

Abstract
Two mutations of IMPDH1 (inosine 5′-monophosphate dehydrogenase type I), R224P and D226N, have recently been found to cause adRP (autosomal dominant retinitis pigmentosa). IMPDH1 catalyses the rate-limiting step in guanine nucleotide biosynthesis and also binds single-stranded nucleic acids. In the present paper, we report the biochemical characterization of the adRP-linked mutations, R224P and D226N, and a potentially pathogenic mutation, V268I. The adRP-linked mutations have no effect on enzyme activity, protein stability or protein aggregation. These results suggest strongly that the mutations do not affect enzyme activity in vivo and thus do not perturb the guanine nucleotide pool. The R224P mutation changes the distribution of enzyme between the nucleus and cytoplasm. This effect was not observed with the D226N mutation, so the relevance of this observation to disease is unclear. In contrast, both mutations decrease the affinity of nucleic acid binding and both fail to co-immunoprecipitate RNA. These observations suggest that nucleic acid binding provides a functional assay for adRP pathogenicity. The putative adRP-linked mutation V268I also disrupts nucleic acid binding, which suggests that this mutation is indeed pathogenic.
Keywords: cystathionine β-synthase domain (CBS domain), inosine 5′-monophosphate dehydrogenase (IMPDH), nucleic acid, retinitis pigmentosa, RNA-binding protein
Abbreviations: adRP, autosomal dominant retinitis pigmentosa; CBS, cystathionine β-synthase; DTT, dithiothreitol; GFP, green fluorescent protein; HEK, human embryonic kidney; IMP, inosine 5′-monophosphate; IMPDH, IMP dehydrogenase; IMPDH1, human IMPDH type I; H1GFP, GFP-tagged human type 1 IMPDH; RP, retinitis pigmentosa; ssDNA, single-stranded DNA
INTRODUCTION
RP (retinitis pigmentosa) is an inherited retinal degeneration that affects more than 1.5 million people worldwide [1]. The disease is characterized by an early onset of night blindness, followed by a progressive loss of the visual field which can lead to blindness [2]. RP is genetically heterogeneous and can be inherited as autosomal dominant (adRP), autosomal recessive and X-linked forms [3]. Fourteen adRP genes have been identified so far, including the gene encoding the enzyme IMPDH1 (inosine 5′-monophosphate dehydrogenase type I) (RetNet, http://www.sph.uth.tmc.edu/RetNet). Two adRP-linked mutations, R224P and D226N, have been identified in IMPDH1 [4,5]. A third mutation, V268I, is present in a member of an adRP family, but insufficient genetic samples are available to demonstrate a correlation between the mutation and disease [5]. At present, mutations have been identified in only ∼50% of adRP cases [6], and it is likely that other alleles of IMPDH1 will be discovered as the search for adRP genes continues. For example, a non-pathogenic allele, A285T, was recently found in the DNA of an unaffected family member (S. J. Bowne, L. S. Sullivan and S. P. Daiger, personal communication). The challenge will be to distinguish pathogenic from non-pathogenic alleles. Since the effects of the adRP-causing mutations on IMPDH function have not been established, the only way to determine the pathogenicity of IMPDH1 mutations is by demonstrating the co-inheritance of a mutation and disease. This demonstration is often not feasible, either due to small family size, lack of records or loss of patient contact. Clearly, there is an urgent need for a functional assay to identify pathogenic IMPDH1 mutations.
AdRP results from the apoptotic loss of photoreceptors, although how mutations of IMPDH1 induce apoptosis is not understood. IMPDH catalyses the rate-limiting step in the de novo pathway of guanine nucleotide biosynthesis and thus controls the size of the guanine nucleotide pool with important consequences for proliferation. As IMPDH inhibitors can induce differentiation and apoptosis, they are used as antitumour, immunosuppressive and antiviral therapies [7–9]. IMPDH also binds single-stranded nucleic acids, although the physiological role of this activity has not been identified [10,11].
In humans, distinct genes encode the IMPDH1 and IMPDH2 isoforms, with expression regulated according to cell type [12,13]. Although IMPDH1 is the predominant isoform in the murine retina, homozygous deletion of IMPDH1 in mice causes only a mild retinopathy [14]. This observation suggests that adRP does not result from the loss of IMPDH1 protein. In addition, the adRP-linked mutations are not in proximity to intron–exon junctions of precursor mRNA and therefore would not be predicted to result in alternative splicing. Instead, the adRP-linked mutations must alter some function of IMPDH1.
The sequence conservation at sites of the adRP-linked mutations supports this hypothesis. Asp226 is completely conserved among IMPDHs and Arg224 is conserved among eukaryotic enzymes, indicating an important role for these residues in protein function. In contrast, Ala285 is variable, which suggests that there are few functional constraints on this residue and this is consistent with the non-pathogenic consequences of mutation at this position. Val268 is less conserved than the adRP-linked positions, but is only replaced with alanine or leucine, which implies that there may also be a functional constraint on the replacement of this residue. Therefore the conservation of Val268 does little to resolve the question of whether the V268I mutation is truly pathogenic. This problem can be expected to extend to additional mutations of IMPDH1.
IMPDH is a homotetramer; each monomer consists of a catalytic domain containing the active site and a subdomain of undefined function (Figure 1). The interface between the two domains is flexible, and different domain orientations are observed in various crystal structures [15]. The non-pathogenic mutation A285T is on the catalytic domain far from both the active site and subdomain (Figure 1). The adRP-linked mutations, R224P and D226N, are located within the subdomain of IMPDH1, whereas V268I is close to the catalytic/subdomain interface. Since deletion of the subdomain does not change enzyme activity, it would be surprising if any of these mutations altered the catalytic properties of IMPDH1 [16,17]. However, both adRP-linked mutations and V268I are well positioned to alter the structure and function of the subdomain.
Figure 1. Structure of IMPDH1.

(A) Location of adRP-linked mutations on IMPDH1. A monomer of the human IMPDH1 crystal structure (Protein Data Bank code 1JCN) is shown with the catalytic domain in grey and the subdomain in blue. The active site is modified by the inhibitor 6-Cl-IMP, shown in purple. Arg224 and Asp226, the residues where mutations cause adRP, are shown in red. Val268, the residue linked to a potentially disease causing mutation, is shown in green. Ala285, the residue linked to a non-pathogenic mutation, is shown in orange. Note that some portions of the subdomain are disordered in the crystal structure. (B) Oligomerization of wild-type and mutant IMPDH1 analysed by gel filtration. Recombinant IMPDH1 was applied to a Superose 6HR column, eluted with 10 mM Tris/HCl, pH 8, 100 mM KCl and 0.5 mM DTT at a flow rate of 0.4 ml/min at 4 °C. The chromatograms show the elution of wild-type (wt) IMPDH1 (blue), A285T (cyan), R224P (green), D226N (red) and V268I (purple), and molecular-mass standards of 670, 450, 240 and 45 kDa (black). A tetramer of IMPDH1 has a molecular mass of 220 kDa. mAU, milli-absorbance units
The subdomain contains two tandem CBS (cystathionine β-synthase) domains. CBS domains are found in a diverse set of proteins, including ClC-chloride channels, CBS and the γ2 subunit of AMP-activated protein kinase. Interestingly, mutations within the CBS domains of these proteins lead to a variety of other hereditary diseases [18]. Although the functions of CBS domains have not been elucidated, they are generally believed to be involved in autoregulation of enzymatic activity, channel gating and/or protein localization [19–21]. The role of the CBS domains in IMPDH is currently under active investigation. Studies in our laboratory have shown that the subdomain binds single-stranded nucleic acids [11]. Others suggest that this region contains an ATP-binding site that allosterically regulates the catalytic domain [22]. A phosphorylation site has also been identified in the subdomain of IMDPH1, although the functional consequences of this modification have not been established [23].
We have characterized the adRP-linked mutants of IMPDH1 in vitro and in cell culture, with the aim of identifying the functional property that correlates with disease. As expected, these mutations do not affect catalytic activity, nor do they affect protein stability or expression in mammalian cells. Contrary to another report [14], we find no evidence that these mutations induce protein aggregation. While some mutations have small effects on the cellular localization of IMPDH1, this property does not appear to correlate with disease. In contrast, the adRP-linked mutations disrupt nucleic acid binding in vitro and in vivo. Nucleic acid binding is also decreased with the potentially disease-causing mutation, V268I, suggesting that this mutation is indeed pathogenic. We propose that nucleic acid binding presents a functional assay for the adRP pathogenicity of IMPDH1 mutations, as well as the first clue to the mechanism of disease progression.
EXPERIMENTAL
Plasmids and mutant construction
Human IMPDH1 was expressed in Escherichia coli using the pH1 vector (derived from pKK223-3) and site-directed mutagenesis performed with the QuikChange kit (Stratagene). The following primers and their complements were used to create the mutations R224P, D226N, V268I and A285T respectively: 5′-GCCATCATCGCCCCGACCGACCTGAAG-3′, 5′-GCCATCATCGCCCGCACCAACCTGAAGAAGAATCG-3′, 5′-GCTCACCCAGGCGGGCATTGATGTCATAGTCTTGG-3′ and 5′-GTGTATCAGATCACCATGGTGCATTAC-3′.
To mutate the stop codon and introduce a restriction enzyme site, pH1 was PCR-amplified with the following primers: 5′-CAGAATTCCATATGGCGGACTACC-3′ and 5′-CCAAAACAGCCAAGCTTAGGTACCCAGTACAGCCG-3′. The product was ligated into pCR4 with Zero Blunt TOPO Kit (Invitrogen) and digested with EcoRI/KpnI. Ligation into pEGFP-N1 (Clontech) resulted in pH1GFP, a C-terminal GFP (green fluorescent protein)-tagged IMPDH1 under control of the mammalian CMV (cytomegalovirus) promoter. To prevent proteolysis, two arginine residues in the IMPDH1–GFP linker region were mutated to glycine using the following primer and its complement: 5′-GTACTGGGTACCGGGGGCCGGGGATCCACCGGTC-3′. For bacterial expression of H1GFP, the coding sequence was cut from pH1GFP with PstI/NotI and ligated into a modified pH1 vector containing an NotI site. The subdomain was deleted from pH1GFP with 5′-GTGCGGAAGGTCAAGAAGTTTGAACAGCGTACTGGTCAGCTGCTCTGTGGGGC-3′ and its complement, resulting in a construct containing residues 1–112 and 242–514, separated by an RTG (Arg-Thr-Gly) linker. The resulting IMPDH cDNAs were sequenced at the Brandeis Sequencing Facility to confirm correct coding sequences.
Protein purification
The protein was purified using Cibacron Blue–Sepharose and IMP (inosine 5′-monophosphate)–Sepharose as described previously [24]. Gel filtration was performed with ∼100 μg of IMPDH1 in 50 mM Tris/HCl, pH 8.0, 0.5 mM DTT (dithiothreitol), 100 mM KCl and the appropriate protein standards on a Superose 6 HR column (Amersham Biosciences).
Enzyme activity
IMPDH activity was assayed as described previously [25]. The assay solution contained 50 mM Tris/HCl, pH 8.0, 1 mM DTT, 100 mM KCl, and various amounts of IMP and NAD+. NADH fluorescence was monitored (λexcitation=340 nm, λemission=460 nm) on a PerSeptive Biosystems Cytofluor II multiwell plate reader at 37 °C. Rates of NADH production were determined by calibration of the instrument with a standard curve of an NADH solution in assay buffer. Initial velocity data were fitted by the Michaelis–Menten equation (eqn 1) or an uncompetitive substrate inhibition equation (eqn 2) using SigmaPlot (SPSS):
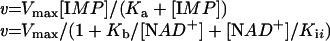 |
1 |
where v is the initial velocity, Vmax is the maximal velocity, Ka and Kb are the Michaelis constants for IMP and NAD+ respectively, and Kii is the substrate inhibition constant for NAD+.
The effect of ATP on activity was assayed under conditions described previously [22]. Activity was monitored in the above assay buffer containing ATP (0.1–3.5 mM), a 5 mM excess of MgCl2 over ATP and the reaction was initiated upon addition of NAD+. The integrity of the ATP preparation was confirmed by monitoring NADH consumption in a coupled ATPase assay using kinesin, pyruvate kinase and lactate dehydrogenase [26].
Cell culture and transfections
HeLa or HEK-293 (human embryonic kidney) cells were cultured in Dulbecco's minimal essential medium supplemented with 10% foetal calf serum, penicillin and streptomycin. Cells were transfected at approx. 70% confluence using Superfect (Qiagen) according to manufacturer's instructions. For live fluorescent microscopy, cells were grown and transfected on Lab-Tek II multi-chamber cover glasses (Nunc).
Cellular localization
Fluorescence signal was viewed 6–30 h post-transfection using an Olympus IX70 inverted fluorescence microscope. DNA was stained with Hoechst dye (Sigma). Images were acquired in multiple focal planes (Δz=0.2–0.3 μm, 20–63 planes) with constant exposure time, deconvolved and subsequently projected in two or three dimensions using a DeltaVision microscopy system and accompanying software. The mean values for nuclear, cytoplasmic and background GFP fluorescence were quantified by projection of the pixel intensity of each cell in a histogram using the DeltaVision™ software. The ratio of nuclear to cytoplasmic fluorescence was determined by background correction and division of the mean pixel intensities of the nucleus by that of the cytoplasm. Similar results were obtained in three independent experiments. Statistical analysis was performed using a two-tailed Student's t test, assuming unequal variance, and significance is defined as P<0.05.
For inhibition of nuclear export, HeLa cells were grown on coverslips in medium supplemented with 10 ng/ml leptomycin B (LC Laboratories), starting 8 h post-transfection. After a 2.5 h incubation, the localization of H1GFP was compared with that in untreated cells.
Filter binding
IMPDH-binding experiments with ssDNA (single-stranded DNA) were performed as described in [11]. The sequence pool of ssDNA was 5′-GGGAATGGATCCACATCTACGAATTCN30TTCACTGCAGACTTGACGAAGCTT-3′, where N30 denotes a random sequence 30 bases long. Protein and 5′-32P-labelled ssDNA (2 nM) were mixed in 10 mM Tris/HCl, pH 8, 50 mM KCl and 1 mM DTT for 20 min at room temperature (25 °C). Proteinbound nucleic acid was separated from free by filtration on a vacuum manifold (Schleicher and Schuell) equipped with a nitrocellulose membrane (to bind protein complexes) over a Hybond membrane (Amersham Biosciences) (to bind nucleic acid) and washed with 100 μl of assay buffer. The radioactivity bound to each filter was quantified by PhosphorImager (Molecular Dynamics). The fraction of nucleic acid bound to the nitrocellulose was fitted to the following equation using Sigmaplot:
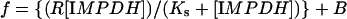 |
3 |
where f is the fraction of nucleic acid bound, R is the maximum specific bound fraction, Ks is the binding constant, and B is the fraction bound to the membrane in the absence of protein. The effect of ATP (0.77 mM) on nucleic acid binding was examined in binding buffer containing 5.7 mM MgCl2.
Immunoprecipitation and digestion of nucleic acid
Nucleic acid bound to H1GFP was immunoprecipitated from HeLa cells with affinity-purified anti-GFP antibodies (Molecular Probes) as described previously [11]. Immunoprecipitates were divided into two for quantification of H1GFP by SDS/PAGE (10% gels) and Coomassie Blue staining or quantification of nucleic acid by polynucleotide kinase labelling with [γ-32P]ATP. Nucleic acid was isolated by filter binding or treated with proteinase K and separated on a 6 M urea/6% polyacrylamide gel. The incorporated radioactivity was quantified by PhosphorImager analysis. Nucleic acid radiolabel intensity was adjusted for H1GFP concentration.
RESULTS AND DISCUSSION
Catalytic activity is not changed by adRP-linked mutations
Wild-type IMPDH1, and R224P, D226N, V268I and A285T mutants were expressed in E. coli strain H712, which lacks endogenous IMPDH, and were purified to >95% homogeneity. The specific activities of the wild-type and mutant enzymes are similar, as observed by others [14]. Contrary to this report [14], no difference in the solubility of mutant and wild-type proteins was observed in our experiments. Gel-filtration analysis demonstrated that the enzymes are tetrameric, indicating that the mutations do not affect oligomerization (Figure 1B). The steady-state kinetic properties of the wild-type enzyme are similar to those previously reported (Table 1) [27]. The kinetic properties of the adRP-linked mutant enzymes are similar to those of the wild-type. Taken together, these results indicate that adRP-linked mutations do not alter enzyme activity.
Table 1. IMPDH1 steady-state kinetic parameters.
Initial velocities were measured in 50 mM Tris/HCl, pH 8, 100 mM KCl and 1 mM DTT at 37 °C at various concentrations of IMP and NAD+. The kinetic parameters are derived by fitting the initial velocities to eqns 1 and 2, and are given as means±S.E.M. n.d., not determined.
| IMPDH1 | kcat (s−1) | Km (IMP) (μM) | Km (NAD) (μM) | Kii (NAD) (mM) |
|---|---|---|---|---|
| Wild-type | 1.2±0.4 | 17±2 | 70±10 | 2.0±0.6 |
| R224P | 1.3±0.3 | 7±2 | 56±15 | 1.0±0.4 |
| D226N | 0.7±0.2 | 12±2 | 50±10 | 4±3 |
| V268I | 0.8±0.3 | 11±2 | 46±9 | 4±2 |
| A285T | 0.9±0.3 | n.d. | n.d. | n.d. |
| H1GFP | 0.9±0.3 | n.d. | n.d. | n.d. |
ATP does not regulate IMPDH1
Another laboratory group has reported that ATP induces a 5-fold increase in activity of the human type II isoform [22]. The R224P mutation abrogated this effect, which suggests that the adRP-linked mutations could perturb the regulation of IMPDH1 by ATP. Therefore we investigated the effect of ATP on IMPDH1 activity. We observed no change in the activity of IMPDH1 or the mutant enzymes in the presence of various concentrations of ATP up to 3.5 mM (4-fold above the reported Ks; results not shown). The ATP concentration was confirmed by a coupled ATPase assay. In addition, ATP has no effect on the ability of IMPDH1 to bind nucleic acid (various concentrations up to 0.77 mM; results not shown), which casts further doubt on the proposal that the subdomain binds ATP. Therefore adRP does not result from a change in regulation of IMPDH1 by ATP. While it is possible that ATP regulation is specific to IMPDH2, we note that researchers in other laboratories have failed to observe this phenomenon [28]. Moreover, an apparent ‘stimulation’ of IMPDH activity by various nucleotides has previously been traced to the presence of contaminating nucleotidases [29]. Therefore ATP regulation seems likely to result from the incomplete purification of the recombinant protein. In any case, ATP regulation does not provide a functional assay for the adRP pathogenicity of IMPDH1 mutations.
Expression of IMPDH1 in mammalian cells
IMPDH1 was C-terminally tagged with GFP. This fusion protein, H1GFP, was expressed in bacteria, purified and shown to have enzymatic activity indistinguishable from that of the wild-type protein (Table 1). HeLa cells were transiently transfected with H1GFP, and conditions were established for expression of the tagged protein at similar levels to endogenous IMPDH (Figure 2A). Both GFP fluorescence and Western analysis demonstrate that similar amounts of wild-type H1GFP and mutant proteins are expressed, which indicates that the mutations do not affect protein stability or turnover. In contrast with another report, no aggregation was observed when R224P and D226N were expressed in HeLa cells [14]. We also did not observe aggregation when these proteins were expressed in HEK-293 cells (results not shown). Therefore protein solubility is unlikely to be the cause of adRP. While it is possible that the GFP tag increases the solubility of the IMPDH1 mutants, we believe that this discrepancy is more readily explained by the overexpression of protein and/or use of a His-tag in the previous work.
Figure 2. Localization of wild-type and mutant H1GFP.

(A) H1GFP (84 kDa) expression following transfection into HeLa cells was compared with endogenous IMPDH (55 kDa) by Western blot with anti-GFP antibodies and anti-IMPDH serum (26 h post-transfection shown). Additional experiments confirm that detection is linear within this range (results not shown). (B) Live fluorescence localization of wild-type (wt), R224P or V268I H1GFP protein in HeLa cells shown as GFP fluorescence. (C) The ratio of nuclear to cytoplasmic H1GFP. As described in the Experimental section, the mean GFP fluorescence intensities for the nucleus and cytoplasm were determined for >20 cells of each type. The results are means±S.D., and those significantly different from wild-type are denoted by an asterisk (*P<1×10−5 and 0.001 for R224P and V268I respectively).
We investigated the localization of wild-type and mutant IMPDH expressed in HeLa cells by monitoring GFP fluorescence and using Hoechst stain to identify the nucleus (Figure 2B). Wild-type H1GFP is found in both the nucleus and cytoplasm. The distribution is similar to that observed with the endogenous enzyme in immunocytology experiments [11]. IMPDH1 does not contain a standard nuclear localization motif, so the signal directing the enzyme to the nucleus is unknown. The nuclear/cytosolic distribution of H1GFP did not change in the presence of the nuclear export inhibitor leptomycin B (results not shown). Deletion of the subdomain also does not affect protein localization, suggesting that this region is not required to direct the protein into the nucleus (results not shown).
The localization of the adRP-linked D226N and non-pathogenic A285T proteins were similar to that of wild-type. In contrast, a larger fraction of the adRP-linked R224P protein was consistently found in the nucleus, while the fraction of nuclear V268I was variable (Figures 2B and 2C). However, since the change in cellular localization was not consistent for both adRP-linked mutants, the relevance of these observations to adRP is unclear. Therefore protein localization does not provide a functional assay for adRP pathogenicity.
AdRP-linked mutations decrease affinity for nucleic acid
We have discovered previously that IMPDH binds single-stranded nucleic acids with nanomolar affinity, although the physiological role of this activity is unknown [11]. The subdomain is part of the nucleic-acid-binding site. Therefore we investigated whether the adRP-linked mutations perturbed nucleic acid binding. As reported previously, IMPDH1 binds a random pool of ssDNA oligonucleotides with Ks=6 nM (Figure 3 and Table 2; IMPDH concentrations refer to tetramers). The GFP tag has no effect on ssDNA binding (Ks=7±2 nM). Also, the non-pathogenic mutant A285T has no effect on the affinity for ssDNA. In sharp contrast, the adRP-linked R224P and D226N mutations display significantly decreased affinities for ssDNA, by factors of 7 and 32 respectively. These observations suggest that nucleic acid binding provides a straightforward functional assay for the adRP pathogenicity of IMPDH1 mutations.
Figure 3. AdRP-linked mutations decrease affinity for a random pool of ssDNA and increase the fraction bound in a filter-binding assay.

The fraction bound was determined by various amounts of IMPDH1 (shown as concentration of tetramer) incubated with a labelled pool of random ssDNA sequences as described in the Experimental section. Results are representative of three experiments, and the best fit of a simple binding model is represented by the lines (eqn 3).
Table 2. Dissociation constants for IMPDH1 tetramers binding to a random pool of ssDNA.
Data from filter binding assays (Figure 3) were fitted with eqn 3 to determine the Ks and maximum percentage bound. Results are means±S.E.M. (n=3)
| IMPDH1 | Ks (nM) | Maximum bound (%) |
|---|---|---|
| Wild-type | 6±2 | 4.0±0.2 |
| R224P | 43±5 | 6.5±0.3 |
| D226N | 190±70 | 70±20 |
| V268I | 300±100 | 50±10 |
| A285T | 3±1 | 4.0±0.3 |
The D226N mutation also appears to decrease the specificity of nucleic acid binding. IMPDH1 binds approx. 4% of the random oligonucleotide pool with high affinity, consistent with our previous observations (Figure 3; note that this represents approx. 1011 sequences) [11]. The non-pathogenic A285T and R224P enzymes also bind approx. 4–7% of the random pool. In contrast, D226N binds 70% of the random pool. This change in the specificity of nucleic acid binding could represent a gain-of-function that is often associated with autosomal dominant mutations. However, since this change in specificity is observed in only one of the adRP-linked mutations, its relevance to disease is unclear.
AdRP-linked mutations decrease RNA association in vivo
We previously demonstrated that RNA co-immunoprecipitates with IMPDH [11]. To determine if the adRP-linked mutations perturbed RNA binding in vivo, we expressed GFP-tagged proteins in HeLa cells. H1GFP was isolated by immunoprecipitation with an anti-GFP antibody, and nucleic acid was identified by labelling with polynucleotide kinase and [γ-32P]ATP (Figures 4A and 4B). Significantly more nucleic acid was observed in immunoprecipitations from cells expressing H1GFP than in the untransfected cells or cells expressing GFP alone. This immunoprecipitated nucleic acid is degraded by RNase, but not by DNase (Figure 4B). Therefore, like endogenous IMPDH, the H1GFP binds RNA in vivo. The bound RNA appears to be heterogeneous and primarily less than 100 nucleotides in length (Figure 4C). The non-pathogenic A285T mutation had no effect on the immunoprecipitation of RNA (Figure 4C). In contrast, much less RNA is present in immunoprecipitations of the adRP-linked R224P and D226N proteins. These results demonstrate that the pathogenic mutations decrease the association of IMPDH1 and RNA in vivo.
Figure 4. AdRP-linked mutations decrease the association of IMPDH with RNA in vivo.

HeLa cells transfected with H1GFP were cross-linked with formaldehyde and immunoprecipitated with anti-GFP antibody. (A) Coomassie-Blue-stained SDS/PAGE (10% gels) of the immunoprecipitate demonstrating that similar amounts of H1GFP are precipitated in each sample. Arrows denote H1GFP (84 kDa), GFP (29 kDa) and antibody (Ab; 50 and 75 kDa). (B) The immunoprecipitates were treated with phosphatase followed by [γ-32P]ATP and polynucleotide kinase to label nucleic acids. The filter-bound radioactivity is shown relative to untransfected cells (control). The 32P-labelled immunoprecipitates were treated with either RNase or DNase. Asterisks mark samples significantly different from wild-type (P<0.02 for both control and RNase). (C) RNA co-immunoprecipitated with GFP, wild-type H1GFP, A285T, R224P, D226N and V268I. Samples were treated as described in (B), with the addition of a proteinase K digestion. 32P-Labelled co-immunoprecipitated nucleic acid was analysed on a 6% denaturing polyacrylamide gel with ssDNA of known molecular mass as general markers. ATP denotes a mock labelling reaction in the absence of immunoprecipitate. wt, wild-type.
V268I is a pathogenic mutation
At present, genetic linkage cannot establish the pathogenicity of the V268I mutation, and this problem is likely to extend to additional mutations of IMPDH1. The above observations indicate that adRP-linked mutations of IMPDH1 cause a defect in nucleic acid binding that can serve as a functional assay for pathogenicity. By this criterion, the V268I mutation is also likely to be pathogenic. An even larger decrease in ssDNA affinity was observed in the V268I protein than either of the other adRP-linked enzymes (50-fold). The V268I mutation increased the maximum fraction bound to ∼50% of the random pool. This demonstrates that the specificity of nucleic acid binding had also changed (Figure 3 and Table 2). Lastly, only background amounts of RNA are present in immunoprecipitations of the V268I protein, confirming that this mutation disrupts RNA binding in vivo (Figure 4C).
Pathological mechanism of IMPDH1-linked adRP
The mechanisms that trigger the apoptotic loss of photoreceptor cells in adRP have not yet been elucidated in detail [30]. Most of the adRP-linked genes encode proteins such as rhodopsin that are expressed exclusively in the retina. These proteins are featured in photoreceptor-specific functions, so it is not surprising that mutations in these genes lead specifically to photoreceptor death. In contrast, IMPDH1 plays an essentially housekeeping role in many tissues, so the photoreceptor-specific effects of these mutations are perplexing. The experiments described above strongly suggest that the adRP-linked mutations of IMPDH1 do not alter protein stability or enzymatic function and thus probably will not perturb the guanine nucleotide pool. Our previous work indicated that IMPDH binds nucleic acids in vitro and in vivo and therefore has an unappreciated role in transcription, post-transcriptional modification, translation, localization and/or some other aspect of RNA metabolism [11]. This unidentified function is mediated by the subdomain [11]. The adRP-linked mutations R224P and D226N lower the affinity of IMPDH1 for single-stranded nucleic acid in vitro and decrease the association with RNA in vivo, suggesting that adRP results from a perturbation in RNA metabolism. While this work was in progress, a third adRP-linked mutation was identified, R231P [31]. This mutation is in close proximity to Arg224 and Asp226, and is thus also likely to perturb nucleic acid binding.
Interestingly, adRP is also linked to four proteins that are involved in pre-mRNA splicing, and other retinopathies are caused by mutations in mitochondrial tRNAs [32–39]. IMPDH associates with proteins that are involved in transcription regulation, splicing and rRNA processing in yeast [40–43]. Perhaps photoreceptor cells are uniquely dependent on some aspect of RNA metabolism that is mediated by both IMPDH1 and these other retinopathy-linked genes.
Summary
The discovery of each new adRP-linked gene rapidly fuels the identification of additional alleles of unknown pathogenicity. In many instances, this issue cannot be resolved because insufficient samples exist to determine the co-segregation of the mutation with disease. In the case of IMPDH1, the inability to predict a functional consequence for the V268I mutation further frustrated the assignment of pathogenicity. Our experiments identify a defect in nucleic acid binding as a common functional phenotype for the adRP-linked mutations of IMPDH1. The V268I mutation also disrupts nucleic acid binding, which suggests strongly that this mutation causes disease. While this work was in progress, five more potentially disease-causing mutations of IMPDH1 were identified: T116M, G324D, H372P, R105W and N198K (S. J. Bowne, L. S. Sullivan and S. P. Daiger, personal communication), some of which are also located within the catalytic domain–subdomain interface. We propose that nucleic acid binding can provide a functional assay to establish the pathogenicity of these new alleles.
Acknowledgments
We thank Dr Melissa J. Moore for helpful discussion and use of equipment. Also, we thank Dr Stephen P. Daiger, Dr Sara J. Bowne and Dr Lori S. Sullivan of University of Texas, Houston, TX, U.S.A., for sharing recent RP mutant data, and Dr Beverly Mitchell of University of North Carolina, Chapel Hill, NC, U.S.A., for IMPDH1 cDNA. This research was supported by grant GM54403 (L.H.), NIH (National Institutes of Health) Macromolecular Structure and Mechanism Training Grant GM07956 (S.E.M.), and a grant from the Markey Charitable Trust to Brandeis University.
References
- 1.Bunker C. H., Berson E. L., Bromley W. C., Hayes R. P., Roderick T. H. Prevalence of retinitis pigmentosa in Maine. Am. J. Ophthalmol. 1984;97:357–365. doi: 10.1016/0002-9394(84)90636-6. [DOI] [PubMed] [Google Scholar]
- 2.Van Soest S., Westerveld A., De Jong P. T., Bleeker-Wagemakers E. M., Bergen A. A. Retinitis pigmentosa: defined from a molecular point of view. Surv. Ophthalmol. 1999;43:321–334. doi: 10.1016/s0039-6257(98)00046-0. [DOI] [PubMed] [Google Scholar]
- 3.Phelan J. K., Bok D. A brief review of retinitis pigmentosa and the identified retinitis pigmentosa genes. Mol. Vision. 2000;6:116–124. [PubMed] [Google Scholar]
- 4.Kennan A., Aherne A., Palfi A., Humphries M., McKee A., Stitt A., Simpson D. A., Demtroder K., Orntoft T., Ayuso C., et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho−/− mice. Hum. Mol. Genet. 2002;11:547–558. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- 5.Bowne S. J., Sullivan L. S., Blanton S. H., Cepko C. L., Blackshaw S., Birch D. G., Hughbanks-Wheaton D., Heckenlively J. R., Daiger S. P. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivolta C., Sharon D., DeAngelis M. M., Dryja T. P. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum. Mol. Genet. 2002;11:1219–1227. doi: 10.1093/hmg/11.10.1219. [DOI] [PubMed] [Google Scholar]
- 7.Franchetti P., Grifantini M. Nucleoside and non-nucleoside IMP dehydrogenase inhibitors as antitumor and antiviral agents. Curr. Med. Chem. 1999;6:599–614. [PubMed] [Google Scholar]
- 8.Allison A. C., Eugui E. M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47:85–118. doi: 10.1016/s0162-3109(00)00188-0. [DOI] [PubMed] [Google Scholar]
- 9.Gu J. J., Gathy K., Santiago L., Chen E., Huang M., Graves L. M., Mitchell B. S. Induction of apoptosis in IL-3-dependent hematopoietic cell lines by guanine nucleotide depletion. Blood. 2003;101:4958–4965. doi: 10.1182/blood-2002-08-2547. [DOI] [PubMed] [Google Scholar]
- 10.Cornuel J., Moraillon A., Gueron M. Participation of yeast inosine 5′-monophosphate dehydrogenase in an in vitro complex with a fragment of the C-rich telomeric strand. Biochimie. 2002;84:279–289. doi: 10.1016/s0300-9084(02)01400-1. [DOI] [PubMed] [Google Scholar]
- 11.McLean J. E., Hamaguchi N., Belenky P., Mortimer S. E., Stanton M., Hedstrom L. Inosine 5′-monophosphate dehydrogenase binds nucleic acids in vitro and in vivo. Biochem. J. 2004;379:243–251. doi: 10.1042/BJ20031585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain J., Almquist S. J., Ford P. J., Shlyakhter D., Wang Y., Nimmesgern E., Germann U. A. Regulation of inosine monophosphate dehydrogenase type I and type II isoforms in human lymphocytes. Biochem. Pharmacol. 2004;67:767–776. doi: 10.1016/j.bcp.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 13.Gu J. J., Spychala J., Mitchell B. S. Regulation of the human inosine monophosphate dehydrogenase type I gene: utilization of alternative promoters. J. Biol. Chem. 1997;272:4458–4466. doi: 10.1074/jbc.272.7.4458. [DOI] [PubMed] [Google Scholar]
- 14.Aherne A., Kennan A., Kenna P. F., McNally N., Lloyd D. G., Alberts I. L., Kiang A. S., Humphries M. M., Ayuso C., Engel P. C., et al. On the molecular pathology of neurodegeneration in IMPDH1-based retinitis pigmentosa. Hum. Mol. Genet. 2004;13:641–650. doi: 10.1093/hmg/ddh061. [DOI] [PubMed] [Google Scholar]
- 15.Colby T. D., Vanderveen K., Strickler M. D., Markham G. D., Goldstein B. M. Crystal structure of human type II inosine monophosphate dehydrogenase: implications for ligand binding and drug design. Proc. Natl. Acad. Sci. U.S.A. 1999;96:3531–3536. doi: 10.1073/pnas.96.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nimmesgern E., Black J., Futer O., Fulghum J. R., Chambers S. P., Brummel C. L., Raybuck S. A., Sintchak M. D. Biochemical analysis of the modular enzyme inosine 5′-monophosphate dehydrogenase. Protein Expression Purif. 1999;17:282–289. doi: 10.1006/prep.1999.1136. [DOI] [PubMed] [Google Scholar]
- 17.Gan L., Petsko G. A., Hedstrom L. Crystal structure of a ternary complex of Tritrichomonas foetus inosine 5′-monophosphate dehydrogenase: NAD+ orients the active site loop for catalysis. Biochemistry. 2002;41:13309–13317. doi: 10.1021/bi0203785. [DOI] [PubMed] [Google Scholar]
- 18.Estevez R., Jentsch T. CLC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 2002;12:531–539. doi: 10.1016/s0959-440x(02)00358-5. [DOI] [PubMed] [Google Scholar]
- 19.Janosik M., Kery V., Gaustadnes M., Maclean K. N., Kraus J. P. Regulation of human cystathionine β-synthase by S-adenosyl-L-methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry. 2001;40:10625–10633. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- 20.Schwappach B., Stobrawa S., Hechenberger M., Steinmeyer K., Jentsch T. J. Golgi localization and functionally important domains in the NH2 and COOH terminus of the yeast CLC putative chloride channel Gef1p. J. Biol. Chem. 1998;273:15110–15118. doi: 10.1074/jbc.273.24.15110. [DOI] [PubMed] [Google Scholar]
- 21.Adams J., Chen Z. P., Van Denderen B. J., Morton C. J., Parker M. W., Witters L. A., Stapleton D., Kemp B. E. Intrasteric control of AMPK via the gamma1 subunit AMP allosteric regulatory site. Protein Sci. 2004;13:155–165. doi: 10.1110/ps.03340004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott J. W., Hawley S. A., Green K. A., Anis M., Stewart G., Scullion G. A., Norman D. G., Hardie D. G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitehead J. P., Simpson F., Hill M. M., Thomas E. C., Connolly L. M., Collart F., Simpson R. J., James D. E. Insulin and oleate promote translocation of inosine-5′ monophosphate dehydrogenase to lipid bodies. Traffic. 2004;5:739–749. doi: 10.1111/j.1600-0854.2004.00217.x. [DOI] [PubMed] [Google Scholar]
- 24.Farazi T., Leichman J., Harris T., Cahoon M., Hedstrom L. Isolation and characterization of mycophenolic acid-resistant mutants of inosine-5′-monophosphate dehydrogenase. J. Biol. Chem. 1997;272:961–965. doi: 10.1074/jbc.272.2.961. [DOI] [PubMed] [Google Scholar]
- 25.Guillen Schlippe Y. V., Riera T. V., Seyedsayamdost M. R., Hedstrom L. Substitution of the conserved Arg-Tyr dyad selectively disrupts the hydrolysis phase of the IMP dehydrogenase reaction. Biochemistry. 2004;43:4511–4521. doi: 10.1021/bi035823q. [DOI] [PubMed] [Google Scholar]
- 26.Berliner E., Mahtani H., Karki S., Chu L., Cronan J., Jr, Gelles J. Microtubule movement by a biotinated kinesin bound to streptavidin-coated surface. J. Biol. Chem. 1994;269:8610–8615. [PubMed] [Google Scholar]
- 27.Hager P. W., Collart F. R., Huberman E., Mitchell B. S. Recombinant human inosine monophosphate dehydrogenase type I and type II protein. Biochem. Pharmacol. 1995;49:1323–1329. doi: 10.1016/0006-2952(95)00026-v. [DOI] [PubMed] [Google Scholar]
- 28.Carr S., Papp E., Wu J., Natsumeda Y. Characterization of human type I and type II IMP dehydrogenases. J. Biol. Chem. 1993;268:27286–27290. [PubMed] [Google Scholar]
- 29.Holmes E. W., Pehlke D. M., Kelley W. N. Human IMP dehydrogenase: kinetics and regulatory properties. Biochim. Biophys. Acta. 1974;364:209–217. doi: 10.1016/0005-2744(74)90006-0. [DOI] [PubMed] [Google Scholar]
- 30.Portera-Cailliau C., Sung C. H., Nathans J., Adler R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 1994;91:974–978. doi: 10.1073/pnas.91.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grover S., Fishman G. A., Stone E. M. A novel IMPDH1 mutation (Arg231Pro) in a family with a severe form of autosomal dominant retinitis pigmentosa. Ophthalmology. 2004;111:1910–1916. doi: 10.1016/j.ophtha.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 32.Crimi M., Galbiati S., Perini M. P., Bordoni A., Malferrari G., Sciacco M., Biunno I., Strazzer S., Moggio M., Bresolin N., Comi G. P. A mitochondrial tRNAHis gene mutation causing pigmentary retinopathy and neurosensorial deafness. Neurology. 2003;60:1200–1203. doi: 10.1212/01.wnl.0000055865.30580.39. [DOI] [PubMed] [Google Scholar]
- 33.Chakarova C. F., Hims M. M., Bolz H., Abu-Safieh L., Patel R. J., Papaioannou M. G., Inglehearn C. F., Keen T. J., Willis C., Moore A. T., et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- 34.Maita H., Kitaura H., Keen T. J., Inglehearn C. F., Ariga H., Iguchi-Ariga S. M. PAP-1, the mutated gene underlying the RP9 form of dominant retinitis pigmentosa, is a splicing factor. Exp. Cell Res. 2004;300:283–296. doi: 10.1016/j.yexcr.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 35.McKie A. B., McHale J. C., Keen T. J., Tarttelin E. E., Goliath R., van Lith-Verhoeven J. J., Greenberg J., Ramesar R. S., Hoyng C. B., Cremers F. P., et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum. Mol. Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- 36.Vithana E. N., Abu-Safieh L., Allen M. J., Carey A., Papaioannou M., Chakarova C., Al-Maghtheh M., Ebenezer N. D., Willis C., Moore A. T. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol. Cell. 2001;8:375–381. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- 37.Mansergh F. C., Millington-Ward S., Kennan A., Kiang A. S., Humphries M., Farrar G. J., Humphries P., Kenna P. F. Retinitis pigmentosa and progressive sensorineural hearing loss caused by a C12258A mutation in the mitochondrial MTTS2 gene. Am. J. Hum. Genet. 1999;64:971–985. doi: 10.1086/302344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sacconi S., Salviati L., Gooch C., Bonilla E., Shanske S., DiMauro S. Complex neurologic syndrome associated with the G1606A mutation of mitochondrial DNA. Arch. Neurol. 2002;59:1013–1015. doi: 10.1001/archneur.59.6.1013. [DOI] [PubMed] [Google Scholar]
- 39.Smith P. R., Bain S. C., Good P. A., Hattersley A. T., Barnett A. H., Gibson J. M., Dodson P. M. Pigmentary retinal dystrophy and the syndrome of maternally inherited diabetes and deafness caused by the mitochondrial DNA 3243 tRNALeu A to G mutation. Ophthalmology. 1999;106:1101–1108. doi: 10.1016/S0161-6420(99)90244-0. [DOI] [PubMed] [Google Scholar]
- 40.Lindstrom D. L., Squazzo S. L., Muster N., Burckin T. A., Wachter K. C., Emigh C. A., McCleery J. A., Yates J. R., 3rd, Hartzog G. A. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol. Cell. Biol. 2003;23:1368–1378. doi: 10.1128/MCB.23.4.1368-1378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevens S. W., Ryan D. E., Ge H. Y., Moore R. E., Young M. K., Lee T. D., Abelson J. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol. Cell. 2002;9:31–44. doi: 10.1016/s1097-2765(02)00436-7. [DOI] [PubMed] [Google Scholar]
- 42.Krogan N. J., Peng W. T., Cagney G., Robinson M. D., Haw R., Zhong G., Guo X., Zhang X., Canadien V., Richards D. P., et al. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell. 2004;13:225–239. doi: 10.1016/s1097-2765(04)00003-6. [DOI] [PubMed] [Google Scholar]
- 43.Ho Y., Gruhler A., Heilbut A., Bader G. D., Moore L., Adams S. L., Millar A., Taylor P., Bennett K., Boutilier K., et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature (London) 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]