

Abstract
Cytotoxins from cobra venom are known to manifest cytotoxicity in various cell types. It is widely accepted that the plasma membrane is a target of cytotoxins, but the mechanism of their action remains obscure. Using the confocal spectral imaging technique, we show for the first time that cytotoxins from cobra venom penetrate readily into living cancer cells and accumulate markedly in lysosomes. Cytotoxins CT1 and CT2 from Naja oxiana, CT3 from Naja kaouthia and CT1 from Naja haje are demonstrated to possess this property with respect to human lung adenocarcinoma A549 and promyelocytic leukaemia HL60 cells. Immobilized plasma membrane binding accompanies the internalization of CT3 from Naja kaouthia in the HL60 cells, but it is very weak for other cytotoxins. Detectable membrane binding is not a property of any of the cytotoxins tested in A549 cells. The kinetics and concentration-dependence of cytotoxin accumulation in lysosomes correlate well with their cytotoxic effects. On the basis of the results obtained, we propose that lysosomes are a primary target of the lytic action of cytotoxins. Plasma membrane permeabilization seems to be a downstream event relative to lysosome rupture. Direct damage to the plasma membrane may be a complementary mechanism, but its relative contribution to the cytotoxic action depends on the cytotoxin structure and cell type.
Keywords: cobra venom, cytotoxicity, cytotoxin, lysosome, confocal spectral imaging
Abbreviations: AO, Acridine Orange; CSI, confocal spectral imaging; CT, cytotoxin; the suffixes No, Nk, Nh and Na denote cytotoxins from Naja oxiana, Naja kaouthia, Naja haje and Naja atra respectively; NTII, neurotoxin II from Naja oxiana; Rh, tetramethylrhodamine; Rh-CT, tetramethylrhodamine-labelled cytotoxin
INTRODUCTION
CTs (cytotoxins) from cobra venom are known to manifest cytotoxicity in various cell types, including erythrocytes, lymphocytes, myocytes, spleen cells and various tumour cells [1–6]. Several mechanisms of cytotoxicity have been reported for different cell types, but all of these mechanisms were proposed to be realized at the external plasma membrane surface. Both membrane proteins and phospholipids may serve as a target for CTs [7], thus accounting for the variety of biological activities reported for CTs.
The property of some CTs to stimulate the depolarization of myocytes [8,9] was ascribed to CT-induced opening of voltagedependent Ca2+ channels, blocking of inwardly rectifying background K+ channels and the formation of new ion conductive pathways [6,10–13]. Accordingly, CTs from cobra venom that are able to cause heart arrest are also known as cardiotoxins. CTs were proposed to have protein targets in the membranes of myocytes [14,15], but these have not yet been identified. Proteins of 92, 77 and 68 kDa that were able to bind CT3Na (CT3 from Naja atra) were found in the plasma membrane of phytohaemagglutinin-stimulated peripheral blood mononuclear cells [16]. It was proposed that CT3Na displays its immunomodulatory properties by means of interactions with some of these proteins.
CTs from Naja atra and Naja kaouthia are able to inhibit protein kinase C efficiently [17–19], and this molecular mechanism was concluded to be responsible, at least partially, for the inhibition both of the proliferation of several cancer cell types and of the PMA-induced differentiation of HL60 cells. More recently, the inhibitory effects of CTs with respect to protein kinase C were explained by the ability of CTs to bind NTPs [20].
Cytolysis is considered to be a general mechanism of CT action in many cell types [1,4,8,21–24]. The ability of water-soluble, highly basic CTs to bind at the lipid bilayer surface is considered to be a basic requirement for the cytolytic activity of CTs. CTs possess a high affinity for binding to negatively charged lipids [7,22,25,26], and at least some CTs undergo lipid-induced oligomerization [27]. It was proposed that CT oligomerization may serve as a basis for toxin-indcued pore formation and/or a direct lytic action on membranes [27,28].
Various characteristics of the interaction of CTs with cells have been studied using radiolabelled CTs: the kinetics and temperature-dependence of binding, competition with unlabelled CTs, association constants and numbers of high-affinity binding sites [1,21,22]. CT from Naja siamensis adsorbed on a solid support or attached to 100 μm-diameter agarose beads was demonstrated to preserve its cytotoxicity against L1210 tumour T-lymphocytes [23]. All of these data were interpreted and analysed on the basis of the proposed plasma membrane binding of CTs without their internalization. At the same time, neither experiments with radiolabelled CTs, nor other studies determined the exact cellular localization of bound CTs.
To our knowledge, there has been only one attempt to visualize the cellular distribution of CTs: the membrane localization of CT3Na in monocytes and lymphocytes was visualized with immunofluorescence staining [16]. Describing their results, the authors mentioned casually that the CT “was seen as a diffuse staining of the membrane and as aggregates within the cells (phytohaemagglutinin-activated lymphocytes), indicating that it bound to and entered the cells”. The assumed intracellular penetration of CT3Na was ignored in the abstract and discussion sections of the paper, and attention was focused on membrane-bound CT3Na.
In the present work, we have compared the cytotoxic effects of CT1No (CT1 from Naja oxiana), CT2No, CT3Nk (CT3 from Naja kaouthia), CT1Nh (CT1 from Naja haje) and CT2Nh on human lung adenocarcinoma A549 and promyelocytic leukaemia HL60 cells, and have used fluorescently labelled CT1No, CT2No, CT3Nk and CT1Nh to study the interactions of CTs with the cells. Using the CSI (confocal spectral imaging) technique [29–32], we prove that CTs penetrate readily inside A549 and HL60 cells, and show substantial accumulation within lysosomes. Evidently, this pathway assumes interaction of CTs with the plasma membrane, but this interaction is transient, and is not accompanied by retention of CTs at the plasma membrane in most cases studied by us. Immobilized cell surface binding was observed between CT3Nk and HL60 cells only, and it was complemented by distinct internalization of CT3Nk. The kinetics and concentration-dependence of CT accumulation in lysosomes correlated well with cytotoxic effects of the CTs. We propose that lysosomes are the major targets of the lytic action of CTs, whereas plasma membrane injury is a downstream event relative to lysosome rupture.
MATERIAL AND METHODS
Chemicals
AO (Acridine Orange), Rh (tetramethylrhodamine) isothiocyanate, carboxymethylrhodamine N-succinimidyl ester and Triton X-100 were purchased from ICN (Costa Mesa, CA, U.S.A.). Tris, propidium iodide and Hoechst33342 were supplied by Sigma (St. Louis, MO, U.S.A.). Other chemicals were of analytical-reagent grade.
Toxins and their fluorescent derivatives
CT1No, CT2No, CT3Nk, CT1Nh and CT2Nh were isolated from cobra venoms as described previously [33]. NTII (neurotoxin II from Naja oxiana) was isolated as described elsewhere [34].
To obtain fluorescent derivatives, a solution of Rh isothiocyanate (0.78 mg in 1.5 ml of DMSO) was added slowly to 1 ml of CT solution (7 mg/ml) in 0.2 M sodium phosphate buffer, pH 10.5, containing 0.175 mM EDTA with continuous stirring. The solution of carboxymethylrhodamine N-succinimidyl ester (0.56 mg in 0.1 ml of DMSO) was added to 1 ml of NTII solution (4.1 mg/ml) in 0.2 M sodium phosphate buffer (pH 8.0) with continuous stirring. The reaction mixture was incubated for 105 min (or 18 h in the case of NTII) at room temperature and then desalted by gel filtration on a Sepadex G-15 column (2.5 cm×90 cm) equilibrated with 0.1 M acetic acid. The protein fraction was freeze-dried and separated by reverse-phase HPLC on a Vydac C18 column (10 mm×250 mm) with a gradient of aqueous acetonitrile from 20 to 60% (v/v) (20–40% for NTII) in 0.1% (v/v) trifluoroacetic acid over 40 min (20 min for NTII) at a flow rate of 2 ml/min. The fractions containing singly labelled CTs and NTII (as determined by matrix-assisted laser-desorption ionization MS) were freeze-dried and used for further studies. The cytotoxicity of each Rh-CT (Rh-labelled CT) was compared with the cytotoxic effect of the corresponding native protein at the concentration of the native CT resulting in 70–80% cell death. The cytotoxicity of Rh-NTII was tested at 10 μM after 3 and 24 h of incubation of the cells with Rh-NTII.
The most cytotoxic fractions of labelled CT1No, CT2No, CT3Nk and CT1Nh were characterized using CD spectroscopy in aqueous solution. The shapes of the CD spectra of the labelled toxins were the same as those of their native precursors, indicating the absence of label-induced conformational changes.
Pyridylethylation of Rh-CTs and mass spectra recording
Selected fractions of Rh-CTs (0.2–0.3 mg) were dissolved in 300 μl of 0.2 M Tris/HCl buffer (pH 8.5; containing 6 M guanidinium hydrochloride), then 10 μl of 100 mg/ml dithiothreitol in the same buffer was added, and the mixture was incubated for 18 h at room temperature. After that, 4-vinylpyridine (3 μl) was added, and the incubation was continued for 3 h. The pyridylethylated derivative was isolated by reverse-phase HPLC on a Jupiter C18 column (4.6 mm×250 mm; Phenomenex) with a gradient of aqueous acetonitrile from 20 to 50% (v/v) in 0.1% trifluoroacetic acid for 30 min, and finally lyophilized.
For direct matrix-assisted laser-desorption ionization MS measurements of tryptic digests, 25 μg of pyridylethylated Rh-CT was incubated with 0.5 μg of porcine trypsin in sodium bicarbonate solution (25 μl; 50 mM) for 2 h at 37 °C. The mass spectra were recorded with a REFLEX III spectrometer (Bruker Daltonics) using 2,5-dihydroxybenzoic acid as a matrix. The samples were prepared using the dried droplet technique. The spectra were recorded in positive-ion mode.
For the tryptic digest of Rh-CT2No (the fraction having the highest cytotoxicity), a peak of 1978.6 Da was detected, which corresponded to the toxin fragment comprising residues 51–60 containing a label. Therefore the label was attached to Lys-58 of CT2No.
We failed to find the mass peaks of labelled fragments for the tryptic digests of Rh-CT3Nk, Rh-CT1No and Rh-CT1Nh. Even in the case of Rh-CT2No the detected peak was very weak, which might indicate either instability of label under MS conditions after chemical treatment or low volatility of the labelled fragments.
Cells and cytotoxicity measurements
A549 human lung adenocarcinoma cells and HL60 promyelocytic leukemia cells were cultured as described previously [33,35]. All incubations of cells with CTs, NTII, Rh-CTs and Rh-NTII were performed in complete medium, i.e. a proper culture medium supplemented with 7% (v/v) fetal calf serum and 2 mM L-glutamine, at 37 °C. The concentration of CT and incubation time varied as indicated in the Figures. For efflux experiments, A549 cells were incubated with Rh-CT1No or Rh-CT3Nk (1 μM for 1 h) in complete medium, rinsed twice and placed for 1 h in CT-free complete medium (37 °C).
To identify lysosomes, cells were stained with AO (50 ng/ml or 0.5 μg/ml for 15 min at 37 °C) after incubation with Rh-CT (0.2–2 μM for 1 or 2 h at 37 °C).
The cytotoxicity of CTs and NTII was determined after incubation of cells (1×106 cells/ml HL60 cells or a confluent monolayer of A549 cells on the cover glass) with different concentrations of CTs for 3 h (NTII for 3 and 24 h). Toxin-treated cells were stained (15 min, 37 °C) with propidium iodide (10 μM) and Hoechst33342 (10 μM) and examined under a microscope (model BH2; Olympus Europa GmbH, Hamburg, Germany) with a 10× objective. The U-MSWG epifluorescence filter unit (480–550 nm excitation filter; 570 nm dichroic mirror; 590 nm barrier emission filter) was used to count dead cells having propidium iodide fluorescence in the nucleus, whereas the U-MWU filter unit (330–385 nm excitation filter; 400 nm dichroic mirror; 420 nm barrier emission filter) was used to count all cells showing nuclear fluorescence of Hoechst33342. Approx. 500–700 cells were counted per experimental point, and titration was performed in triplicate for each CT.
The experimental dependence of cytotoxicity on CT concentration was fitted using the Hill formula:
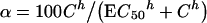 |
1 |
where α is the percentage of dead cells, C is the CT concentration in the medium, EC50 is the CT concentration that induces 50% cell death, and h is the Hill coefficient. This fitting was used to define EC50 and h values for each CT.
CSI measurements
CSI measurements were performed using an installation described elsewhere [29] with a UPLAPO60×W objective (Olympus Europa GmbH). The lateral, axial and spectral resolutions were ∼0.5 μm, ∼3 μm and 1 nm respectively. The CSI technique is based on measurement of the fluorescence spectrum at each point of a specimen [29–32]. Excitation of spectra was performed with an Ar+ laser (514 nm, 10 μW; model 164-45; Spectra-Physics GmbH, Darmstadt, Germany) or a Nd3+-YAG-laser (532 nm, 2 μW; model LCS-T-11; Laser-Compact, Moscow, Russia). Each experimental spectrum recorded in confocal mode from the specimen was deconvolved as the sum of the reference spectra with weighting coefficients. Taken in a combination appropriate for a particular experiment, the fluorescence spectra of Rh-CT, free Rh and AO (two to three model spectra) were used as the reference spectra. The integrated intensity of a reference spectrum multiplied by the weighting coefficient for each point produced a two-dimensional map that described the relative distribution of the corresponding dye along the optical section of a specimen.
The concentration of Rh-CT within the cytoplasm of cells was deduced from the integrated intensities of the discriminated intracellular fluorescence spectra of Rh-CT. The dependence of fluorescence intensity on Rh-CT concentration was calibrated with a solution of Rh-CT in 1% Triton X-100 and 20 mM Tris/HCl, pH 7.4, under the same experimental conditions (optical pathway, light collection geometry, lateral and axial resolution, laser power and integration time) that were used for the CSI measurements. The quantitative two-dimensional maps of Rh-CT distribution were reconstructed for each cell measured, and the cytoplasmic concentration of Rh-CT was averaged over 30–50 cells.
It should be mentioned that the spectral analysis employed in the CSI technique allowed us to discriminate between the overlapping signals of Rh-CT and free Rh (resulting from conjugate deterioration) due to the shifted maxima of their fluorescence spectra. Free Rh penetrates quickly inside cells and accumulates primarily in mitochondria. This means that a trace amount of free Rh in the extracellular medium provides a perceptible intracellular concentration of Rh. Accordingly, correction for the contribution of free Rh was performed in our experiments, in order to avoid (i) overestimation of the intracellular concentration of Rh-CTs, and (ii) incorrect conclusions concerning the intracellular localization of Rh-CTs.
RESULTS
Cytotoxicity of CTs
The cytotoxicity of CTs towards adherent A549 cells decreased in the order CT2No≥CT3Nk≫CT1No≫CT2Nh≈CT1Nh (Table 1, Figure 1); CT2No was 78-fold more toxic towards A549 cells than CT1Nh. Hill coefficients for CTs varied from 1.5 to 2.6 (Table 1), thus indicating a co-operative mode of interaction between the CTs and the cells.
Table 1. Cytotoxicity of CTs for A549 and HL60 cells.
The cells were incubated with the CTs for 3 h. The Hill coefficient (h; see eqn 1) characterizes the co-operativity of the interaction between the CT and the cells. The EC50(Rh-CT) value represents the estimated concentration of a Rh-CT that is equally as effective in causing cell death as the EC50 of the respective CT.
| A549 cells | HL60 cells | ||||
|---|---|---|---|---|---|
| CT | EC50 (μM) | h | EC50 (μM) | h | EEC50(Rh-CT) (μM) |
| CT1No | 16.6±0.6 | 1.8±0.1 | 0.58±0.03 | 1.5±0.1 | 1.5±0.3 |
| CT2No | 1.7±0.1 | 1.5±0.2 | 0.33±0.02 | 2.8±0.2 | 0.36±0.08 |
| CT3Nk | 2.6±0.2 | 2.1±0.3 | 0.18±0.01 | 3.2±0.1 | 0.27±0.05 |
| CT1Nh | 132±9 | 2.6±0.5 | 2.6±0.1 | 2.4±0.1 | 4.2±0.8 |
| CT2Nh | 116±6 | 2.3±0.3 | 1.9±0.1 | 2.6±0.2 | – |
Figure 1. Dependence of cell death on extracellular CT concentration.

Cells were incubated with CTs for 3 h.
HL60 cells were more sensitive to the CTs than were A549 cells (Table 1, Figure 1), whereas the range of differences in cytotoxicity was relatively small, in the order CT3Nk>CT2No>CT1No≫CT2Nh≥CT1Nh; CT3Nk was just 14-fold more toxic than CT1Nh. Interactions between CTs and HL60 cells have a co-operative character: the Hill coefficients varied from 2.4 to 3.2 for all CTs except CT1No (Table 1).
On the basis of the cytotoxicity data, CT1No, CT2No, CT3Nk and CT1Nh were selected and labelled with Rh for investigation of their localization in cells. The concentrations inducing 80% cell death after a 3 h incubation of HL60 cells with the native CT were chosen in order to compare the cytotoxicity of Rh-CTs with that of the corresponding non-modified CTs. Under these conditions, Rh-CTs killed 1.2–4-fold fewer cells than the respective native proteins. Using these data, the concentrations of Rh-CTs resulting in approx. 50% cell death were calculated (Table 1). The similar values of these concentrations of the Rh-CTs and the respective native CTs (Table 1) allow us to assume that the mode of interaction between Rh-CTs and cells is unchanged.
NTII and Rh-NTII were used for comparison. Incubation of A549 or HL60 cells with NTII at concentrations ranging from 10 nM to 10 μM for 24 h did not result in any cytotoxicity. Incubation with Rh-NT at 10 μM for 3 or 24 h was not toxic to the cells.
Localization of Rh-CTs in A549 cells
The CSI technique revealed a distinct cytoplasmic vesicular localization of all Rh-CTs in A549 cells (Figures 2a–2d). No plasma membrane staining was observed even in the area of intercellular contacts (Figure 2c). The staining pattern was the same whether Rh-CTs were present in the extracellular medium during observation (Figure 2a) or washed out before observation (Figures 2b–2d). Different fractions of Rh-CTs (eluting from a chromatographic column at different times and apparently having a different location of the Rh label) stained the cells in a similar manner (results not shown). There were no qualitative changes in the cellular localization of Rh-CTs during incubation (30 min–4 h) at CT concentrations ranging from 0.1 μM to the lethal level. Nuclear penetration of Rh-CT and overall cytoplasmic staining was observed in dying and dead cells (Figures 2e–2g). Analogous experiments with Rh-NTII revealed neither internalization nor membrane binding of this toxin.
Figure 2. Typical distributions of Rh-CT1No (a), Rh-CT1Nh (b), Rh-CT2No (c) and Rh-CT3Nk (d) in living A549 cells, and Rh-CT3Nk distribution in dying (e, f) and dead (g) A549 cells.

The left column shows conventional bright-field micrographs of cells, whereas the right column shows confocal spectral images of Rh-CT distribution at the equatorial optical section of cells. The intensity scale corresponds to the intracellular concentration of Rh-CTs in μM. Cells were incubated with Rh-CT1No (5 μM for 30 min), Rh-CT1Nh (1 μM for 1 h), Rh-CT2No (1 μM for 1 h) or Rh-CT3Nk (1 μM for 1 h) and placed under the microscope without washing (b–d) or after thorough washing (a, e–g). Dying (e, f) and dead (g) A549 cells were identified by characteristic morphological changes in the shape of a cell and in the state of a nucleus and cytoplasm.
Incubation of the cells with AO, the vital fluorescent probe of lysosomes, revealed co-localization of Rh-CTs and AO in lysosomes. Two sets of experiments were performed, and both showed lysosomal localization of Rh-CTs. The first was carried out at an excitation wavelength of 514 nm and with a low AO concentration (50 ng/ml). In these experiments it was possible to distinguish the dominant fluorescent signal of monomeric AO (emission maximum 530 nm) and the minor signal of aggregated AO (emission maximum 640 nm), which co-localized with the Rh-CT signal in lysosomes (Figures 3a–3c). The second experiment was performed at an excitation wavelength of 532 nm and with a high AO concentration (0.5 μg/ml). In this case, the AO concentration was high enough to form predominantly aggregates in lysosomes (640 and 665 nm emission maxima), and the ‘spotty’ pattern of the Rh-CT distribution was entirely replaced with the spotty pattern of the fluorescence of aggregated AO (results not shown). This phenomenon can be explained by fluorescence resonance energy transfer from Rh-CT to the aggregates of AO, which indicates exact co-localization between Rh-CT and AO.
Figure 3. Co-localization of Rh-CT3Nk and lysosome probe AO in A549 (a–d) and HL60 (e–h) cells.

(a, e) Conventional bright-field micrographs of cells. Bar corresponds to 5 μm. The broken line indicates the nucleus region. (b, f) Distribution of Rh-CT3Nk. (c, g) AO staining of lysosomes. (d, h) Merged images of RhCT3Nk and AO distribution in cells. Yellow indicates co-localization of RhCT3Nk and AO in lysosomes. A549 (HL60) cells were incubated with 2 μM (0.2 μM) Rh-CT3Nk for 2 h.
Localization of Rh-CTs in HL60 cells
A spotted distribution of Rh-CTs was also observed in HL60 cells, and CSI measurements confirmed both intracellular localization of the Rh-CT-containing vesicles (Figure 4) and their lysosomal origin (Figures 3d–3f). Plasma membrane staining was a distinct feature with Rh-CT3Nk, which occurred in addition to the intracellular lysosomal accumulation of the toxin (Figures 3–5). Membrane staining was detected in 30–50% of cells. Usually it was non-uniform, with a tendency to form caps and/or clusters on the membrane. Membrane-bound Rh-CT3Nk was immobilized, since no dissociation of Rh-CT3Nk from the membrane occurred over a 2 h incubation of cells in Rh-CT3Nk-free medium, and no lateral diffusion of the CT was detected using the FRAP (fluorescence recovering after photobleaching) technique (Figure 5). Membrane staining of HL60 cells with Rh-CT1No, Rh-CT2No and Rh-CT1Nh was very weak, and was found only in 3–5% of cells. No staining of HL60 cells with Rh-NTII was observed.
Figure 4. Typical distributions of Rh-CT1No (a), Rh-CT1Nh (b), Rh-CT2No (c) and Rh-CT3Nk (d) in living HL60 cells.

The left column shows conventional bright-field micrographs of cells (bar=5 μm). The broken line indicates the nucleus region. The right column shows confocal spectral images of Rh-CT distribution at the equatorial optical section of cells. The intensity scale corresponds to the intracellular concentration of Rh-CTs in μM. Cells were incubated with Rh-CT1No (0.5 μM), Rh-CT1Nh (1 μM), Rh-CT2No (0.5 μM) or Rh-CT3Nk (0.2 μM) for 1 h and placed under the microscope after thorough washing.
Figure 5. Rh-CT3Nk immobilized on the plasma membrane of HL60 cells shows no lateral diffusion.

The fluorescence of membrane-bound Rh-CT3Nk (a) was bleached with a laser in the restricted region indicated by the arrow (b). No fluorescence recovery due to Rh-CT3Nk lateral diffusion was observed 15 min (c) or 25 min (d) after photobleaching. Cells were incubated with 0.2 μM Rh-CT3Nk for 1 h.
Accumulation and retention of Rh-CT3Nk and Rh-CT1No in A549 cells
The kinetics of the intracellular accumulation of CTs in A549 cells were studied using Rh-CT3Nk and Rh-CT1No as examples of CTs with high and intermediate toxicity respectively. Intracellular accumulation of Rh-CT3Nk and Rh-CT1No in A549 cells was very fast: the average cytoplasmic concentration (i.e. the concentration of CT in lysosomes averaged over the cytoplasmic region of cells) of CTs approached saturation during a 1 h incubation (Figure 6). The intracellular accumulation of the CTs was reversible (at least partially): cells loaded with Rh-CT1No or Rh-CT3Nk released 45±9% and 43±8% respectively of the CT during a 1 h incubation in CT-free medium at 37 °C (Figure 6). Exposure of the cells to equimolar concentrations of Rh-CT3Nk and Rh-CT1No resulted in rather similar intracellular levels of CTs (Figure 6), which is in contrast with a large difference in the cytotoxicity of these toxins (Table 1, Figure 1). An increase in the extracellular concentration of the CTs from 1 to 10 μM (1 h incubation) was accompanied by a noticeable increase in the average cytoplasmic concentrations (from 2.6±0.2 to 11.4±0.2 μM for Rh-CT1No, and from 4.2±0.1 to 16.8±0.8 μM for Rh-CT3Nk), although the accumulation factors (ratio of cytoplasmic/extracellular CT concentration) decreased. The lysosomal concentration of Rh-CT cannot be measured accurately with the CSI technique, since the diameter of the lysosomes is considerably smaller than the thickness of the optical section (approx. 3 μm) that is discriminated by the confocal filtration of the signal. At the same time, we can provide a low estimate of the lysosomal concentration of a Rh-CT: it is at least 20–100-fold higher than the extracellular concentration of the Rh-CT (Figure 2).
Figure 6. Accumulation and retention of Rh-CT3Nk and Rh-CT1No in A549 cells.

Uptake (average cytoplasmic concentration; ACC) of Rh-CTs is shown for cells incubated with 1 μM Rh-CT. Also shown is retention of Rh-CTs in cells pre-incubated with the corresponding Rh-CT (1 μM for 1 h) and placed in Rh-CT-free medium for 1 h. Bars indicate S.D.
When the extracellular concentration of Rh-CT3Nk was within the cytotoxic range, dying and dead cells were recognized by bright-field illumination due to characteristic morphological changes in the shape of the cell and in the state of the nucleus and cytoplasm (e.g. Figures 2e–2g). The concentrations of Rh-CT1No used in the experiments were outside the cytotoxic range, and so no dying cells were observed.
An examination of dying cells allowed us to assess the development of changes in the intracellular distribution of Rh-CT3Nk (Figures 2e–2g). Initial signs of cytotoxicity (nucleus rounding, condensation of perinuclear cytoplasmic structures) were not accompanied by noticeable changes in the intracellular distribution of Rh-CT3Nk. The lysosomal localization of the toxin was preserved, at least partially; the plasma membrane was impermeable with respect to the diffusion of extracellular Rh-CT in the cytoplasm (Figure 2e). The development of a cytotoxic effect (Figure 2f) was accompanied by penetration of Rh-CT into the nucleus (diffuse staining of nucleus) and its enhanced accumulation in nucleoli (bright spots in nucleus); weak diffuse staining of the cytoplasm and persistent localization of Rh-CT in some lysosomes were also observed. The loss of plasma membrane integrity seems to be a late event of CT action, and resulted in the bright staining of the cytoplasm and nucleus with Rh-CT that had diffused from the extracellular medium (Figure 2g); the lysosomal localization of Rh-CT disappeared or was masked by the intense Rh-CT signal in the cytoplasm.
DISCUSSION
Our experiments have revealed that efficient intracellular penetration of CTs and indistinguishable or very low CT accumulation at the plasma membrane are common features of CTs from different cobra venoms when they are added to adherent cells and cells in suspension. Only one exception to the common distribution pattern was found among four Rh-labelled CTs studied by us: CT3Nk demonstrated both internalization and noticeable membrane binding with HL60 cells. To our knowledge, this is the first time that the intracellular penetration of CTs has been demonstrated unambiguously.
For a long time, the action of CTs has been considered to occur at the plasma membrane, and internalization was not even hypothesized. Reasonably, questions arise as to whether our data are artifacts, and whether they contradict experimental data published previously. We examined the cellular localization of several Rh-CT derivatives. The Rh label was most probably attached to different amino groups in these derivatives, but its exact position was not clear, except for the most active Rh-CT2No derivative. The location of the label affected the cytotoxicity of the derivatives, but did not change their cellular distribution. Finally, for our experiments we selected Rh-CTs whose cytotoxicity was similar to that of the native toxins and whose conformation was not altered by the label. For example, Rh-CT2No was as toxic as CT2No. It is extremely unlikely that labelling could change the localization of the CT and yet still provide the same level of cytotoxicity. With regard to Rh-CT2No, the label was located (Lys-58) far from the sites known to be essential for interaction of the CT with membranes [36] and for cytotoxicity [37]. Moroever, conjugation with Rh did not change the conformation of the toxins. These properties of Rh-CT2No are a good basis to assume that labelling changed neither the interaction of CT2No with cells nor its ability to be internalized. Structurally similar (but functionally different) NTII labelled with Rh was not able to accumulate in lysosomes; therefore the presence of Rh label is not sufficient in itself to cause the internalization of toxins.
The origin of the intracellular signal was verified by spectral analysis, and the contribution of the signal from deteriorated (free) Rh was subtracted. It is worth mentioning that the staining pattern of free Rh corresponded to the mitochondria (results not shown), and not to lysosomes as in the case of Rh-CTs. It is important that the CSI measurements were performed on living cells, thus eliminating artifacts of fixation, and incubation of cells with Rh-CTs was carried out under biologically relevant conditions, namely in cell culture medium supplemented with fetal calf serum at 37 °C.
In fact, our data do not contradict experimental results obtained previously. Earlier radiolabelling experiments showed that both the binding equilibrium and the development of cytotoxic effects were saturated after a 1 h incubation of fetal lung cells (cultured human amnion cells) with CT3Na [22]. This time course of CT binding and cytotoxicity development correlates well with the kinetics of CT accumulation in lysosomes, which approaches saturation during a 1 h incubation (Figure 6), as revealed with Rh-CTs by quantitative CSI analysis. Our cytotoxicity measurements confirmed that the number of dead cells increased between 0.5 and 2 h of incubation (results not shown), and thus a 3 h incubation of cells with CTs was selected in order to provide analysis when the cytotoxic effect had reached a plateau (Table 1, Figure 1).
Using radiolabelled CTs, it was concluded that CTs were able to dissociate from cell membrane [1]. Keeping in mind that the exact localization of cell-associated CTs could not be identified using radiolabelled CTs, a decrease in the amount of cell-associated CTs can be explained by efflux, which, according to our data (Figure 6), releases as much as 50% of intracellular (lysosomal) CT over 1 h.
Internalization of CTs may explain the higher sensitivity of tumour cells to CTs as compared with normal cells [2,4,21]. It is well known that uptake of exogenous ligands by tumour cells is very high, and this fact is widely employed in chemotherapy to provide selectivity for tumour treatment. Enhanced internalization of CTs by tumour cells as an explanation for the increased toxic effect seems to resolve a dilemma of supposed CT-specific receptors or CT-specific binding sites [21,22], which had been neither identified nor observed, as targets of the toxic action at the membrane of tumour cells.
Pronounced concentration- and time-dependent accumulation of CTs in lysosomes without detectable binding at the plasma membrane (except for CT3Nk binding on HL60 cells) indicates that lysosomes are probable targets of CT action. On varying the incubation time and the extracellular concentration of Rh-CTs, we observed a 20–100-fold increase in the lysosomal concentration of Rh-CTs, but we were not able to recognize increased plasma membrane binding in different optical sections of cells (Z-stacks) even under conditions when cytotoxic effects were already evident. No staining of the plasma membrane was detected irrespective of whether the Rh-CTs were present in the external medium during observation or were washed out of the cells.
Evidently, internalization of CTs and their consequent transport to lysosomes assume an interaction of the CTs with the plasma membrane, but according to our data this interaction is transient and not accompanied by retention of CTs at the plasmalemma. It is unlikely that very low (undetectable) CT binding is able to injure the plasma membrane.
In theory, even a low amount of CT bound to some ionic channels could inhibit/activate them and ultimately induce cell death. Given this assumption, we have to also suppose that (i) such channels are present in morphologically different cells, (ii) they are vitally important, and (iii) the affinity of CTs for these channels is rather low (micromolar to millimolar concentrations of CTs are required to disturb these channels and kill cells). Currently, this model of CT action is rather speculative for cancer cells and requires experimental evidence.
At the same time, binding to zwitterionic and negatively charged phospholipids is a general property of CTs [7,11,22,25–27,36], and a very high concentration of CTs in lysosomes is a prerequisite for the interaction of CTs with the lysosomal membrane and its lysis. When released into the cytosol, both CTs and lysosomal factors such as proteases can initiate necrosis, which will result in plasma membrane permeabilization. Figures 2(e)–2(g) show the proposed chain of events as traced by the Rh-CT3Nk fluorescence distribution. According to data on molecular interactions [20], CTs can bind ATP in the cytosol and hence disturb ATP-dependent cellular processes. It is clearly seen (Figure 2f) that released CT penetrates into the nucleus, and binds with chromatin and extensively with relaxed DNA in nucleoli. This nuclear binding could also contribute to the toxic effect.
The cytotoxicity measurements revealed a co-operative mode of interaction of the CTs with cells (1.5<h<3.2; Table 1). Previously, positive co-operativity in the binding of CT3Na to heart cells and human leukaemic T-cells (CEM cells), as well as of CT1No, CT2No, CT3Nk, CT1Nh and CT2Nh to murine WEHI-3 cells, was reported [1,33]. Analysing the data presented in these papers, one can recognize positive co-operativity in the interactions of CTs 1, 3 and 4 from Naja atra with K562 and HL60 cells [17], as well as in that of CT2 from Naja naja with Yoshida sarcoma cells [4]. One cannot exclude the possibility that this co-operative mode of action (at least in our case) is related to the formation of pore-inducing dimers or higher aggregates of CTs on the lysosomal membrane. It has been shown previously that the interaction of CTs with negatively charged lipids can cause CT dimerization, an important intermediate for eventual oligomerization during the formation of a pore in membrane bilayers [27]. High concentrations of CTs in lysosomes promote both interaction of toxins with lipids and self-association. It should be stressed that, according to our lower estimate, the lysosomal concentrations of CTs may be 20–100 times higher than their concentration in the extracellular medium.
The phospholipid binding activity of CTs containing a His-4 residue was reported to be pH-dependent and to decrease at low pH, with an apparent pKa of approx. 6.0 [38]. pH-dependent binding was attributed to protonation of His-4 at the membrane–water interface and to protonation-related local conformational changes in CT structure. Among five CTs studied by us, CT1Nh and CT2Nh contain His-4 and demonstrate low cytotoxicity. Since the lysosomal accumulation of Rh-CT1Nh (low-toxicity CT) and Rh-CT1No (intermediate-toxicity CT) occurred equally efficiently, it is reasonable to suppose that low pH in lysosomes is a factor that may reduce the binding of CT1Nh and CT2Nh to the lysosomal membrane, in this way diminishing the cytotoxic effect.
Considering the lipid mode of CT action, one cannot exclude the possibility that some functionally important interactions of CTs with proteins occur in lysosomes that contribute to the overall cytotoxic effect, but these remain to be identified.
In spite of the fact that the immobilized binding of CT3Nk at the plasma membrane of HL60 cells (Figures 3–5) accords well with the generally accepted model of CT action, this pattern was cell specific: HL60 cells, but not A549 cells, were membrane-stained. Since three other Rh-CTs studied by us demonstrated only very weak binding to HL60 cell membranes, this binding mode seems to be defined by the individual structural features of CT3Nk. The molecular and structural basis of plasma membrane binding requires further extensive investigation. Membrane binding seems to contribute to the overall toxicity of CT3Nk: CT3Nk is more toxic for HL60 cells than is CT2No, whereas for the A549 cells, where no membrane binding was detected, the relative cytotoxicities of CT3Nk and CT2No were reversed (Table 1). It is possible that the membrane staining is related to the binding of CT3Nk to some membrane proteins that are highly expressed in some cells, as was revealed for CT3Na in phytohaemagglutinin-activated lymphocytes [16]. Indirect support for this hypothesis comes from the non-uniform distribution of membrane-bound CT3Nk observed in the HL60 cells (Figures 3 and 6), which is qualitatively consistent with the capping of CT3Na on the activated lymphocytes [16].
In conclusion, the internalization and lysosome-targeted action of CTs seem to be an important component of CT-mediated cytotoxicity. Bearing in mind that about 90 CT analogues have been identified so far, extended experiments are required to verify whether all groups of known CTs possess this property. The immobilization and activity of CTs at the plasma membrane is an additional cytotoxic factor. Its contribution to cytotoxicity is proposed to depend on both cell type and CT structure. The molecular basis of the interaction of the CT with the membrane, which results in both membrane immobilization and intracellular transport, requires detailed investigation.
Acknowledgments
This work was supported by grants from the Russian Federation Federal Agency for Science and Innovations (01.106.11.0006), Program RAS MCB and the Russian Foundation for Basic Research (03-04-48496).
References
- 1.Stevens-Truss R., Messer W. S., Hinman C. L. Heart and T-lymphocyte cell surfaces both exhibit positive cooperativity in binding a membrane-lytic toxin. J. Membr. Biol. 1996;150:113–122. doi: 10.1007/s002329900035. [DOI] [PubMed] [Google Scholar]
- 2.Chaim-Matyas A., Borkow G., Ovadia M. Isolation and characterization of a cytotoxin P4 from the venom of Naja nigricollis nigricollis preferentially active on tumor cells. Biochem. Int. 1991;24:415–421. [PubMed] [Google Scholar]
- 3.Harvey A. L., Marshall R. J., Karlsson E. Effects of purified cardiotoxins from the Thailand cobra (Naja naja siamensis) on isolated skeletal and cardiac muscle preparations. Toxicon. 1982;20:379–396. doi: 10.1016/0041-0101(82)90001-0. [DOI] [PubMed] [Google Scholar]
- 4.Iwaguchi T., Takechi M., Hayashi K. Cytolytic activity of cytotoxin isolated from Indian cobra venom against experimental tumor cells. Biochem. Int. 1985;10:343–349. [PubMed] [Google Scholar]
- 5.Chen Y. H., Hu C. T., Yang J. T. Membrane disintegration and hemolysis of human erythrocytes by snake venom cardiotoxin (a membrane-disruptive polypeptide) Biochem. Int. 1984;8:329–338. [PubMed] [Google Scholar]
- 6.Tzeng W. F., Chen Y. H. Suppression of snake-venom cardiotoxin-induced cardiomyocyte degeneration by blockage of Ca2+ influx or inhibition of non-lysosomal proteinases. Biochem. J. 1988;256:89–95. doi: 10.1042/bj2560089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dufton M. J., Hider R. C. Structure and pharmacology of elapid cytotoxins. Pharmacol. Ther. 1988;36:1–40. doi: 10.1016/0163-7258(88)90111-8. [DOI] [PubMed] [Google Scholar]
- 8.Harvey A. L. Cardiotoxins from cobra venom: possible mechanisms of action. J. Toxicol. Toxin Rev. 1985;4:41–69. [Google Scholar]
- 9.Hodges S. J., Agbaji A. S., Harvey A. L., Hider R. C. Cobra cardiotoxins. Purification, effects on skeletal muscle and structure/activity relationships. Eur. J. Biochem. 1987;165:373–383. doi: 10.1111/j.1432-1033.1987.tb11450.x. [DOI] [PubMed] [Google Scholar]
- 10.Rees B., Bilwes A. Three-dimensional structures of neurotoxins and cardiotoxins. Chem. Res. Toxicol. 1993;6:385–406. doi: 10.1021/tx00034a001. [DOI] [PubMed] [Google Scholar]
- 11.Chien K. Y., Chiang C. M., Hseu Y. C., Vyas A. A., Rule G. S., Wu W. G. Two distinct types of cardiotoxin as revealed by the structure and activity relationship of their interaction with zwitterionic phospholipid dispersions. J. Biol. Chem. 1994;269:14473–14483. [PubMed] [Google Scholar]
- 12.Bilwes A., Rees B., Moras D., Menez R., Menez A. X-ray structure at 1.55 A of toxin gamma, a cardiotoxin from Naja nigricollis venom. Crystal packing reveals a model for insertion into membranes. J. Mol. Biol. 1994;239:122–136. doi: 10.1006/jmbi.1994.1357. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher J. E., Jiang M. S. Possible mechanisms of action of cobra snake venom cardiotoxins and bee venom melittin. Toxicon. 1993;31:669–695. doi: 10.1016/0041-0101(93)90375-s. [DOI] [PubMed] [Google Scholar]
- 14.Dufton M.-J., Hider R. C. The structure and pharmacology of elapid cytotoxins. In: Harvey A. L., editor. Snake Toxins. New York: Pergamon Press; 1991. pp. 259–302. [Google Scholar]
- 15.Harvey A. L. Cardiotoxins from snake venoms. In: Tu A. T., editor. Handbook of Natural Toxins. New York: Marcel Dekker Inc.; 1991. pp. 85–106. [Google Scholar]
- 16.Su S. H., Su S. J., Lin S. R., Chang K. L. Cardiotoxin-III selectively enhances activation-induced apoptosis of human CD8+ T lymphocytes. Toxicol. Appl. Pharmacol. 2003;193:97–105. doi: 10.1016/s0041-008x(03)00327-2. [DOI] [PubMed] [Google Scholar]
- 17.Chiou S. H., Raynor R. L., Zheng B., Chambers T. C., Kuo J. F. Cobra venom cardiotoxin (cytotoxin) isoforms and neurotoxin: comparative potency of protein kinase C inhibition and cancer cell cytotoxicity and modes of enzyme inhibition. Biochemistry. 1993;32:2062–2067. doi: 10.1021/bi00059a025. [DOI] [PubMed] [Google Scholar]
- 18.Kuo J. F., Raynor R. L., Mazzei G. J., Schatzman R. C., Turner R. S., Kem W. R. Cobra polypeptide cytotoxin I and marine worm polypeptide cytotoxin A-IV are potent and selective inhibitors of phospholipid-sensitive Ca2+-dependent protein kinase. FEBS Lett. 1983;153:183–186. doi: 10.1016/0014-5793(83)80144-6. [DOI] [PubMed] [Google Scholar]
- 19.Chiou S. H., Chuang M. H., Hung C. C., Huang H. C., Chen S. T., Wang K. T., Ho C. L. Inhibition of protein kinase C by snake venom toxins: comparison of enzyme inhibition, lethality and hemolysis among different cardiotoxin isoforms. Biochem. Mol. Biol. Int. 1995;35:1103–1112. [PubMed] [Google Scholar]
- 20.Jayaraman G., Krishnaswamy T., Kumar S., Yu C. Binding of nucleotide triphosphates to cardiotoxin analogue II from the Taiwan cobra venom (Naja naja atra). Elucidation of the structural interactions in the dATP-cardiotoxin analogue ii complex. J. Biol. Chem. 1999;274:17869–17875. doi: 10.1074/jbc.274.25.17869. [DOI] [PubMed] [Google Scholar]
- 21.Borkow G., Chaim-Matyas A., Ovadia M. Binding of cytotoxin P4 from Naja nigricollis nigricollis to B16F10 melanoma and WEHI-3B leukemia cells. FEMS Microbiol. Immunol. 1992;5:139–145. doi: 10.1111/j.1574-6968.1992.tb05896.x. [DOI] [PubMed] [Google Scholar]
- 22.Takechi M., Tanaka Y., Hayashi K. Binding of cardiotoxin analogue III from Formosan cobra venom to FL cells. FEBS Lett. 1986;205:143–146. doi: 10.1016/0014-5793(86)80882-1. [DOI] [PubMed] [Google Scholar]
- 23.Hinman C. L., Jiang X. L., Tang H. P. Selective cytolysis by a protein toxin as a consequence of direct interaction with the lymphocyte plasma membrane. Toxicol. Appl. Pharmacol. 1990;104:290–300. doi: 10.1016/0041-008x(90)90303-c. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X. L., Hinman C. L. Ablation of natural killer cell function by soluble cardiotoxin. Int. J. Immunopharmacol. 1990;12:247–254. doi: 10.1016/0192-0561(90)90079-3. [DOI] [PubMed] [Google Scholar]
- 25.Vincent J. P., Balerna M., Lazdunski M. Properties of association of cardiotoxin with lipid vesicles and natural membranes. A fluorescence study. FEBS Lett. 1978;85:103–107. doi: 10.1016/0014-5793(78)81258-7. [DOI] [PubMed] [Google Scholar]
- 26.Dufourcq J., Faucon J. F. Specific binding of a cardiotoxin from Naja mossambica mossambica to charged phospholipids detected by intrinsic fluorescence. Biochemistry. 1978;17:1170–1176. doi: 10.1021/bi00600a006. [DOI] [PubMed] [Google Scholar]
- 27.Forouhar F., Huang W. N., Liu J. H., Chien K. Y., Wu W. G., Hsiao C. D. Structural basis of membrane-induced cardiotoxin A3 oligomerization. J. Biol. Chem. 2003;278:21980–21988. doi: 10.1074/jbc.M208650200. [DOI] [PubMed] [Google Scholar]
- 28.Sue S. C., Rajan P. K., Chen T. S., Hsieh C. H., Wu W. Action of Taiwan cobra cardiotoxin on membranes: binding modes of a beta-sheet polypeptide with phosphatidylcholine bilayers. Biochemistry. 1997;36:9826–9836. doi: 10.1021/bi970413l. [DOI] [PubMed] [Google Scholar]
- 29.Feofanov A. V., Nazarova A. I., Karmakova T. A., Pljutinskaya A. D., Grichine A. I., Yakubovskaya R. I., Lebedeva V. S., Ruziyev R. D., Mironov A. F., Maurizot J.-C., Vigny P. Photobiological properties of 13,15-N-(carboxymethyl)- and 13,15-N-(2-carboxyethyl)-cycloimide derivatives of chlorin p6. Russ. J. Bioorg. Chem. 2004;30:374–384. [PubMed] [Google Scholar]
- 30.Feofanov A., Sharonov G., Grichine A., Karmakova T., Pljutinskaya A., Lebedeva V., Ruziyev R., Yakubovskaya R., Mironov A., Refregier M., et al. Comparative study of photodynamic properties of 13,15-N-cycloimide derivatives of chlorin p6. Photochem. Photobiol. 2004;79:172–188. doi: 10.1562/0031-8655(2004)079<0172:csoppo>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 31.Feofanov A., Charonov S., Kudelina I., Fleury F., Nabiev I. Localization and molecular interactions of mitoxantrone within living K562 cells as probed by confocal spectral imaging analysis. Biophys. J. 1997;73:3317–3327. doi: 10.1016/S0006-3495(97)78356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feofanov A., Charonov S., Fleury F., Kudelina I., Nabiev I. Quantitative confocal spectral imaging analysis of mitoxantrone within living K562 cells: intracellular accumulation and distribution of monomers, aggregates, naphtoquinoxaline metabolite and drug-target complexes. Biophys. J. 1997;73:3328–3336. doi: 10.1016/S0006-3495(97)78357-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feofanov A. V., Sharonov G. V., Dubinnyi M. A., Astapova M. V., Kudelina I. A., Dubovskii P. V., Rodionov I. D., Utkin Y. N., Arseniev A. S. Comparative study of structure and activity of cytotoxins from Naja oxiana, Naja kaouthia and Naja haje cobra venom. Biokhimya (Moscow) 2004;69:1410–1421. doi: 10.1023/b:biry.0000046890.46901.7e. [DOI] [PubMed] [Google Scholar]
- 34.Utkin Y. N., Kukhtina V. V., Kryukova E. V., Chiodini F., Bertrand D., Methfessel C., Tsetlin V. V. “Weak toxin” from Naja kaouthia is a nontoxic antagonist of alpha 7 and muscle-type nicotinic acetylcholine receptors. J. Biol. Chem. 2001;276:15810–15815. doi: 10.1074/jbc.M100788200. [DOI] [PubMed] [Google Scholar]
- 35.Grichine A., Feofanov A., Karmakova T., Kazachkina N., Pecherskih E., Yakubovskaya R., Mironov A., Egret-Charlier M., Vigny P. Influence of the substitution of 3-vinyl by 3-formyl group on the photodynamic properties of chlorin p6: molecular, cellular and in vivo studies. Photochem. Photobiol. 2001;73:267–277. doi: 10.1562/0031-8655(2001)073<0267:iotsov>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 36.Dubovskii P. V., Lesovoy D. M., Dubinnyi M. A., Utkin Y. N., Arseniev A. S. Interaction of the P-type cardiotoxin with phospholipid membranes. Eur. J. Biochem. 2003;270:2038–2046. doi: 10.1046/j.1432-1033.2003.03580.x. [DOI] [PubMed] [Google Scholar]
- 37.Kini R. M. Molecular moulds with multiple missions: functional sites in three-finger toxins. Clin. Exp. Pharmacol. Physiol. 2002;29:815–822. doi: 10.1046/j.1440-1681.2002.03725.x. [DOI] [PubMed] [Google Scholar]
- 38.Chiang C. M., Chien K. Y., Lin H. J., Lin J. F., Yeh H. C., Ho P. L., Wu W. G. Conformational change and inactivation of membrane phospholipid-related activity of cardiotoxin V from Taiwan cobra venom at acidic pH. Biochemistry. 1996;35:9167–9176. doi: 10.1021/bi952823k. [DOI] [PubMed] [Google Scholar]