imaging,[ 29 , 39 , 40 ] and live animal imaging.[ 41 , 42 ] For example, BF2 formazanates have been combined with an N‐annulated perylene diimide (9) creating a π‐conjugated system with panchromatic absorption leading to applications in organic solar cells.[ 32 ] BF2 formazanates have also been incorporated into π‐conjugated polymers, whereby the absorption spectra of the polymer tend to be significantly red‐shifted compared to the respective monomer.[ 34 , 35 ] Polymer 10, which is comprised of electron‐deficient BF2 formazanate units and electron‐rich Pt(II) diyne units, exhibits reversible electron‐accepting abilities upon reduction using cobaltocene.[ 35 ] BF2 formazanates have also been combined with thiophene moieties leading to polymer 11, which possess a low optical band gap of 1.48 eV. Furthermore, BF2 formazanates have been incorporated into donor‐acceptor π‐conjugated polymers with fluorene units (12), where broad absorption of visible light as well as near‐infrared emission is achieved.[ 33 ]
Figure 2.

Examples of π‐conjugated systems incorporating BF2 formazanate dyes.
In this work, we report a series 𝜋‐conjugated compounds and copolymers, comprised of BF2 formazanate (BF2 ),[ 43 ] 9,9‐dihexylfluorene (FL),[ 44 ] and/or 1,4‐bis(2‐ethylhexyloxy)benzene (Bn)[ 45 ] units (Figure 1), synthesized using Glaser‐Hay coupling and linked by alkyne dimer (C4) units. The optical and redox properties of these copolymers are investigated in relation to their appropriate homopolymers and/or model compounds. Additionally, the alkyne linkages within the copolymers are converted into cobalt carbonyl clusters when reacted with dicobalt octacarbonyl [Co2(CO)8], demonstrating the ability to tune the optical properties of these polymers through post‐polymerization functionalization.
2. Results and Discussion
2.1. Synthesis of Model Compounds
A series of 𝜋‐conjugated model compounds and polymers comprised of BF2 formazanate (BF2 ),[ 43 ] 9,9‐dihexylfluorene (FL),[ 44 ] and/or 1,4‐bis(2‐ethylhexyloxy)benzene (Bn)[ 45 ] linked by C4 units were synthesized using Glaser‐Hay coupling conditions. Model compounds were synthesized to help rationalize the properties observed for the polymers discussed in this paper. A solution of HCC‐BF2‐CCH, HCC‐FL‐CCH, or HCC‐Bn‐CCH and excess phenylacetylene was added to a solution of CuCl and TMEDA and stirred for 3 h before purification by column chromatography, affording Ph‐BF2‐Ph, Ph‐FL‐Ph, and Ph‐Bn‐Ph as dark purple or yellow solids in 34%, 46%, and 50% yields, respectively (Scheme 1a). Another model compound, BF2‐BF2‐BF2 was synthesized to mimic the properties of a BF2 homopolymer, since homopolymerization of the HCC‐BF2‐CCH monomer was unsuccessful due to the extreme insolubility of the resulting polymer beyond only a few repeating units. A solution of HCC‐BF2‐CCH and excess BF2‐CCH was added to a solution of CuCl and TMEDA and stirred for 3 h before purification by column chromatography, affording BF2‐BF2‐BF2 as a dark purple solid in 36% yield (Scheme 1b). The relatively low yields of the model compounds can be attributed to the production of various by‐products under Glaser‐Hay conditions, including BF2 , FL, or Bn‐based oligomers of differing lengths, dimerized phenylacetylene, and/or dimerized BF2‐CCH units. The structures of these model compounds were confirmed by mass spectrometry, multinuclear NMR spectroscopy, and for Ph‐BF2‐Ph and Ph‐FL‐Ph by X‐ray crystallography.
Scheme 1.

Synthesis of model compounds, Ph‐BF2‐Ph, Ph‐FL‐Ph, Ph‐Bn‐Ph, and BF2‐BF2‐BF2 and their cobalt carbonyl derivatives, Ph‐Co‐BF2‐Co‐Ph, Ph‐Co‐FL‐Co‐Ph, and Ph‐Co‐Bn‐Co‐Ph. Monomers and building blocks are defined within the dashed box.
Alkyne units can react with [Co2(CO)8], whereby two equivalents of CO are displaced and a [Co2(CO)6] cluster bridges the carbon atoms of the alkyne functional group, forming a Co2C2 tetrahedron in a trans configuration along the alkyne backbone.[ 46 , 47 ] Depending on the steric bulk of the neighboring substituents of the alkyne units, it is possible for two linked alkynes to each react with one mole of [Co2(CO)8].[ 47 ] Upon coordination of the cobalt carbonyl complexes, the rigidity and electronic delocalization of the molecular system can be maintained and interesting properties can be achieved.[ 46 ] In this work, model compounds Ph‐BF2‐Ph, Ph‐FL‐Ph, and Ph‐Bn‐Ph were reacted with 5 equivalent of [Co2(CO)8] for 4 h before purification by column chromatography, affording Ph‐Co‐BF2‐Co‐Ph, Ph‐Co‐FL‐Co‐Ph, and Ph‐Co‐Bn‐Co‐Ph as dark blue or brown solids in 83%, 45%, and 12% yields, respectively (Scheme 1a). The conversion of the alkyne units to cobalt carbonyl clusters within these model compounds was confirmed using mass spectrometry, multinuclear NMR, and FT‐IR spectroscopy.
2.2. NMR and FT‐IR Spectroscopy for Model Compounds
1H, 11B, 13C{1H}, and 19F{1H} NMR (Figures S1–S10, S23–S29, Supporting Information) and FT‐IR spectroscopy were used to characterize the model compounds and their cobalt carbonyl functionalized derivatives. For the model compounds, the 1H NMR spectra were comprised of signals corresponding to aryl and/or alkyl proton environments in the proposed structures. Upon reaction with [Co2(CO)8], the chemical shifts of the aryl protons remained relatively unchanged, however, the chemical shifts of the alkyl protons shifted slightly upfield (e.g., multiplet at 1.97–1.93 ppm in Ph‐FL‐Ph (Figure S4, Supporting Information) shifted to 1.72–1.68 ppm in Ph‐Co‐FL‐Co‐Ph (Figure S26, Supporting Information)). In the 11B NMR spectra, the Ph‐BF2‐Ph (Figure S3, Supporting Information) and BF2‐BF2‐BF2 (Figure S10, Supporting Information) compounds both exhibit triplets at ca. −0.5 ppm. The 19F{1H} NMR spectrum for Ph‐BF2‐Ph (Figure S3, Supporting Information) contains a quartet at −142.0 ppm, however, BF2‐BF2‐BF2 (Figure S3, Supporting Information) exhibits two quartets, one at −141.6 ppm corresponding to the center BF2 unit and one at –142.7 ppm, with doubled intensity, corresponding to the outer BF2 units. Conversion of Ph‐BF2‐Ph to Ph‐Co‐BF2‐Co‐Ph resulted in the 19F{1H} NMR signal shifting slightly upfield to −143.4 ppm (Figure S25, Supporting Information), while no change was observed in the 11B NMR spectra. The 13C{1H} NMR spectra for all model compounds include signals corresponding to the unique aryl, alkynyl, and/or alkyl carbons. 13C{1H} NMR spectroscopy was a key tool for monitoring the conversion of alkyne units to cobalt carbonyl clusters after reacting the models, Ph‐BF2‐Ph, Ph‐FL‐Ph, and Ph‐Bn‐Ph, with [Co2(CO)8]. The 13C{1H} NMR spectra of the models, Ph‐BF2‐Ph, Ph‐FL‐Ph, and Ph‐Bn‐Ph, contain four signals in the region of ca. 85–70 ppm corresponding to the four unique alkyne carbons of each compound (Figures S2, S5, and S7, Supporting Information). Whereas in the 13C{1H} NMR spectra of the Co‐functionalized derivatives, Ph‐Co‐BF2‐Co‐Ph, Ph‐Co‐FL‐Co‐Ph, and Ph‐Co‐Bn‐Co‐Ph there are four signals in the region of ca. 100–90 ppm corresponding to the four unique carbons coordinated to the cobalt carbonyl clusters (Figures S24, S27, and S29, Supporting Information). Furthermore, the 13C{1H} NMR spectra of Ph‐Co‐BF2‐Co‐Ph, Ph‐Co‐FL‐Co‐Ph, and Ph‐Co‐Bn‐Co‐Ph contain two signals ≈200 ppm, corresponding to the carbonyl groups in different chemical environments.[ 48 ] The downfield shift associated with the alkyne carbon signals and the appearance of carbonyl carbon signals indicate that the alkyne units have been converted into cobalt carbonyl clusters.[ 46 , 48 ] Based on these findings as well as mass spectrometry data, each alkyne unit within the model compounds was transformed into a [C2Co2(CO)6] cluster.
FT‐IR spectroscopy can be used to monitor the conversion of alkyne units to cobalt carbonyl clusters. Upon Co functionalization, the alkyne stretch at ≈2200 cm−1 and the bridging carbonyl stretch of [Co2(CO)8] at ≈1860 cm−1 disappear and stretches at ca. 2090, 2050, and 2025 cm−1 appear, corresponding to the terminal carbonyl groups of the dicobalt hexacarbonyl [Co2(CO)6] complex.[ 47 ] When comparing the IR spectra of the model compounds, Ph‐BF2‐Ph, Ph‐FL‐Ph, and Ph‐Bn‐Ph, to Ph‐Co‐BF2‐Co‐Ph, Ph‐Co‐FL‐Co‐Ph, and Ph‐Co‐Bn‐Co‐Ph, the alkyne and bridging carbonyl stretches of the respective reactants disappear and the terminal CO stretches of the [C2Co2(CO)6] cluster appear, indicating conversion of the alkyne units to cobalt carbonyl clusters (Figures S38–S40, Supporting Information).
2.3. X‐Ray Crystallography for Model Compounds
Single crystals of Ph‐BF2‐Ph and Ph‐FL‐Ph (Figure 3 ) suitable for X‐ray diffraction were obtained by vapor diffusion of pentane into a saturated solution of Ph‐BF2‐Ph in CH2Cl2 or by slow evaporation of a saturated solution of Ph‐FL‐Ph in toluene. Relevant bond lengths and angles for each solid‐state structure have been summarized in Tables S6 and S7 (Supporting Information). The solid‐state structures of Ph‐BF2‐Ph and Ph‐FL‐Ph have alternating triple (Ph‐BF2‐Ph: 1.204(2) and 1.205(2) Å, Ph‐FL‐Ph: 1.2117(10) and 1.2135(11) Å) and single (Ph‐BF2‐Ph: 1.374(2) Å, Ph‐FL‐Ph: 1.3672(11) Å) carbon‐carbon bond lengths which are not statistically different compared to other alkynes bound to BF2 formazanate and fluorene based compounds with conjugated alkyne units.[ 49 − 51 ] For Ph‐BF2‐Ph, the average C─N (1.3449(18) Å) and N─N (1.3185(16) Å) bond lengths associated with the BF2 formazanate units are intermediate of typical single and double bonds for the atoms involved, indicating a high degree of electronic delocalization within the formazanate ring. For Ph‐FL‐Ph, the carbon‐carbon bond lengths associated with the fluorene unit are intermediate of typical single and double carbon–carbon bonds, also demonstrating a high degree of electronic delocalization within the fluorene unit. The boron atom in Ph‐BF2‐Ph has a tetrahedral geometry and is displaced from the N1–N2–C1–N3–N4 plane by 0.823(2) Å. The dihedral angles between the planes defined by the N‐aryl substituents and the formazanate backbone of Ph‐BF2‐Ph are offset by 25.29(7)° and 27.08(7)°. The dihedral angles between the planes defined by the N‐aryl substituents and the external phenyl substituents of Ph‐BF2‐Ph are offset by 45.86(5)° and 78.14(5)°. The dihedral angle between the plane defined by the fluorene unit and the external phenyl substituents of Ph‐FL‐Ph are offset by 56.29(3)° and 23.42(4)°.
Figure 3.

Solid‐state structures of model compounds Ph‐BF2‐Ph and Ph‐FL‐Ph. Hydrogen atoms are omitted for clarity and anisotropic displacement parameter ellipsoids are shown at a 50% probability level.
2.4. Synthesis of Glaser‐Hay‐Coupled Polymers
Random copolymers comprised of BF2 , FL, and/or Bn units were synthesized by Glaser‐Hay coupling using a 1:1 molar ratio of monomers. Polymerization by Glaser‐Hay coupling is classified as step‐growth, thus molecular weight evolution as a function of time had to be considered. Polymerizations of copolymers as well as homopolymers were monitored by removing aliquots from the reaction mixtures at different time points to determine the optimal timeframe to achieve high average molecular weights (>10000 g mol−1) and high yields of soluble polymer. These studies were necessary since it was determined that as the reaction time increased, the average molecular weight of the polymer increased, but the solubility of the polymer decreased, affecting the amount of soluble polymer that could be isolated with high average molecular weight. Based on the findings from the molecular weight versus time studies (Tables S1–S5, Supporting Information), samples of random copolymers and homopolymers were synthesized. A 1:1 molar ratio of HCC‐BF2‐CCH and HCC‐FL‐CCH, HCC‐BF2‐CCH and HCC‐Bn‐CCH, or HCC‐FL‐CCH and HCC‐Bn‐CCH were combined, added to a solution of CuCl and TMEDA, and stirred for 1.5 h, 0.75 h, or 8 h, respectively, before purification by trituration in pentane, affording P‐BF2‐FL, P‐BF2‐Bn, or P‐FL‐Bn as dark navy, dark green‐blue, or yellow solids in 55%, 37%, or 61% yields, respectively (Scheme 2 ). For the homopolymers, a solution of HCC‐FL‐CCH or HCC‐Bn‐CCH monomer units was added to a solution of CuCl and TMEDA and stirred for 12 h or 6 h, respectively, before purification by trituration in pentane, affording P‐FL or P‐Bn as yellow solids in 52% or 36% yields, respectively (Scheme 2). P‐FL and P‐Bn homopolymers have been previously synthesized using Glaser coupling,[ 25 , 52 ] however, here we report quicker and more efficient syntheses and purification processes, while achieving similar Mn values. The structures of all polymers synthesized were confirmed using multinuclear NMR spectroscopy and their molecular weight distributions were analyzed using gel permeation chromatography (GPC) (Table 1 ; Figure S44, Supporting Information).
Scheme 2.

Synthesis of polymers, P‐FL, P‐Bn, P‐BF2‐FL, P‐BF2‐Bn, and P‐FL‐Bn, and their cobalt carbonyl derivatives, Co‐P‐BF2‐FL, Co‐P‐BF2‐Bn, and Co‐P‐FL‐Bn. Monomers and building blocks are defined within the dashed box.
Table 1.
Molecular weight and polydispersity values were determined by GPC for each polymer.
| Compound | Mn [g mol‒1] a) | Mw [g mol‒1] a) | Đ a ) |
|---|---|---|---|
| P‐FL | 19800 | 42300 | 2.14 |
| P‐Bn | 32300 | 50100 | 1.55 |
| P‐BF2‐FL | 14800 | 54900 | 3.71 |
| P‐BF2‐Bn | 15500 | 48100 | 3.10 |
| P‐FL‐Bn | 20800 | 33200 | 1.59 |
All values were estimated using conventional calibration methods relative to polystyrene standards.
The random copolymers were also reacted with [Co2(CO)8], to convert the alkyne moieties to cobalt carbonyl clusters. Solutions of P‐BF2‐FL, P‐BF2‐Bn, or P‐FL‐Bn were reacted with excess [Co2(CO)8] for 4 h before purification by column chromatography or trituration in pentane, affording Co‐P‐BF2‐FL, Co‐P‐BF2‐Bn, and Co‐P‐FL‐Bn as dark green‐blue, blue or yellow‐brown solids in 74%, 60%, and 33% yields, respectively (Scheme 2). The conversion of the alkyne units to cobalt carbonyl clusters within these polymers was analyzed using multinuclear NMR and FT‐IR spectroscopy.
2.5. NMR and FT‐IR Spectroscopy for Polymers
1H, 11B, 13C{1H}, and 19F{1H} NMR (Figures S11–S22, S30–S37, Supporting Information) and FT‐IR spectroscopy were used to characterize the polymers and their cobalt carbonyl functionalized derivatives. For all of the polymers, the 1H NMR spectra had appropriate signals corresponding to the aryl, terminal alkyne, and/or alkyl proton environments of the predicted structures, but they were broadened in comparison to the model compounds. The 1H NMR spectra were used to determine the ratio of the monomer units incorporated into the backbone of the random copolymers. The relative integrations of signals unique to each monomer unit were compared and it was found that the ratio of BF2 :FL in P‐BF2‐FL was 1:1.1 (Figure S15, Supporting Information), the ratio of BF2 :Bn in P‐BF2‐Bn was 1:1.2 (Figure S18, Supporting Information), and the ratio of FL:Bn in P‐FL‐Bn was 1:1.3 (Figure S21, Supporting Information). Due to the insolubility of BF2 oligomers beyond only a few repeating units, it is likely that more FL or Bn monomers would be available to react in solution, leading to a slightly greater percentage of FL and Bn units being incorporated into the P‐BF2‐FL and P‐BF2‐Bn copolymers. 1H NMR spectra were also a key tool to determine if all the alkyne units within the polymer chain were converted to cobalt carbonyl clusters, after reacting with [Co2(CO)8], based on the presence of signals corresponding to terminal C─H protons bound the cobalt carbonyl cluster [(CO)6Co2C2]─H. The conversion of P‐BF2‐FL to Co‐P‐BF2‐FL resulted in the disappearance of the terminal alkyne proton signal at 3.16 ppm (Figure S15, Supporting Information) and the appearance of a signal at 6.45 ppm (Figure S30, Supporting Information) associated with the proton of [(CO)6Co2C2]─H, indicating that all terminal alkyne linkages were Co functionalized. However, in the 1H NMR spectra of Co‐P‐BF2‐Bn (Figure S33, Supporting Information) and Co‐P‐FL‐Bn (Figure S36, Supporting Information), terminal alkyne proton signals remained, along with the appearance of [(CO)6Co2C2]─H signals, indicating that not all of the terminal alkyne units were converted into cobalt carbonyl clusters. This was the case even after reacting P‐BF2‐Bn and P‐FL‐Bn with a 10‐fold excess of [Co2(CO)8]. Upon coordination of the cobalt carbonyl complexes, the chemical shifts of the aryl protons remained relatively unchanged, however, the chemical shifts of some of the alkyl protons shifted slightly.
In the 11B and 19F{1H} NMR spectra, the P‐BF2‐FL (Figure S17, Supporting Information) and P‐BF2‐Bn (Figure S20, Supporting Information) polymers both exhibit triplets at −0.6 ppm and quartets at 142.1 ppm, respectively. In comparison, the 11B and 19F{1H}NMR spectra of Co‐P‐BF2‐FL (Figure S32, Supporting Information) and Co‐P‐BF2‐Bn (Figure S35, Supporting Information) exhibit downfield shifted triplets at −0.5 ppm and upfield shifted quartets at ca. −143 ppm, respectively. The 13C{1H} NMR spectra for all polymers display signals corresponding to the unique aryl, alkynyl, and/or alkyl carbons. In comparison to the model compounds, the copolymers generally had a greater number of signals in the 13C{1H} NMR spectra due to a greater number of carbon environments associated with the various possible connectivities of the repeating units of the polymer. 13C{1H} NMR spectroscopy was also used to monitor the conversion of alkyne units to cobalt carbonyl clusters after reacting P‐BF2‐FL, P‐BF2‐Bn, and P‐FL‐Bn with [Co2(CO)8]. For Co‐P‐BF2‐FL, Co‐P‐BF2‐Bn, and Co‐P‐FL‐Bn, the alkyne carbon signals in the region of 85–74 ppm disappeared (Figures S16, S19, and S22, Supporting Information) and signals in the region of 100–90 ppm appeared (Figures S31, S34, and S37, Supporting Information). For Co‐P‐BF2‐FL, the signals in the 100–90 ppm region represent carbons bound to the [Co(CO)6] clusters. However, for Co‐P‐BF2‐Bn and Co‐P‐FL‐Bn, the signals in the 100–90 ppm region are assumed to be a combination of carbons bound to [Co(CO)6] clusters and unfunctionalized alkyne carbons neighboring [Co(CO)6] clusters.[ 46 ] The 13C{1H} NMR spectra of Co‐P‐BF2‐FL, Co‐P‐BF2‐Bn, and Co‐P‐FL‐Bn also contain two signals ≈200 ppm, corresponding to the carbonyl groups.[ 48 ]
Based on the NMR spectroscopy findings, we propose that the alkyne linkages of Co‐P‐BF2‐FL have been completely converted into cobalt carbonyl clusters. However, the alkyne linkages of Co‐P‐BF2‐Bn and Co‐P‐FL‐Bn have not been completely converted into cobalt carbonyl clusters. This is most likely due to the presence of bulky Bn units which could sterically hinder the conversion of alkynes adjacent to these groups into their corresponding clusters.
FT‐IR spectroscopy was also used to monitor the conversion of alkyne units to cobalt carbonyl clusters within the copolymers. When comparing the IR spectra of P‐BF2‐FL, P‐BF2‐Bn, and P‐FL‐Bn to Co‐P‐BF2‐FL, Co‐P‐BF2‐Bn, and Co‐P‐FL‐Bn, the alkyne and bridging carbonyl stretches of the respective reactants disappear and the terminal CO stretches of the [C2Co2(CO)6] cluster appear, qualitatively indicating that the majority of the alkyne units have been converted to cobalt carbonyl clusters (Figures S41–S43, Supporting Information).
2.6. Thermal Analysis of Polymers
The thermal properties of the polymers, P‐BF2‐FL, P‐BF2‐Bn, P‐FL‐Bn, P‐FL, and P‐Bn as well as their cobalt carbonyl functionalized derivatives, Co‐P‐BF2‐FL, Co‐P‐BF2‐Bn, and Co‐P‐FL‐Bn, were analyzed by thermogravimetric analysis (Figure S45, Supporting Information) and differential scanning calorimetry (DSC) (Figure S46, Supporting Information), under an atmosphere of N2. The decomposition onset temperatures (Td), taken at 2% weight loss, and char yields for all polymers are reported in Table 2 . The homopolymers, P‐FL and P‐Bn, had lower Td values and higher char yields compared to the copolymers. Compared to the unfunctionalized copolymers, the cobalt carbonyl derivatives had lower Td values, likely due to the loss of carbonyl groups at relatively low temperatures, and higher char yields, likely due to the presence of cobalt in the char. Based on DSC analysis, no glass transition temperatures (Tg) were observed within the thermal stability window of all the polymers.
Table 2.
Thermal gravimetric analysis data.
| Compound | Decomposition onset temperature [Td] [°C] a , b) | Char yield at 1000 °C [%] a) |
|---|---|---|
| P‐FL | 188 | 33 |
| P‐Bn | 172 | 23 |
| P‐BF2‐FL | 265 | 12 |
| P‐BF2‐Bn | 259 | 7 |
| P‐FL‐Bn | 293 | 9 |
| Co‐P‐BF2‐FL | 108 | 34 |
| Co‐P‐BF2‐Bn | 115 | 33 |
| Co‐P‐FL‐Bn | 112 | 35 |
Measured under an atmosphere of N2; and
Td taken at 2% weight loss.
2.7. Cyclic Voltammetry
Cyclic voltammograms (CVs) for each model compound and polymer in CH2Cl2 are shown in Figure 4 and the data are summarized in Table 3 . The Ph‐BF2‐Ph model exhibits two reversible, one‐electron reduction waves, corresponding to the reduction of the BF2 formazanate unit to a radical anion (E red1) and then a dianion (E red2). The CV of BF2‐BF2‐BF2 displays overlapping reduction waves in the E red1 and E red2 regions, resulting in four reduction events. The first reduction wave at ‒0.74 V is a one‐electron event, associated with the reduction of the center BF2 formazanate unit. The second reduction at ‒0.85 V is a two‐electron event which represents the reduction of the two outer BF2 formazanate units. The third, one‐electron, reduction at ‒1.66 V, and the fourth, two‐electron, reduction at ‒1.85 V correspond to the reduction of the center and outer BF2 formazanate units, respectively. The Ph‐FL‐Ph and Ph‐Bn‐Ph model compounds each exhibit one irreversible reduction event at ‒2.35 V and ‒2.34 V, respectively, corresponding to the reduction of the FL and Bn units (E red3). The P‐FL homopolymer does not undergo any reductions within the solvent stability window, while P‐Bn possesses a reversible reduction wave at ‒2.05 V. The P‐Bn homopolymer exhibits a less negative reduction potential and is thus easier to reduce compared to the Ph‐Bn‐Ph model since it is easier to add electrons to a larger 𝜋‐system. Interestingly, no reduction events are observed for the P‐FL‐Bn copolymer. The CVs for the copolymers, P‐BF2‐FL and P‐BF2‐Bn, each have two reversible reduction events corresponding to the formation of radical anions and dianions of the BF2 units. The reduction waves of these copolymers appear at similar or less negative reduction potentials compared to the reduction waves of previously reported π‐conjugated polymers containing BF2 formazanate units.[ 35 , 43 ] The P‐BF2‐FL and P‐BF2‐Bn copolymers also each possess one irreversible reduction event corresponding to the reduction of the FL or Bn unit. The reduction waves of these copolymers occur at about the same potentials as the Ph‐BF2‐Ph, Ph‐FL‐Ph, and Ph‐Bn‐Ph model compounds, indicating that the polymers retain redox activity. However, the current response for these copolymers is smaller than observed for the models, likely due to poor diffusion of the polymer at the working electrode. For all model compounds and polymers, no oxidation events were observed within the solvent stability window.
Figure 4.

Cyclic voltammograms of a) model compounds and b) polymers as 1 mm, dry, degassed CH2Cl2 solutions containing 0.1 m [nBu4N][PF6] as the supporting electrolyte, recorded at 500 mV s−1. The initial scan direction is denoted by the arrows.
Table 3.
Solution phase cyclic voltammetry data obtained in CH2Cl2.
| Compound | BF2 Reductions a) , b) | FL/Bn Reduction a) , b) | |
|---|---|---|---|
| E red1 [V] | E red2 [V] | E red3 [V] | |
| Ph‐BF2‐Ph | ‒0.75 | ‒1.63 | ‒ |
| Ph‐FL‐Ph | ‒ | ‒ | ‒2.35 c) |
| Ph‐Bn‐Ph | ‒ | ‒ | ‒2.34 c) |
| BF2‐BF2‐BF2 | ‒0.74, ‒0.85 | ‒1.66, ‒1.85 | ‒ |
| P‐FL | ‒ | ‒ | ‒ |
| P‐Bn | ‒ | ‒ | ‒2.05 |
| P‐BF2‐FL | ‒0.75 | ‒1.62 | ‒2.35 c) |
| P‐BF2‐Bn | ‒0.75 | ‒1.62 | ‒2.32 c) |
| P‐FL‐Bn | ‒ | ‒ | ‒ |
All potentials are reported relative to the Fc/Fc+ redox couple;
Hyphens indicate that no redox activity was observed; and
Irreversible process, potential at maximum cathodic current reported.
2.8. UV–Vis Absorption Spectroscopy
The UV–vis absorption spectra of the models, Ph‐BF2‐Ph, Ph‐FL‐Ph, Ph‐Bn‐Ph, BF2‐BF2‐BF2 , and polymers, P‐FL, P‐Bn, P‐BF2‐FL, P‐BF2‐Bn, and P‐FL‐Bn, in CH2Cl2 and as thin films are shown in Figure 5 and Figure S47 (Supporting Information) and the data are summarized in Table 4 . The model compounds and polymers have regions of high‐intensity absorption associated with BF2 (), FL (λFL), or Bn (λBn) based transitions. Comparing the Ph‐BF2‐Ph ( = 575 nm, = 39000 M−1 cm−1) and BF2‐BF2‐BF2 ( = 586 nm, = 93250 M−1 cm−1) model compounds in solution, BF2‐BF2‐BF2 exhibits a lower energy and higher molar absorptivity. The homopolymers, P‐FL and P‐Bn, also have lower energy λFL or λBn in comparison to their respective model compounds, Ph‐FL‐Ph and Ph‐Bn‐Ph. The P‐BF2‐FL and P‐BF2‐Bn copolymers have red‐shifted compared to the Ph‐BF2‐Ph and BF2‐BF2‐BF2 model compounds, and similar λFL to the Ph‐FL‐Ph model or red‐shifted λBn compared to the Ph‐Bn‐Ph model. These observations are consistent with extended 𝜋‐conjugation upon polymerization and agree with previously reported 𝜋‐conjugated polymers containing BF2 units, where the BF2 ‐based transition () red‐shifts in comparison to the respective BF2 monomer unit.[ 33 − 35 , 43 ] The modest redshift of ca. 25 nm when comparing the BF2 formazanate absorption bands of the Ph‐BF2‐Ph model compound to those of the P‐BF2‐FL and P‐BF2‐Bn copolymers points to the fact that the cross‐conjugation associated with the meta‐substitution pattern of the BF2 formazanate ring may limit extended conjugation. Both P‐BF2‐FL and P‐BF2‐Bn copolymers have higher energy λFL or λBn compared to their relevant homopolymer, P‐FL, and P‐Bn. The P‐BF2‐FL and P‐BF2‐Bn copolymers also have lower molar absorptivities ( and FL/Bn) compared to the applicable model compound or homopolymer. Interestingly, for the P‐FL‐Bn copolymer, only one absorption band is observed which exhibits intermediate λFL/Bn (424 nm) and εFL/Bn (42250 M−1 cm−1) values of the P‐FL and P‐Bn homopolymers. For all model compounds and polymers, thin film absorption profiles (Figure S47, Supporting Information) were broadened and generally red‐shifted in comparison to solution absorption profiles.
Figure 5.
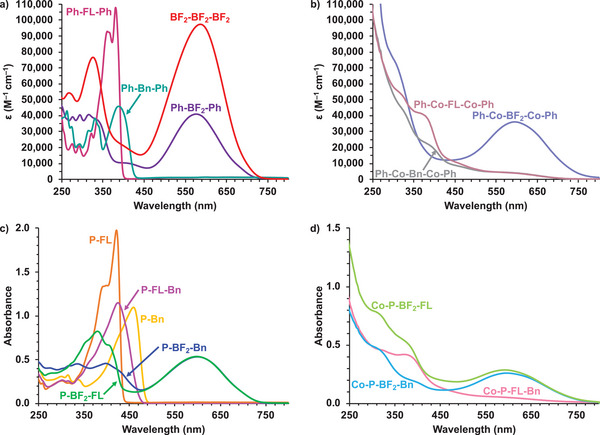
UV–vis absorption spectra recorded for a) CH2Cl2 solutions of model compounds, b) CH2Cl2 solutions of cobalt carbonyl derivatives of models, c) 27 µm CH2Cl2 solutions of homopolymers and copolymers, and d) CH2Cl2 solutions of cobalt carbonyl derivatives of copolymers: Co‐P‐BF2‐FL (20 µm), Co‐P‐BF2‐Bn (22 µm), and Co‐P‐FL‐Bn (21 µm).
Table 4.
Experimental UV/vis absorption spectral data.
| Solution: CH2Cl2 a) | Thin Film a) | ||||||
|---|---|---|---|---|---|---|---|
| Compound | λBF2 [nm] |
εBF2 b) [M−1 cm−1] |
λFL/Bn [nm] |
εFL/Bn b) [M−1 cm−1] |
λBF2 [nm] | λFL/Bn [nm] | |
| Ph‐BF2‐Ph | 575 | 39000 | ‒ | ‒ | 595 | ‒ | |
| Ph‐FL‐Ph | ‒ | ‒ | 380 c) | 100000 | ‒ | 390 c) | |
| Ph‐Bn‐Ph | ‒ | ‒ | 387 | 44000 | ‒ | 416 | |
| BF2‐BF2‐BF2 | 586 | 93250 | ‒ | ‒ | 617 | ‒ | |
| P‐FL | ‒ | ‒ | 421 c) | 72000 | ‒ | 390 c) | |
| P‐Bn | ‒ | ‒ | 459 | 40250 | ‒ | 461 | |
| P‐BF2‐FL | 598 | 19750 | 379 | 30750 | 608 | 379 | |
| P‐BF2‐Bn | 600 | 19750 | 397 | 17000 | 614 | 398 | |
| P‐FL‐Bn | ‒ | ‒ | 424 | 42250 | ‒ | 420 | |
| Ph‐Co‐BF2‐Co‐Ph | 595 | 32750 | ‒ | ‒ | ‒ | ‒ | |
| Ph‐Co‐FL‐Co‐Ph | ‒ | ‒ | 374 d) | 38500 | ‒ | ‒ | |
| Ph‐Co‐Bn‐Co‐Ph | ‒ | ‒ | 384 d) | 19750 | ‒ | ‒ | |
| Co‐P‐BF2‐FL | 592 | 13000 | 375 d) | 24750 | ‒ | ‒ | |
| Co‐P‐BF2‐Bn | 595 | 11750 | 390 d) | 9000 | ‒ | ‒ | |
| Co‐P‐FL‐Bn | ‒ | ‒ | 370 | 19250 | ‒ | ‒ | |
Hyphens indicate that no absorption band was observed or measured;
Molar absorptivity values reported for polymers were calculated using the average molecular weight of the repeating units based on the NMR ratio of the repeating units;
λ FL represents fluorene‐based transition with maximum absorbance; and
The UV–vis absorption spectra of the cobalt carbonyl analogs of the model compounds and copolymers are shown in Figure 5 and the data are summarized in Table 4. The cobalt functionalized derivatives have qualitatively similar features ( and λFL/Bn) as the unfunctionalized versions, except the broad absorption band observed in the UV region is associated with the [C2Co2(CO)6] clusters.[ 47 ] In comparison to the Ph‐BF2‐Ph model compound, Ph‐Co‐BF2‐Co‐Ph has red‐shifted , indicating that electronic delocalization is not significantly disrupted. However, for Ph‐Co‐FL‐Co‐Ph and Ph‐Co‐Bn‐Co‐Ph, λFL and λBn are blue‐shifted compared to the Ph‐FL‐Ph and Ph‐Bn‐Ph model compounds, indicating that the cobalt carbonyl complexes decrease π‐conjugation within the molecules. The cobalt carbonyl copolymers, Co‐P‐BF2‐FL, Co‐P‐BF2‐Bn, and Co‐P‐FL‐Bn also display blue‐shifted , λFL and/or λBn compared to the unfunctionalized copolymers, likely due to a decrease in π‐conjugation that accompanies the reaction of the alkyne units. All of the cobalt carbonyl analogs have lower molar absorptivities compared to their respective unfunctionalized model compounds and copolymers.
2.9. Photoluminescence Spectroscopy
The photoluminescence spectra of the model compounds Ph‐BF2‐Ph, Ph‐FL‐Ph, Ph‐Bn‐Ph, BF2‐BF2‐BF2 , and polymers P‐FL, P‐Bn, P‐BF2‐FL, P‐BF2‐Bn, and P‐FL‐Bn in CH2Cl2 and as thin films are shown in Figure S48 (Supporting Information) and the data are summarized in Table S10 (Supporting Information). Only the purely organic model compounds and polymers were found to be photoluminescent. In solution, the Ph‐FL‐Ph and Ph‐Bn‐Ph models possessed maximum wavelengths of photoluminescence (λPL) of 413 and 423 nm and photoluminescent quantum yields (ΦPL) of 31% and 32%, respectively. The homopolymers, P‐FL and P‐Bn, were the most photoluminescent materials studied with λPL values of 428 and 476 nm and ΦPL values of 75% and 77%, respectively, which are similar to previously reported values.[ 25 , 52 ] The P‐FL‐Bn copolymer exhibits a λPL of 469 nm, which is intermediate of the related homopolymers, however, P‐FL‐Bn has a lower ΦPL of 68%. For all of these organic materials, thin films were found to have lower energy λPL and significantly lower ΦPL values (Table S10, Supporting Information). The Ph‐BF2‐Ph and BF2‐BF2‐BF2 model compounds exhibit quenched photoluminescence in solution and as thin films. The P‐BF2‐FL and P‐BF2‐Bn copolymers likely exhibit quenched photoluminescence due to donor‐excited photoinduced electron transfer (d‐PET). In the case of d‐PET, the BF2 unit, which is strongly electron‐accepting, would accept a photoexcited electron from the FL or Bn unit upon photoexcitation, if the LUMO of BF2 lies in between the HOMO and LUMO of the FL or Bn units.[ 43 , 53 , 54 , 55 ] After the FL or Bn unit is excited, an electron is transferred from the HOMO to the LUMO of the FL or Bn unit and then decays non‐radiatively within the formazanate chromophore.[ 43 , 53 , 54 , 55 ] The photoluminescence spectra of the cobalt carbonyl functionalized derivatives of the model compounds and copolymers in CH2Cl2 are shown in Figure S49 (Supporting Information) and the data are summarized in Table S10 (Supporting Information). The cobalt carbonyl analogs of the organic models and polymers, Ph‐Co‐FL‐Co‐Ph, Ph‐Co‐Bn‐Co‐Ph, and Co‐P‐FL‐Bn, display quenched emission, which has been previously reported for other cobalt carbonyl complexed fluorescent organic molecules.[ 56 , 57 , 58 ]
3. Conclusion
𝜋‐Conjugated model compounds and polymers, with Mn values greater than 10000 g mol−1, comprised of BF2 , FL, and/or Bn units bridged by alkyne dimers were successfully synthesized using Glaser‐Hay‐coupling conditions and their redox and optical properties were explored. This is the first time Glaser‐Hay coupling has been utilized to efficiently synthesize 𝜋‐conjugated polymers containing BF2 formazanate dyes. The Ph‐BF2‐Ph and BF2‐BF2‐BF2 models and the P‐BF2‐FL and P‐BF2‐Bn copolymers exhibited redox activity associated with the reversible reduction of the BF2 units. The Ph‐FL‐Ph and Ph‐Bn‐Ph models and the P‐BF2‐FL and P‐BF2‐Bn copolymers also exhibited irreversible reduction waves corresponding to the reduction of the FL and Bn units. Compared to relevant model compounds, the homopolymers and BF2 ‐containing copolymers had red‐shifted absorption maxima corresponding to the BF2 and FL/Bn units, due to extended π‐conjugation. The P‐FL‐Bn copolymer had an absorption profile intermediate of the P‐FL and P‐Bn homopolymers. Only the purely organic model compounds and polymers were found to be photoluminescent, since BF2 units quench photoluminescence. The model compounds and copolymers were reacted with [Co2(CO)8] to convert the alkyne linkages into cobalt carbonyl clusters to determine the effect on optical properties. The conversion of alkyne units was monitored by mass spectrometry, NMR, and FT‐IR spectroscopy and it was found that the copolymers containing Bn units could only be partially functionalized. The cobalt carbonyl functionalized derivatives of the model compounds and the copolymers exhibited blue‐shifted absorption maxima corresponding to the BF2 and FL/Bn units and lower molar absorptivities compared to their non‐cobalt counterparts due to a decrease in π‐conjugation. Furthermore, the cobalt carbonyl complexes quench the photoluminescence of the related unfunctionalized organic materials. Due to the reversible electron‐accepting abilities and the broad absorption of UV–vis light, the P‐BF2‐FL and P‐BF2‐Bn copolymers have the potential to be used in organic electronics, such as photovoltaic devices. The reported series of model compounds and polymers contribute new knowledge regarding the structure‐property relationships of Glaser‐coupled 𝜋‐conjugated materials that will inform the design of new materials for use in the 𝜋‐conjugated materials arena.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the Natural Sciences and Engineering Council (NSERC) of Canada (E.L.C.: CGS‐M Scholarship; J.S.D.: CGS‐D Scholarship; J.B.G.: DG, RGPIN‐2023‐03318), the Ontario Ministry for Research and Innovation (E.L.C.: Ontario Graduate Scholarship), and the Canadian Foundation for Innovation (J.B.G.: JELF, 33977). We would like to thank Dr. Mat Willans at the JB Stothers NMR Facility at The University of Western Ontario for his help with the high temperature 13C{1H} NMR experiments. We thank Dr. Aruni Pulukkody from The University of Western Ontario for collecting mass spectrometry data.
Cotterill E. L., Jaberi Y., Dhindsa J. S., Boyle P. D., Gilroy J. B., Glaser‐Hay‐Coupled Random Copolymers Containing Boron Difluoride Formazanate Dyes. Macromol. Rapid Commun. 2025, 46, 2400786. 10.1002/marc.202400786
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information material of this article.
References
- 1. Heeger A. J., J. Phys. Chem. B. 2001, 105, 8475. [Google Scholar]
- 2. Dou L., Liu Y., Hong Z., Li G., Yang Y., Chem. Rev. 2015, 115, 12633. [DOI] [PubMed] [Google Scholar]
- 3. Zhao X., Zhan X., Chem. Soc. Rev. 2011, 40, 3728. [DOI] [PubMed] [Google Scholar]
- 4. Koezuka H., Tsumura A., Ando T., Synth. Met. 1987, 18, 699. [Google Scholar]
- 5. Yu G., Gao J., Hummelen J. C., Wudl F., Heeger A. J., Science 1995, 270, 1789. [Google Scholar]
- 6. Chung K., Yu Y., Kwon M. S., Swets J., Kim J., Youk J. H., MRS Commun. 2015, 5, 169. [Google Scholar]
- 7. Brown A. R., Bradley D. D. C., Burroughes J. H., Friend R. H., Greenham N. C., Burn P. L., Holmes A. B., Kraft A., Appl. Phys. Lett. 1992, 61, 2793. [Google Scholar]
- 8. Siemsen P., Livingston R. C., Diederich F., Angew. Chem., Int. Ed. 2000, 39, 2632. [PubMed] [Google Scholar]
- 9. Liu J., Lam J. W. Y., Tang B. Z., Chem. Rev. 2009, 109, 5799. [DOI] [PubMed] [Google Scholar]
- 10. Li W., Michinobu T., Macromol. Chem. Phys. 2016, 217, 863. [Google Scholar]
- 11. Guo X., Watson M. D., Macromolecules 2011, 44, 6711. [Google Scholar]
- 12. Egbe D. A. M., Neugebauer H., Sariciftci N. S., J. Mater. Chem. 2011, 21, 1338. [Google Scholar]
- 13. Egbe D. A. M., Bader C., Klemm E., Ding L., Karasz F. E., Grummt U. W., Birckner E., Macromolecules 2003, 36, 9303. [Google Scholar]
- 14. Akhtar R., Zahoor A. F., Synth. Commun. 2020, 50, 3337. [Google Scholar]
- 15. Xie Z., Wei Y., Zhao X., Li Y., Ding S., Chen L., Mater. Chem. Front. 2017, 1, 867. [Google Scholar]
- 16. Zhou J., Gao X., Liu R., Xie Z., Yang J., Zhang S., Zhang G., Liu H., Li Y., Zhang J., Liu Z., J. Am. Chem. Soc. 2015, 137, 7596. [DOI] [PubMed] [Google Scholar]
- 17. Anderson J. L., Martin S. J., Bradley D. D. C., Angew. Chem., Int. Ed. 1994, 33, 655. [Google Scholar]
- 18. Hu R., Ye R., Lam J. W. Y., Li M., Leung C. W. T., Tang B. Z., Asian J. Chem. 2013, 8, 2436. [DOI] [PubMed] [Google Scholar]
- 19. Sun H., Öner I. H., Wang T., Zhang T., Selyshchev O., Neumann C., Fu Y., Liao Z., Xu S., Hou Y., Turchanin A., Zahn D. R. T., Zschech E., Weidinger I. M., Zhang J., Feng X., Angew. Chem., Int. Ed. 2019, 58, 10368. [DOI] [PubMed] [Google Scholar]
- 20. Shi J., Wu Y., Sun S., Tong B., Zhi J., Dong Y., J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 229. [Google Scholar]
- 21. Liu B., Yu W.‐L., Lai Y.‐H., Huang W., Chem. Mater. 2001, 13, 1984. [Google Scholar]
- 22. Abbel R., Schenning A. P. H. J., Meijer E. W., J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 4215. [Google Scholar]
- 23. Cao K., Shen D. E., Österholm A. M., Kerszulis J. A., Reynolds J. R., Macromolecules 2016, 49, 8498. [Google Scholar]
- 24. Ming S., Zhang H., Lin K., Jiang F., Li Z., Liu P., Xu J., Nie G., Duan X., J. Polym. Sci. 2020, 58, 3370. [Google Scholar]
- 25. Baier M. C., Huber J., Mecking S., J. Am. Chem. Soc. 2009, 131, 14267. [DOI] [PubMed] [Google Scholar]
- 26. Gilroy J. B., Otten E., Chem. Soc. Rev. 2020, 49, 85. [DOI] [PubMed] [Google Scholar]
- 27. Chang M.‐C., Chantzis A., Jacquemin D., Otten E., Dalton Trans. 2016, 45, 9477. [DOI] [PubMed] [Google Scholar]
- 28. Chang M.‐C., Otten E., Chem. Commun. 2014, 50, 7431. [DOI] [PubMed] [Google Scholar]
- 29. Maar R. R., Barbon S. M., Sharma N., Groom H., Luyt L. G., Gilroy J. B., Chem. ‐ Eur. J. 2015, 21, 15589. [DOI] [PubMed] [Google Scholar]
- 30. Barbon S. M., Price J. T., Reinkeluers P. A., Gilroy J. B., Inorg. Chem. 2014, 53, 10585. [DOI] [PubMed] [Google Scholar]
- 31. Barbon S. M., Staroverov V. N., Gilroy J. B., J. Org. Chem. 2015, 80, 5226. [DOI] [PubMed] [Google Scholar]
- 32. Koenig J. D. B., Farahat M. E., Dhindsa J. S., Gilroy J. B., Welch G. C., Mater. Chem. Front. 2020, 4, 1643. [Google Scholar]
- 33. Kawano Y., Ito Y., Ito S., Tanaka K., Chujo Y., Macromolecules 2021, 54, 1934. [Google Scholar]
- 34. Kumar C., Agrawal A. R., Ghosh N. G., Karmakar H. S., Das S., Kumar N. R., Banewar V. W., Zade S. S., Dalton Trans. 2020, 49, 13202. [DOI] [PubMed] [Google Scholar]
- 35. Dhindsa J. S., Maar R. R., Barbon S. M., Avilés M. O, Powell Z. K., Lagugné‐Labarthet F., Gilroy J. B., Chem. Commun. 2018, 54, 6899. [DOI] [PubMed] [Google Scholar]
- 36. Chang M.‐C., Otten E., Inorg. Chem. 2015, 54, 8656. [DOI] [PubMed] [Google Scholar]
- 37. Barbon S. M., Staroverov V. N., Gilroy J. B., Angew. Chem., Int. Ed. 2017, 56, 8173. [DOI] [PubMed] [Google Scholar]
- 38. Xiang H., Zhao L., Yu L., Chen H., Wei C., Chen Y., Zhao Y., Nat. Commun. 2021, 12, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sharma N., Barbon S. M., Lalonde T., Maar R. R., Milne M., Gilroy J. B., Luyt L. G., RSC Adv. 2020, 10, 18970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barbon S. M., Novoa S., Bender D., Groom H., Luyt L. G., Gilroy J. B., Org. Chem. Front. 2017, 4, 178. [Google Scholar]
- 41. Wang S., Shi H., Wang L., Loredo A., Bachilo S. M., Wu W., Tian Z., Chen Y., Weisman R. B., Zhang X., Cheng Z., Xiao H., J. Am. Chem. Soc. 2022, 144, 23668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang S., Lu K., Xiao H., Curr. Opin. Chem. Biol. 2024, 81, 102473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barbon S. M., Gilroy J. B., Polym. Chem. 2016, 7, 3589. [Google Scholar]
- 44. Liu B., Tian Z., Dang F., Zhao J., Yan X., Xu X., Yang X., Zhou G., Wu Y., J. Organomet. Chem. 2016, 804, 80. [Google Scholar]
- 45. Ozdemir M., Choi D., Zorlu Y., Cosut B., Kim H., Kim C., Usta H., New J. Chem. 2017, 41, 6232. [Google Scholar]
- 46. Lewis J., Lin B., Raithby P. R., Transit. Met. Chem. 1995, 20, 569. [Google Scholar]
- 47. Greenfield H., Sternberg H. W., Friedel R. A., Wotiz J. H., Markby R., Wender I., J. Am. Chem. Soc. 1956, 78, 120. [Google Scholar]
- 48. Lewis J., Lin B., Khan M. S., Al‐Mandhary M. R. A., Raithby P. R., J. Organomet. Chem. 1994, 484, 161. [Google Scholar]
- 49. Dhindsa J. S., Cotterill E. L., Buguis F. L., Anghel M., Boyle P. D., Gilroy J. B., Angew. Chem., Int. Ed. 2022, 61, e202208502. [DOI] [PubMed] [Google Scholar]
- 50. Wong W.‐Y., Lu G.‐L., Choi K.‐H., Guo Y.‐H., J. Organomet. Chem. 2005, 690, 177. [Google Scholar]
- 51. Khan M. S., Al‐Mandhary M. R. A., Al‐Suti M. K., Ahrens B., Mahon M. F., Male L., Raithby P. R., Boothby C. E., Köhler A., Dalton Trans. 2003, 1, 74. [Google Scholar]
- 52. Yuan Y., Zhou W., Zhu Y., Tian M., Zheng Y., Shi Z., Feng F., Song Y., Dyes Pigm. 2022, 204, 110423. [Google Scholar]
- 53. Niu H., Liu J., O'Connor H. M., Gunnlaugsson T., James T. D., Zhang H., Chem. Soc. Rev. 2023, 52, 2322. [DOI] [PubMed] [Google Scholar]
- 54. Zhu H., Fan J., Mu H., Zhu T., Zhang Z., Du J., Peng X., Sci. Rep. 2016, 6, 35627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Y., Wang H., Wan L., Bian Y., Jiang J., J. Org. Chem. 2011, 76, 3774. [DOI] [PubMed] [Google Scholar]
- 56. Coleman A., Pryce M. T., Inorg. Chem. 2008, 47, 10980. [DOI] [PubMed] [Google Scholar]
- 57. Wong W.‐Y., Choi K.‐H., Lin Z., Eur. J. Inorg. Chem. 2002, 2112. [Google Scholar]
- 58. Shiotsuka M., Inui Y., Sekioka Y., Yamamoto Y., Onaka S., J. Organomet. Chem. 2007, 692, 2441. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information material of this article.