


Abstract
The identification of methylated sites on bacterial genomic DNA would be a useful tool to study the major roles of DNA methylation in prokaryotes: distinction of self and nonself DNA, direction of post-replicative mismatch repair, control of DNA replication and cell cycle, and regulation of gene expression. Three types of methylated nucleobases are known: N6-methyladenine, 5-methylcytosine and N4-methylcytosine. The aim of this study was to develop a method to detect all three types of DNA methylation in complete genomic DNA. It was previously shown that N6-methyladenine and 5-methylcytosine in plasmid and viral DNA can be detected by intersequence trace comparison of methylated and unmethylated DNA. We extended this method to include N4-methylcytosine detection in both in vitro and in vivo methylated DNA. Furthermore, application of intersequence trace comparison was extended to bacterial genomic DNA. Finally, we present evidence that intrasequence comparison suffices to detect methylated sites in genomic DNA. In conclusion, we present a method to detect all three natural types of DNA methylation in bacterial genomic DNA. This provides the possibility to define the complete methylome of any prokaryote.
INTRODUCTION
In prokaryotes, all three natural types of DNA methylation are observed: N6-methyladenine, N4-methylcytosine and 5-methylcytosine. DNA methylation is introduced enzymatically by DNA methyltransferases after DNA replication. Thus, DNA methylation adds extra information to the DNA; therefore, the methylated bases have been coined using the fifth, sixth and seventh letters of the genetic alphabet (1). Several roles of DNA methylation in prokaryotes have been proposed: distinction of self and nonself DNA, direction of post-replicative mismatch repair, control of DNA replication, cell cycle and level of gene expression. The distinction between self and nonself has traditionally been associated with defence against bacteriophages (2), but was more recently implicated in speciation and evolution in general (3). Different methylation patterns owing to uneven distribution of restriction modification systems have been found in most bacteria (4,5). Unfortunately, the epigenetic information in methylated DNA is lost upon PCR or subcloning as consecutive replication rounds result in the methylation pattern of the host strain.
Rao and Buckler-White (6) previously showed that in automated dye terminator sequencing data N6-methyladenine in template plasmid DNA results in an increase in the complementary T signal in sequence reactions and 5-methylcytosine results in a decrease in the complementary G signal. The peak height variations reflect the disproportionate rate of incorporation of labelled deoxynucleotides versus their analogs, a phenomenon that is highly dependent on template DNA sequence, type of label and neighbouring sequences (6–9). We extended this method to the detection of all three types of methylation and evaluated this method for its application to bacterial genomic DNA.
MATERIALS AND METHODS
Bacterial strains and culture conditions
Escherichia coli DH5α was grown either on solid or in liquid Luria–Bertani medium. For selection, media were supplemented with 100 mg/l ampicillin (Life Technologies) when appropriate. Neisseria meningitidis strains, MC58 and 800615, were cultured on heated blood (chocolate) agar plates at 37°C in a humidified atmosphere with 5% CO2. Helicobacter pylori strain 1061 (10) and its derivative 1061KC1_C17 (11) were cultured on solid Colombia agar plates (Oxoid) containing 7% lysed horse blood (Rottier) at 37°C under micro-aerobic conditions (5% O2, 10% CO2 and 85% N2) using CampyPAK pouches (Fischer Scientific) for 48 h.
DNA manipulation
Chromosomal DNA of N.meningitidis strains was isolated using the Puregene DNA isolation kit (Gentra systems). Chromosomal DNA of H.pylori strain 1061 was isolated as described earlier (11). pUC19 plasmid DNA was isolated from E.coli DH5α using the Wizard plus SV miniprep kit (Promega).
In vitro methylation of pUC19 DNA was performed with M.EcoRI (N6-methyladenine), M.HaeIII (5-methylcytosine) or M.BamHI (N4-methylcytosine) obtained from New England Biolabs according to the manufacturer's instructions. Complete methylation was subsequently confirmed by incubation of methylated DNA with the corresponding restriction endonucleases (Roche), separation by agarose gel electrophoresis and visualization by ethidium bromide staining. Unmethylated pUC19 served as a control for restriction. Protection of DNA against cleavage indicates efficient methylation. Digestion of chromosomal DNA with restriction enzymes Sau3AI, NdeI and PstI (Roche) was performed according to the manufacturer's instructions, but incubation was extended to 16 h. A mock incubation of chromosomal DNA with ultrapure water instead of restriction enzyme served as a control.
Sequencing
Sequencing was performed using primers −21M13 (5′-GTA AAA CGA CGG CCA G-3′), M13Rev (5′-CAG GAA ACA GCT ATG AC-3′), ABPUCF (5′-AGC AGA TTG TAC TGA GAG TGC ACC A-3′), ABPUCR (5′-GCA GCT GGC ACG ACA GGT TTC CCG A-3′), ABfrpC1 (5′-CGA GAG GGC AAC CAT CTT CTT ATC A-3′), ABfrpC2 (5′-GGG CGT CGT TGC CTT CTC CTC CA-3′) or KvAcat01 (5′-CGA AAT ACT CTT TTC GTG TCC -3′) using the ABI PRISM Dye Terminator Cycle Sequencing Kit or the ABI PRISM BigDye Terminators v2.0 or v3.0 Cycle Sequencing Kit (Applied Biosystems) or the DYEnamic ET Terminator Kit (Amersham Pharmacia Biotech).
Analysis
Sequences were analyzed using the ABI 3100 Genetic Analyzer and electropherograms were compared using the TRACE-DIFF program and trace display option of the GAP4 program included in the Staden computer package (12). Relative peak area (RPA) values were generated using the phred (13) component of the CodonCode aligner program. Phred output was imported into Microsoft Excel and SPSS 11.5.1 for further analyses. RPAs from peaks between phred positions 500 and 5000 (corresponding to sequence positions ∼50–400) were compared.
RESULTS
Template N4-cytosine methylation results in increased incorporation of labelled ddGTP
N6-methyladenine and 5-methylcytosine can be detected with the ABI PRISM Dye Terminator Cycle Sequencing kit (6), resulting in a higher T signal if the template contains N6-methyladenine, and a lower G signal if the template contains 5-methylcytosine. We observed previously that N4-methylcytosine could be detected as well by direct sequencing of DNA without a prior amplification step, resulting in a higher G peak if the template contains N4-methylcytosine. This was confirmed by comparison of electropherograms generated using in vitro M.BamHI methylated pUC19 DNA, which results in N4-methylcytosine at the underlined position in the sequence GGATCC, in vitro M.HaeIII methylated pUC19 DNA, which results in C5-methylcytosine at the underlined position in the sequence CCGG, and untreated pUC19 DNA using the DYEnamic ET Terminator Kit. The results are illustrated in Figure 1. C5-methylcytosine results in a decrease in the complementary G signal, whereas N4-methylcytosine results in an increase in the complementary G signal. Thus, the two types of cytosine methylation can both be detected and discriminated using plasmid DNA as a template.
Figure 1.

Differences between G signals complementary to unmethylated C residues, N4-methylcytosine residues and 5-methylcytosine residues using the DYEnamic ET Terminator Kit. A1: untreated pUC19 DNA; A2: M.BamHI N4-cytosine methylated pUC19DNA; A3: trace difference between A2 and A1. B1: untreated pUC19DNA; B2: M.HaeIII 5-cytosine methylated pUC19 DNA; B3: trace difference between B2 and B1. The G residues complementary to the methylated cytosines are boxed. N4-methylcytosine results in an increase in the complementary G signal, whereas 5-methylcytosine results in a decrease in the complementary G signal.
Direct detection of DNA methylation in genomic DNA
Since genomic epigenetic information present in methylated DNA is lost upon subcloning or PCR, we sought to apply the detection of DNA methylation by direct sequencing to prokaryotic total genomic DNA. Sequence data obtained from genomic DNA of two N.meningitidis strains, known to contain different methylation patterns, were compared. Strain 800615 harbors the NmeSI restriction modification system, of which the methyltransferase modifies the cytosine residue in the sequence AGTACT into N4-methylcytosine (5). Strain MC58 lacks the NmeSI restriction modification system. The Neisserial frpC locus contains an AGTACT site in both strains, and was sequenced with specific primers using total genomic DNA as template for both strains. Figure 2 shows the detection of N4-methylcytosine in the sequence AGTACT in strain 800615 genomic DNA.
Figure 2.
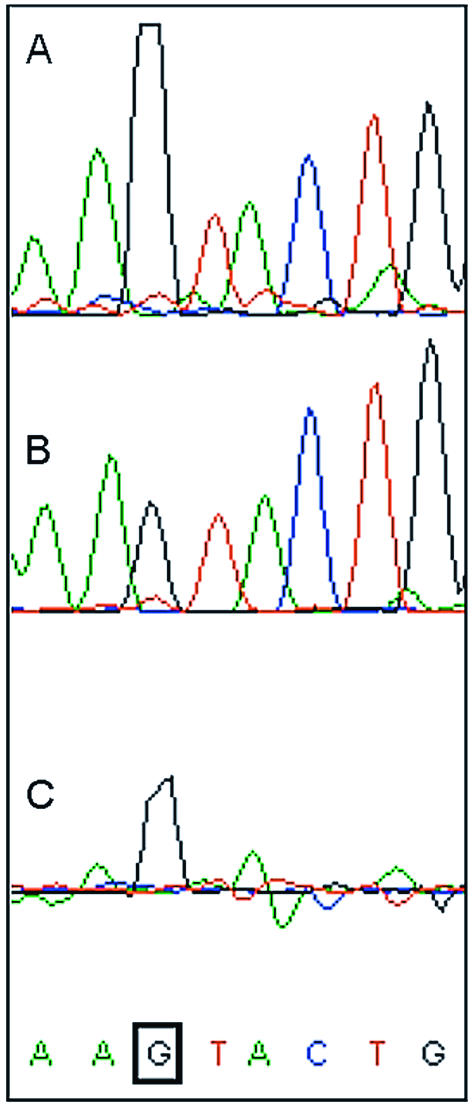
Differences between G signals complementary to unmethylated C residues and N4-methylcytosine residues in genomic N.meningitidis DNA using the DYEnamic ET Terminator Kit. (A) M.NmeSI N4-cytosine methylated strain 800615 DNA; (B) strain MC58 DNA; and (C) trace difference between A and B. The G complementary to the methylated cytosine is boxed. N4-methylcytosine results in an increase in the complementary G signal.
Intrasequence comparisons
As outlined, intersequence comparison of G or T signals may identify sites containing methylated bases in one of the two sequences. However, intrasequence comparison would be favourable, as it reduces costs by a factor of 2. We, therefore, sought to detect DNA methylation by intrasequence comparison. It was previously noted that the 2 nt 5′ to the incorporated base are most important for the so-called positional effects in peak-height differences (8,9). In a random sequence with equal amounts of A, C, G and T, similar triplets are expected on average for every 64 (=43) bp. This implies that in most sequence traces for a given trinucleotide, more than one copy will be present. Difference in trace signal heights would be expected if the same template was methylated for one trinucleotide while it was unmethylated for the other trinucleotide of identical sequence. First, we tested this assumption using a sequence trace obtained from a plasmid isolated from an E.coli strain carrying the Dam and Dcm methyltransferases. Figure 3 suggests that both dam (N6-methyladenine at GATC) and dcm methylation (5-methylcytosine at CCWGG) can be detected this way. A rigorous test of the difference in trace signal was performed by comparison of RPAs of 196 T signals in the sequence GAT from 31 different sequence traces obtained using plasmids isolated from Dam positive E.coli as template. In Figure 4, the significant difference between the 45 T signals in Dam methylated GATC context and the 151 T signals in unmethylated GATD context is illustrated. Although the distribution obviously overlaps, nearly all T signals in GATC sites differed markedly from T signals in GATD sites in individual sequence traces. Of the T signals in 45 GATC sites, the RPA of T signals in 44 GATC sites were higher than the RPA of T signals in all GATD sites in those traces. Of note, it has been reported that a few Dam sites in E.coli are not methylated (14–17).
Figure 3.

Template methylation can be inferred by intrasequence comparisons of base signals in similar 5′ context in plasmid DNA using the ABI PRISM Dye Terminator Cycle Sequencing Kit. Red boxes: comparison of the T signal in the red underlined dam GATC site (template adenine modified) and the T signal in another GAT sequence. The T signal complementary to a modified adenine is increased. Black boxes: comparison of the last G signal in the black underlined dcm CCTGG site (template cytosine modified at C5 position) and the last G signal in another TGG sequence. The G signal complementary to a C5 modified cytosine is decreased.
Figure 4.

Boxplot of RPA values for the T signal in the sequence GATC and the T signal in GATD obtained using plasmid DNA isolated from Dam positive E.coli as a template. The line in the box indicates the median value, the box indicates the interquartile range, the whiskers indicate the minimum and maximum up to 1.5 times the interquartile range, the circle indicates an outlier.
The intrasequence comparison was also applied to sequences obtained using genomic DNA as a template. The sequence runs obtained for the above mentioned N.meningitidis 800615 frpC sequence were used for intrasequence comparisons. The methylated C in AGTACT is present in the sequence context AAAGTACTGG (or on the complementary strand: CCAGTACTTT). The G residue is expected to give a different peak height. Therefore, the trinucleotide sequences AAG and CAG in the forward and reverse sequence runs were compared, as illustrated in Figure 5. The marked difference in G peak height with identical 5′ context confirms methylation of the corresponding C residue in the sequence AGTACT, whereas there was no methylation in the sequence GCCACT or GGTACT.
Figure 5.

Template methylation can be inferred by intrasequence comparisons of base signals in similar 5′ context in genomic DNA. Sequences with and without template N4-methylcytosine are compared in different parts of two single sequence traces derived from the frpC sequence using N.meningitidis strain 800615 genomic DNA as a template in a reaction with the DYEnamic ET Terminator Kit. A1: G peak following 5′ CA in AGTACT context, containing template N4-methylcytosine methylation; A2: G peak following 5′ CA in a sequence context without cytosine methylation; B1: the reverse sequence trace shows increased G peak height following 5′ AA in AGTACT context, resulting in template N4-methylcytosine methylation; B2: G peak following 5′ AA in a sequence context without cytosine methylation.
Successful identification of methylated sites in total genomic DNA without a priori knowledge of methyltransferases present is illustrated in Figure 6. Genomic DNA from H.pylori strain 1061KC1_C17 was used as a template in a sequence reaction, and G and T signal heights were carefully compared for similar 5′ sequence contexts in the resulting sequence trace. As illustrated in Figure 5, two markedly increased T peak heights were observed, complementary to putatively methylated A residues in the sequences GCTGCAGC and AGTGATCCC, respectively. Since most methyltransferases recognize palindromic sequences, possible recognition sites for two putative N6-adenosine methyltransferases are the hexamer CTGCAG and the tetramer GATC.
Figure 6.

Template methylation can be inferred by intrasequence comparisons of base signals in similar 5′ context in genomic DNA. In a sequence trace obtained using chromosomal DNA of H.pylori strain 1061KC1_C17 and the ABI PRISM Dye Terminator Cycle Sequencing Kit, two differences in T peak height were observed. (A) The T peak following 5′ GC is higher in A1 than in A2, suggesting template N6-adenosine methylation in the complementary strand. A palindromic hexamer, which could be a methyltransferase recognition site, is underlined in red. (B) The T peak following 5′ GA is higher in B1 than in B2, suggesting template N6-methyladenosine methylation in the complementary strand. The palindromic tetramer that could be the methyltransferase recognition site, is underlined in red.
Adenosine methylation of these sites would result in resistance of the H.pylori 1061 DNA to the restriction enzymes PstI and NdeII, respectively. To explore this possibility, genomic H.pylori 1061 DNA was incubated with these restriction endonucleases using the unmethylated PCR product ‘SHV’, which contains both NdeII and PstI sites, as a control. Figure 7 illustrates that NdeII and PstI cut the PCR product, but do not cleave H.pylori DNA, confirming the inferred methylation sites.
Figure 7.

Inhibition of restriction of H.pylori 1061 DNA. Lane 1, marker X (Roche); lane 2, H.pylori 1061 DNA and a PCR product ‘SHV’ after digestion with NdeII; lane 3, H.pylori 1061 DNA and a PCR product ‘SHV’ after mock incubation; lane 4, H.pylori 1061 DNA and a PCR product ‘SHV’ after digestion with PstI; lane 5, H.pylori 1061 DNA; lane 6, marker X (Roche); lane 7, PCR product ‘SHV’ after PstI digestion; and lane 8, PCR product ‘SHV’ after NdeII digestion. NdeII cleaves GATC but is inhibited by N6-adenosine methylation. PstI cleaves CTGCAG but is inhibited by N6-adenosine methylation. Note the presence of several plasmid bands in uncleaved H.pylori 1061 DNA.
DISCUSSION
Previous methods for detection of methylated bases in prokaryotic DNA sequences broadly fall into two categories. First, there is detection of the methylation status of a certain nucleotide in a known sequence context, e.g. by restriction analyses. Second, there is detection of a certain methylnucleotide at unknown sites, e.g. methylcytosine detection by bisulphite sequencing. In this study, we describe a comprehensive method that allows detection of the three naturally occurring methylated bases without prior knowledge of the sequence context. Moreover, intersequence and intrasequence trace comparisons yield strand-specific and site-specific DNA methylation patterns in complete genomic DNA. Thus, it theoretically allows the identification of a prokaryotic methylome (18).
Several applications can be envisaged for this relatively simple method. The methylation status of a certain site can be confirmed, even full methylation and hemimethylation can be discriminated by sequencing both strands of a template. This could be of importance in studies of so-called epigenetic switches, where the methylation status of certain sites controls phase variation (14–17).
Recognition sites of DNA methyltransferases can be confirmed by comparison of traces obtained in methyltransferase positive and negative backgrounds. These backgrounds can either be the bacterial species that is the source of the methyltransferase, or an E.coli strain that carries the cloned methyltransferase. Also, the type of methylation of a DNA methyltransferase can be identified, as N6-methyladenine, N4-methylcytosine and 5-methylcytosine can be discriminated.
Even unknown methylation patterns can be detected in bacterial strains of interest, as illustrated by the H.pylori 1061 example. The sequences GATC and CTGCAG were found to contain N6-methyladenosine. A query for restriction and modification enzymes found in strains of the genus Helicobacter in the REBASE database (14–16,19) yielded candidate methyltransferases for both sites. GATC is the recognition site of the N6-adenosine methyltransferase M.HpyAIII/M.Hpy99VI (20–22). This methyltransferase is present in both the sequenced H.pylori genomes (23,24), and GATC methylation was found in all 90 strains tested in a recent study (25). Therefore, its presence in H.pylori strain 1061 would not be surprising. CTGCAG is not listed as a methyltransferase recognition site, but the type III restriction enzymes Hpy790639P, Hpy790545P and HpyAXI are listed with this recognition site (21,22,26). Possibly, a methyltransferase that is part of one of these systems is present in H.pylori strain 1061.
Acknowledgments
We thank Wendy Keijzers and Nico Ponne for expert technical assistance, Nashwan al Naiemi and Kim Schipper for their sequence data, and an anonymous referee for suggesting additional analyses. Funding to pay the Open Access publication charges for this article was provided by the Department of Medical Microbiology at the Academic Medical Center.
Conflict of interest statement. None declared.
REFERENCES
- 1.Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 2.Arber W., Linn S. DNA modification and restriction. Annu. Rev. Biochem. 1969;38:467–500. doi: 10.1146/annurev.bi.38.070169.002343. [DOI] [PubMed] [Google Scholar]
- 3.Jeltsch A. Maintenance of species identity and controlling speciation of bacteria: a new function for restriction/modification systems? Gene. 2003;317:13–16. doi: 10.1016/s0378-1119(03)00652-8. [DOI] [PubMed] [Google Scholar]
- 4.Alm R.A., Trust T.J. Analysis of the genetic diversity of Helicobacter pylori: the tale of two genomes. J. Mol. Med. 1999;77:834–846. doi: 10.1007/s001099900067. [DOI] [PubMed] [Google Scholar]
- 5.Bart A., Pannekoek Y., Dankert J., van der Ende A. NmeSI restriction–modification system identified by representational difference analysis of a hypervirulent Neisseria meningitidis strain. Infect. Immun. 2001;69:1816–1820. doi: 10.1128/IAI.69.3.1816-1820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao B.S., Buckler-White A. Direct visualization of site-specific and strand-specific DNA methylation patterns in automated DNA sequencing data. Nucleic Acids Res. 1998;26:2505–2507. doi: 10.1093/nar/26.10.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandis J.W. Dye structure affects Taq DNA polymerase terminator selectivity. Nucleic Acids Res. 1999;27:1912–1918. doi: 10.1093/nar/27.8.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker L.T., Deng Q., Zakeri H., Carlson C., Nickerson D.A., Kwok P.Y. Peak height variations in automated sequencing of PCR products using Taq dye-terminator chemistry. Biotechniques. 1995;19:116–121. [PubMed] [Google Scholar]
- 9.Parker L.T., Zakeri H., Deng Q., Spurgeon S., Kwok P.Y., Nickerson D.A. AmpliTaq DNA polymerase, FS dye-terminator sequencing: analysis of peak height patterns. Biotechniques. 1996;21:694–699. doi: 10.2144/96214rr02. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin A., Kersulyte D., Sisson G., Veldhuyzen van Zanten S.J., Berg D.E., Hoffman P.S. Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Mol. Microbiol. 1998;28:383–393. doi: 10.1046/j.1365-2958.1998.00806.x. [DOI] [PubMed] [Google Scholar]
- 11.van Amsterdam K., van Vliet A.H., Kusters J.G., Feller M., Dankert J., van der Ende A. Induced Helicobacter pylori vacuolating cytotoxin VacA expression after initial colonisation of human gastric epithelial cells. FEMS Immunol. Med. Microbiol. 2003;39:251–256. doi: 10.1016/S0928-8244(03)00226-8. [DOI] [PubMed] [Google Scholar]
- 12.Bonfield J.K., Rada C., Staden R. Automated detection of point mutations using fluorescent sequence trace subtraction. Nucleic Acids Res. 1998;26:3404–3409. doi: 10.1093/nar/26.14.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ewing B., Hillier L., Wendl M.C., Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 14.Hernday A., Krabbe M., Braaten B., Low D. Self-perpetuating epigenetic pili switches in bacteria. Proc. Natl Acad. Sci. USA. 2002;99:16470–16476. doi: 10.1073/pnas.182427199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernday A., Braaten B., Low D. The intricate workings of a bacterial epigenetic switch. Adv. Exp. Med. Biol. 2004;547:83–89. doi: 10.1007/978-1-4419-8861-4_7. [DOI] [PubMed] [Google Scholar]
- 16.Hernday A.D., Braaten B.A., Low D.A. The mechanism by which DNA adenine methylase and PapI activate the pap epigenetic switch. Mol. Cell. 2003;12:947–957. doi: 10.1016/s1097-2765(03)00383-6. [DOI] [PubMed] [Google Scholar]
- 17.van der Woude M., Braaten B., Low D. Epigenetic phase variation of the pap operon in Escherichia coli. Trends Microbiol. 1996;4:5–9. doi: 10.1016/0966-842x(96)81498-3. [DOI] [PubMed] [Google Scholar]
- 18.Feinberg A.P. Methylation meets genomics. Nature Genet. 2001;27:9–10. doi: 10.1038/83825. [DOI] [PubMed] [Google Scholar]
- 19.Roberts R.J., Vincze T., Posfai J., Macelis D. REBASE—restriction enzymes and DNA methyltransferases. Nucleic Acids Res. 2005;33:D230–D232. doi: 10.1093/nar/gki029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong H., Lin L.F., Porter N., Stickel S., Byrd D., Posfai J., Roberts R.J. Functional analysis of putative restriction–modification system genes in the Helicobacter pylori J99 genome. Nucleic Acids Res. 2000;28:3216–3223. doi: 10.1093/nar/28.17.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin L.F., Posfai J., Roberts R.J., Kong H. Comparative genomics of the restriction–modification systems in Helicobacter pylori. Proc. Natl Acad. Sci. USA. 2001;98:2740–2745. doi: 10.1073/pnas.051612298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitkute J., Stankevicius K., Tamulaitiene G., Maneliene Z., Timinskas A., Berg D.E., Janulaitis A. Specificities of eleven different DNA methyltransferases of Helicobacter pylori strain 26695. J. Bacteriol. 2001;183:443–450. doi: 10.1128/JB.183.2.443-450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alm R.A., Ling L.S., Moir D.T., King B.L., Brown E.D., Doig P.C., Smith D.R., Noonan B., Guild B.C., deJonge B.L., et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 24.Tomb J.F., White O., Kerlavage A.R., Clayton R.A., Sutton G.G., Fleischmann R.D., Ketchum K.A., Klenk H.P., Gill S., Dougherty B.A., et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 25.Simala-Grant J.L., Lam E., Keelan M., Taylor D.E. Characterization of the DNA adenine 5′-GATC-3′ methylase HpyIIIM from Helicobacter pylori. Curr. Microbiol. 2004;49:47–54. doi: 10.1007/s00284-004-4244-4. [DOI] [PubMed] [Google Scholar]
- 26.Legrain P., Rain J.C., Colland F., de Reuse H., Labigne A. WO 02066501 A. Patent. 2002