

Abstract
Identifying effective therapies targeting multi-protein complexes that lack catalytic sites or cofactor pockets remains a long-standing challenge. The proto-oncogene, ubiquitin E3 ligase SCFSkp2, is one such target. SCFSkp2 promotes the proteasomal degradation of the cyclin-dependent kinase inhibitor p27, which controls cell cycle progression. Targeted knockout of Rb1/Trp53 causes metastatic prostate cancer in mice; additional knockout of Skp2 completely blocks tumorigenesis. We compared gene-edited mice that carried two different single amino acid changes in the SCFSkp2 complex, structurally predicted to inhibit the degradation of p27. Mutation of the SCFSkp2 accessory protein Cks1 (Cks1N45R) completely blocked Rb1/Trp53-driven prostate tumorigenesis, phenocopying Skp2 knockout, whereas a mutation directly stabilizing p27 (p27T187A) did not. This was consistent with structural models that predicted the binding of both p27 and p27T187A to the SCFSkp2/Cks1/Cdk2/CyclinA/p27 complex, and their subsequent ubiquitination and degradation, albeit at different rates. Two binding modes, which differ in their dependence on phosphorylated T187, are predicted by the model. Studies confirmed the role of p27 in mediating tumorigenesis in Rb1/Trp53 mutant tumors and revealed a mutually destabilizing Skp2 and p27 feedback loop. The integration of gene editing, drug-surrogate mutations, and mouse tumor models offers a blueprint for studying SCFSkp2 and other multi-subunit biomedical targets.
Subject terms: Targeted therapies, Checkpoints, Small molecules, Tumour-suppressor proteins
The integration of gene editing, drug surrogate mutations, and genetically engineered mouse cancer models offers a blueprint for studying the role of the E3 ubiquitin ligase SCFSkp2 and the CDKI p27 in Rb1/Trp53-driven prostate tumorigenesis.
Introduction
Mutations in the tumor suppressor gene RB1 have been identified as contributing to the aggressiveness of human malignancies, such that the likelihood of its mutation is often increased in more advanced, metastatic, and chemotherapy-resistant cancers1–3. This includes metastatic castration-resistant prostate cancer (mCRPC), where co-deletion of RB1 and TP53 drives tumors to a metastatic and antiandrogen-resistant phenotype in mice and humans1–6. Meta-analyses revealed that while concurrent TP53 and RB1 alterations are present in only 5% of all primary human prostate tumors, they occur in 39% of mCRPC with adenocarcinoma histology and 74% of mCRPC with neuroendocrine histology7. The combined biallelic loss of both RB1 and TP53 also occurs in >95% of small cell lung cancers, an aggressive, fast-growing, and frequently metastasizing form of lung cancer that has short median survival (10 months) and 5-year survival rates of less than 5%8. In addition, RB1 loss with or without TP53 mutation is a distinctive feature of subsets of patients with other advanced cancers, including triple-negative breast cancer and osteosarcomas9,10. Therefore, long-term curative therapeutic strategies must address the loss of RB1 in these advanced cancers.
All the mutations in RB1 identified to date lead to its inactivation and loss of its tumor suppressor activity. RB1 protein (pRb) regulates gene expression by binding to and suppressing the E2F transcription factors. The E2F family are the major transcriptional regulators of cell cycle-dependent gene expression, particularly those genes required for the G1/G0 to S phase transition11. The loss of RB1 de-couples cell cycle regulation from the upstream kinases CDK4/6, and leaves the E2F transcription factors constitutively unblocked to promote cell-cycle initiation. While the ability of pRb to bind to E2Fs has been the focus of much research, protein interaction databases indicate that there are more than 300 proteins that might interact with pRb11. Notably, pRb exerts significant cell cycle control that is transcription-independent, due to its well-characterized regulation of protein stability by direct effects on the ubiquitin-ligase proteasomal degradation pathway11,12. A specific E3 ubiquitin ligase, SCFSkp2, has been identified as a direct repression target of pRb12–14. This E3 ligase belongs to the Skp1-Cul1-F-box class, and contains the F box protein Skp212–14. SCFSkp2 plays an essential role in the ubiquitination, degradation, and regulation of cellular proteins, including the cyclin-dependent kinase inhibitor p27 (CdkN1b), a key cell cycle regulator that prevents progression into S-phase11,14–17. While the levels of p27 mRNA remain relatively constant throughout the cell cycle, p27 protein levels decline dramatically as cells enter S-phase, due to its SCFSkp2-mediated degradation11,15. Both elevated SCFSkp2, acting as an oncogene, and diminished p27 protein expression have been reported in a broad range of human cancers, where their expression is often correlated with poor prognosis18. A prominent function of p27 is to bind to and inhibit the kinase activity of cyclin-dependent kinase (Cdk)- cyclin complexes that control cell division, a function that has increased importance in cells that have one or more other oncogenic drivers19. Thus, pRb directly regulates SCFSkp2 activity, which in turn controls cellular p27 levels, which then regulates the G1/S cell cycle restriction point by inhibiting one or more Cdk/cyclins. A better understanding of this pathway could aid in the discovery of novel inhibitors, as there currently are no targeted therapies for RB1-deficient tumors. The loss of pRb cannot be practically reversed or repaired in a patient’s disseminated tumor. By contrast, exploiting vulnerabilities that result from the RB1-deficiency could offer an efficacious therapeutic strategy that is cancer-specific, which in principle, spares the wildtype RB1-expressing non-transformed cells.
We and others have shown that tumorigenesis driven by the loss of Rb1 in mice is profoundly counteracted by the knockout (KO) and/or downregulation of the Skp2 F-box component of SCFSkp218,20–22. Skp2 participates in the degradation of >20 known cellular protein substrates which, in addition to cell cycle regulation, can affect cell senescence, apoptosis, migration, DNA repair, and epigenetic regulation23. Thus it was uncertain if the antitumor effect of Skp2 KO was solely due to its effect on p27 and the regulation of the cell cycle. Our current understanding of the downstream biology of SCFSkp2 and the potential mechanisms for the anti-tumor effect of Skp2-KO stem from previous studies that have focused on manipulating p27 itself, based on the observation that it has a defined phosphorylation-dependent degradation sequence (phosphodegron). Degradation of p27 is initiated by its phosphorylation on the T187 degron, which promotes its subsequent binding to, and ubiquitination by SCFSkp2 in the nucleus. A non-phosphorylatable variant of p27, T187A, cannot interact with the phosphodegron-binding pocket of SCFSkp2, and therefore is thought not to be subject to proteasomal degradation in cells or in cell-free systems24,25. However, in contrast to genetic-engineered mouse models with Skp2 KO, the substitution of the wild type p27 with the T187A variant only weakly inhibited tumorigenesis in multiple mouse tumor models10,21,26. Consistent with these findings, expression of p27T187A had only a modest effect on cell proliferation both in vitro and in vivo27. These observations question the role of p27 stabilization as the sole mechanism of the anti-tumor effects of Skp2 KO, and they potentially confound the identification of the optimal approach to pharmacologically modulate SCFSkp2 activity28.
Unlike other F-box proteins that define other SCF E3 ligases, Skp2 requires a small accessory protein, Cks1 (Cdk subunit 1) for the recognition and ubiquitination of p2723,29,30. Cks1 belongs to a highly conserved family of cell cycle regulatory proteins, and while all members of the family can bind to Cdks, only Cks1 is also able to bind to Skp229–31. In vitro reconstitution experiments have shown that Cks1 is essential for p27 ubiquitination, while gene KO approaches, i.e., Cks1−/− cells and mice, have been used to determine Cks1’s physiologic functions23,30. Cks1 KO impairs the proliferation of cells, while Cks1-null mice were smaller than wild-type animals30,32. Whether these phenotypes are strictly attributable to the impairment of SCFSkp2/p27 interactions is uncertain, as Cks1 has multiple functions in cell division32.
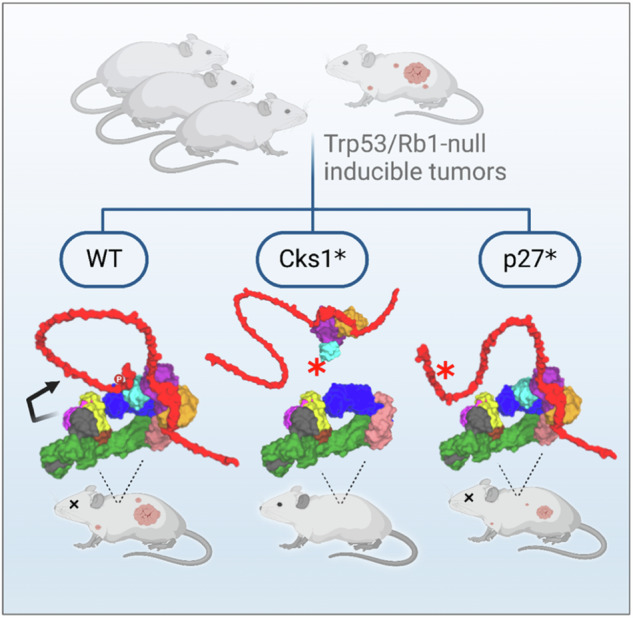
To better understand the molecular actions of SCFSkp2, we reconstructed CryoEM structures into a complete three-dimensional model of the eleven subunit higher-order complex of SCFSkp2, including Cks1, Cdk2, cyclin A, and p27 (Fig. 1). The models were assembled using Schrödinger Maestro and PyMol software suite applied to CryoEM and X-ray co-crystal data in PDB codes 6TTU, 7B5R, 2 AST, and 1JSU33–35. We also note the contemporaneous publication of a relevant CryoEM structure corresponding to the Skp1-Skp2-Cks1-Ckd2-Cyclin A subunits of p27-bond SCFSkp2-Cks1, which is fully consistent with the previous CryoEM structures and the models presented in Fig. 136. The model illustrates the stepwise binding and post-translational events involved in the ubiquitin-dependent proteasomal degradation of p27, and also conveys the critical differences among the numerous modes of inhibition that may be envisioned to inhibit the activity or assembly of this E3 ligase (Fig. 2a–d; S1A)28,37,38. When the accessory protein Cks1 associates with SCFSkp2, it defines the phosphodegron-binding site that recruits this distinct subset of substrates for ubiquitination (Table S1)31. Thus, higher-order complexes of SCFSkp2 with or without Cks1 can be conceptualized as distinct E3 ligases because SCFSkp2-Cks1 can engage a repertoire of ubiquitination substrates that is distinct from SCFSkp223,31. Cks1 has a different surface that interacts with the C-terminal domain of Cdk2, bridging the assembly of SCFSkp2-Cks1 with Cdk2-cyclinA. Therefore cyclin A provides a second binding site in higher order SCFSkp2 to engage substrates that have a cyclin-binding motif, such as p27 (Fig. 2a)37,38. This model also suggests why the anti-tumorigenic effects of Skp2-KO (represented by Fig. 2b) have been inconsistent with the outcomes of p27T187A knock-in (KI, Fig. 2c)10,21,26,27.
Fig. 1. Stepwise binding and post-translational events involved in the ubiquitin-dependent proteasomal degradation of p27 mediated by SCFSkp2.

A SCFSkp2 has a strong binding affinity for Cks1 to form SCFSkp2-Cks1 in early S-phase when CDK2-Cyclin complexes are predominantly bound to the N-terminus of p27 cyclin-binding motif31,34. The intrinsic disorder of the N- and C-termini of p27 are highlighted with a circular arrow over the crystallographically unresolved residues 1–26 and 94–19837,38. B The CDK2-Cyclin-p27 complexes require Cks1 to associate with SCFSkp2-Cks1 (or may together recruit free p27 after step G, not shown). C Y88/Y89 and Y74 are tyrosine residues known to require phosphorylation by non-receptor tyrosine kinases to enable the maximum rate of T187 phosphorylation by a proximal CDK2 enzyme bound to Cks1 or to the N-terminus of Skp2. D The phosphorylation of pT187 provides a critical negative charge enabling the C-terminus of p27 to associate with the phosphodegron site of SCFSkp2-Cks1, which is a binding groove comprised of Skp2 and Cks1 residues31. E Binding of the p27(pT187) phosphodegron (residues 181–190) brings the intrinsically disordered residues of p27 in proximity to the E2 enzyme to facilitate the first ubiquitin transfer; for example, as shown here on K165, among other p27 Lysine residues. F Subsequent ubiquitin-K48-ubiquitination (Ub’ and Ub”) events may occur independently of the phosphodegron site, but the p27 C-terminal region is likely to be sufficiently flexible to associate with the phosphodegron site in the presence of oligo-ubiquitin chains as shown by this model. G Finally, poly-ubiquitinated p27 is recruited and loaded into the proteasome for proteolytic degradation (shown in a cartoon). The posttranslational change occurring in each step is highlighted with boldface.
Fig. 2. Distinct outcomes from targeted genetic perturbations of the multi-subunit SCFSkp2 E3 ligase in Rb1-loss driven tumorigenesis models.

a–d Three-dimensional models of the higher order complexes of SCFSkp2/Cks1/Cdk2/cyclinA/p27 having all wildtype proteins (a); and representations of a complex that cannot form due to lack of Skp2 (b); of a complex binding the p27T187A variant (c); and of the effect of the Cks1N45R mutation on complex formation (d). The intrinsically disordered sections of p27 are represented as loops (Fig. 1). e Interface of Skp2-Cks1 highlighting the pocket occupied by the residue N45 that is changed in the gene-edited subunit Cks1(N45R)31,34. f, g Kaplan-Meier survival curves in mice with Rb1/Trp53 double knock out-driven prostate cancers (f), and with Rb1-knockout driven pituitary cancers (g), when combined with the respectively indicated genotypes and targeted perturbations. Includes some data for Skp2 KO and p27T187A mice that have been previously published by our group21. The size of each cohort is noted in Table 1. f All groups were significantly different from Rb1/Trp53-DKO, p < 0.0001 by Log-rank test. g POMC-Cre;Rb1lox/lox was significantly different POMC-Cre;Rb1lox/lox; Cks1N45R/N45R.
High-resolution X-ray co-crystal structures of the Skp2-Cks1 subunits show that Asparagine 45 of Cks1 occupies a tight pocket at the interface of the protein-protein interaction31,39,40. The interface of Skp2-Cks1 highlighting the pocket occupied by the residue N45 is shown in Fig. 2e. The substitution of N45R in Cks1 abrogates its association with Skp2 and prevents the assembly of higher-order complexes of SCFSkp2 with Cdk2 and cyclin A that are required for p27 ubiquitination (Figs. 2a, d, and S1)31,39,40. Importantly, the N45R mutation leaves unperturbed the other functions of Skp2 as an F-box protein in SCFSkp2. At the same time, while the N45R mutant of Cks1 was nearly inactive in SCFSkp2-related functions, it had close to wild-type activity in other processes that the wild-type Cks1 promotes41. For this reason, we selected the Cks1N45R mutant to interrogate the consequences of Cks1-targeted perturbation on tumorigenesis.
We hypothesize that the Cks1N45R mutation could mimic the inhibition of the protein-protein interaction between Skp2 and Cks1 (Fig. 2d), offering insights into possible therapeutic approaches to leverage the SCFSkp2-dependent vulnerabilities that result from RB1 deficiency. We tested this hypothesis using CRISPR/Cas9-mediated homologous recombination gene editing in mice to introduce the Cks1N45R variant to prevent the assembly of SCFSkp2-Cks1 higher order complexes, without perturbing SCFSkp2 (Fig. 2d). Herein, we show that Cks1N45R blocked tumorigenesis, equally effectively as Skp2 KO, in highly aggressive Rb1 KO and Trp53/Rb1 DKO inducible tumors. This is noteworthy as alternative strategies have fallen short. These findings define a path toward a treatment strategy for metastatic advanced prostate cancer and other RB1-deficient malignancies. More broadly, the integration of gene editing and structurally predicted drug-surrogate mutations offers a blueprint for studying other multi-subunit biomedical targets in vivo.
Results
Cks1N45R KI, but not p27T187A KI, phenocopies the antitumor effect of Skp2 KO
In our genetic mouse model studies, we previously reported that the prostate-specific co-deletion of Rb1 and Trp53 (PB-Cre4;Rb1lox/lox;Trp53lox/lox) (RP-DKO mice) developed prostate cancer21. The carcinomas were highly metastatic, resistant to androgen depletion, and had gene expression signatures commonly found in human prostate carcinomas. When crossed with Skp2−/− mice (Skp2-KO) to remove the SCFSkp2 ligase, however, none of the mice had gross prostate carcinomas; rather tumorigenesis did not progress past the prostatic intraepithelial neoplasia stage5,21. As noted above, the Skp2-KO predictably affects all SCFSkp2 substrates due to the complete disassembly of the complex; hence it is an imperfect model of pharmacological intervention. On the other hand, the effect of Cks1-targeted perturbations would be expected to be narrower in scope, as Cks1 is required for recruitment and ubiquitination of only a subset of SCFSkp2 substrates, including p27, p21, and p57 (Table S1)32,42,43.
The ubiquitination-competent SCFSkp2 E3 ligase is an assembly of up to 11 proteins, which includes cyclin A and Cdk2; all of which may be required for full ubiquitination activity for substrates like p2726,39. The reconstruction of the three-dimensional models of higher-order SCFSkp2 complexes from complementary CryoEM structures (Fig. 2a–e) predicts a striking difference in the functional impacts to the sequential assembly of subunits and of p27 engagement as a consequence of KO of the central Skp2 subunit (Fig. 2b) or the KI of Cks1N45R (Fig. 2d), when compared to the KI of p27T187A (Fig. 2c)36,37. While the p27T187A mutation specifically disables phosphorylation and binding of the C-terminus sequence of p27, the N-terminus region of p27T187A can still bind and inhibit cyclin-Cdk2 kinase activity (Fig. 2c)25. Therefore, p27T187A remains able to associate with SCFSkp2, albeit indirectly and only through its N-terminus cyclin-binding motif that is distal to the E2 enzyme. In contrast, analyses of complexes containing either Skp2-KO or Cks1N45R predict that SCFSkp2 and Cdk2-CyclinA-p27 cannot assemble into higher-order complexes to facilitate p27 ubiquitination. At the same time, p27 can bind to, and presumably inhibit, Cdk2/cyclin A (Fig. 2b, d).
To determine if these structural differences have differential consequences on tumorigenesis, Cks1N45R/N45R KI mice were generated by CRISPR/Cas9 homology-directed repair. These mice appeared healthy, and their lifespan, fertility, and lower body weight were consistent with Skp2-KO and Cks1-KO mice (Fig. S2A, B)30,44. We crossed the gene-edited Cks1N45R mice with the prostate-specific RP-DKO mice, and compared prostate tumorigenesis and life-span to the RP-DKO, RP-DKO-Skp2-KO, and RP-DKO-p27T187A mice (Fig. 2f; Table 1). In this tumor model, RP-DKO mice with wild-type p27, Cks1, and Skp2, developed prostate tumors with 100% penetrance as early as 5 months, and had a median survival of 7.8 months (Fig. 2f)5. The RP-DKO-p27T187A mice also had fully penetrant prostate tumorigenesis, and although the rate of tumor growth was statistically slower compared to RP-DKO controls, overall survival was only minimally increased21. In contrast, only one of 39 RP-DKO-Cks1N45R mice developed a prostate tumor, and survival in these mice was the same as in RP-DKO-Skp2-KO mice (>90% at 20 months) (Fig. 2f; Table 1). MRI imaging of a RP-DKO-p27T187A mouse showed a large prostate tumor at the age of 8.5 months (Fig. S2C, left panels), while no prostate tumors were visible in an RP-DKO-Cks1N45R mouse at 13.2 months (Fig. S2C, right panels). Prostate epitheliums of RP-DKO-p27T187A, RP-DKO-Cks1N45R, and RP-DKO-Skp2-KO mice expressed higher levels of p27 and lower levels of Ki67 than that in RP-DKO mice, as determined by immunohistochemistry (Fig. S2D). In contrast, both the RP-DKO-Cks1N45R and RP-DKO-Skp2-KO prostate epitheliums expressed higher levels of cleaved caspase 3, an indicator of apoptosis, than the RP-DKO and RP-DKO-p27T187A mice (Fig. S2D).
Table 1.
Incidence of prostate and pituitary mouse tumors from targeted genetic perturbations of the multi-subunit SCFSkp2 E3 ligase in Rb1-loss driven tumorigenesis models, as shown in Fig. 2f, g
| Genotype | Mice per group | Mice with prostate tumors | Mice with pituitary tumors |
|---|---|---|---|
| PB-Cre4;Rb1lox/lox;p53lox/lox | 58 | 58 | - |
| PB-Cre4;Rb1lox/lox;p53lox/lox;Skp2−/− | 32 | 0 | - |
| PB-Cre4;Rb1lox/lox;p53lox/lox;Cks1N45R/N45R | 39 | 1 | - |
| PB-Cre4;Rb1lox/lox;p53lox/lox;p27T187A/T187A | 38 | 38 | - |
| PB-Cre4;Rb1lox/lox;p53lox/lox;Cks1N45R/N45R;p27−/− | 12 | 11 | - |
| POMC-Cre;Rb1lox/lox | 41 | - | 41 |
| POMC-Cre;Rb1lox/lox;Cks1N45R/N45R | 29 | - | 0 |
| POMC-Cre;Rb1lox/lox;Cks1N45R/N45R;p27−/− | 13 | - | 11 |
Next, we tested the protective effect of Cks1N45R in the cancer model that originally led to the discovery of the vulnerability of Rb1-deficient tumors to SCFSkp2 perturbation16. Whereas human patients carrying a heterozygous RB1 deletion are predisposed to retinal cancer, mice with germline heterozygous Rb1 mutation develop pituitary and thyroid tumors, which occur when the corresponding wildtype Rb1 allele is lost19. As we had previously shown, the pituitary-specific deletion of Rb1 (POMC-Cre;Rb1lox/lox mice) caused 100% incidence of large pituitary tumors, with a median survival of 8 months (Fig. 2g, Table 1)16. In comparison, none of the POMC-Cre;Rb1lox/lox Cks1N45R mice developed pituitary tumors, and these mice had a normal lifespan. It is noteworthy that additional deletion of the endogenous copy of p27 completely reversed the inhibitory effect of Cks1N45R in both the RP-DKO prostate and the Rb1-null pituitary cancer models (Fig. 2f, g, Table 1). These data demonstrate that the hypothesized anti-tumorigenic therapeutic benefit of disruption of the SCFSkp2 E3 ligase could be distilled to a single-residue mutation in gene-edited Cks1N45R mice, which displayed equally effective protection from tumorigenesis driven by Rb1/Trp53 double-deletion, and tumors initiated by Rb1 loss of heterozygosity alone.
Cks1 N45R abolishes its Skp2-binding ability, and inhibits p27 degradation and cell cycle progression
We next sought to determine the molecular mechanisms for the antitumor effect of targeted Cks1 disruption in prostate tumorigenesis. We first confirmed that the N45R substitution in Cks1 blocked its ability to interact with Skp2, using coimmunoprecipitation assays in 293 T cells overexpressing Flag-tagged Skp2 together with Myc-tagged Cks1N45R, compared to wildtype Cks1 control (Fig. 3a; compare lane 6 with lane 3). In contrast, in agreement with the model proposed in Fig. 2a, c, both WT-p27 and p27T187A were immunoprecipitated by HA-Cullin1 (Fig. 3b), cyclin A, and Cdk2 (Fig. 3c). This observation does not minimize the importance of T187 phosphorylation for optimal p27-Skp2 binding, but rather suggests alternative mechanisms of binding could occur via a protein complex that also contains Skp2.
Fig. 3. Comparison of the binding of Cks1N45R and p27T187A to Skp2, Cyclin A, and Cdk2.

a Immunoprecipitation of Myc-Cks1 from 293 T cells overexpressing Flag-Skp2 and Myc-Cks1-wild type or mutant Myc-Cks1N45R. Western blot shows loss of coimmunoprecipitated Skp2 in cells expressing Cks1N45R (compare lanes 6–3). b 293 T cells were transfected with HA-Cullin1 and either wildtype p27 or p27T187A. IP was with HA-Cullin1 followed by Western blot with HA-Cullin1 and myc-p27. c 293 T cells were transfected with either wildtype p27 or p27T187A, with or without Myc-cyclin A. IP was with Myc-cyclin A, followed by Western blot for p27, Myc-Cyclin A or Cdk2.
We then used mouse embryonic fibroblasts (MEFs), harvested from the Rb1lox/lox;Trp53lox/lox founder mice and those crossed with the various p27, Cks1, and Skp2 genotypes (Fig. 4a). The MEFs were incubated with Adeno-Cre to delete Rb1 and Trp53 genes (RP-DKO-MEFs) in tissue culture. Cks1N45R KI in RP-DKO-MEFs displayed a significant nine-fold increase in p27 protein levels, nearly as large as the 11-fold increase seen with Skp2 KO (Fig. 4b, c). In contrast, p27T187A RP-DKO-MEFs had a significantly smaller 3.5-fold increase in p27, consistent with the diminished anti-tumor effect of this genotype. As a control, we determined the levels of p21 (CdkN1a), another CIP/KIP protein that has redundant degradation pathways and is a significantly less robust substrate of SCFSkp2, and found there were no differences between the different RP-DKO-MEFs (Fig. 4d).
Fig. 4. Cks1N45R knock-in is significantly more effective than p27T187A knock-in in blocking p27 proteasomal degradation.

a Derivation of Rb1/Trp53 double-knockout mouse embryonic fibroblast cells (RP-DKO-MEFs) used in this study. Image was created in BioRender (Maianti J., 2025 https://BioRender.com/n34z903). b Western blot of p27 in RP-DKO-MEFs of four genotypes, as indicated. c Quantitation of p27 protein levels as shown in (b) (n = five biologically independent western blots). * Indicates significantly different from control, ** significantly different than control and p27-T187A. Mean ± SEM, n = 3–5. d Western blot of the CIP/KIP protein p21 in RP-DKO-MEFs. e p27 mRNA levels (relative to GAPDH) of RP-DKO-MEFs corresponding to western in (d). ns, not significant. Mean ± SEM, n = 6 biologically independent samples. f p27 levels in RP-DKO-MEFs of four genotypes treated with or without MG132 (20 μM) for 6 hours. g Quantitation of levels of p27 protein (corrected for tubulin) as shown in (f). * Indicates significant effect of MG132. Mean ± SEM, n = 4 biologically independent samples. h, i Early passage primary prostate tumor cells from Rb1 and Trp53 double knockout mice (RP-DKO-PrCa cells) were transfected with a pTripZ-shCks1 construct. Cks1 knockdown was induced by doxycycline. h Cks1 mRNA levels were measured by RT-qPCR. Mean ± SEM, n = 3 biologically independent samples. i Western blot showing an increase in p27 protein level upon Cks1 knockdown. j Cell proliferation in vitro of RP-DKO-PrCa cells with or without doxycycline treatment to knock down Cks1. Mean ± SEM, n = 3 biologically independent samples. k Cell growth in vitro of RP-DKO-MEFs with wild type Cks1 or Cks1N45R. Mean ± SEM, n = 3 biologically independent samples.
We observed that p27 mRNA levels were essentially unchanged across the panel of RP-DKO MEFs, indicating increases in p27 protein were post-transcriptional (Fig. 4e). This was confirmed using the proteasome inhibitor MG132, which as expected, caused a three-fold increase in p27 protein in the wild type RP-DKO (Fig. 4f, g). While MG132 treatment caused a two-fold increase in p27 in RP-DKO-p27T187A cells, this was not statistically significant. MG132 had no effect on p27 in the RP-DKO-Cks1N45R and RP-DKO-Skp2-KO MEFs, indicating that the degradation of p27 protein was not occurring in these cells (Fig. 4f, g). These data confirmed, both in vivo and in tissue culture, that while the outcomes of p27T187A expression differ from controls, they do not phenocopy Skp2-KO nor Cks1N45R. The data raised two possibilities: that p27T187A may have residual binding affinity to SCFSkp2-Cks1 in spite of the lack of a phosphodegron (phospho-T187); or alternatively, that engagement of p27T187A by its N-terminal cyclin-binding motif supports slow ubiquitination by SCFSkp2-Cks1 assembled with Cdk2-cyclinA (Fig. 2c) or higher-order E3-E3 complexes33.
The effect of Cks1N45R KI was phenocopied when Cks1 was knocked down (KD) with a pTripZ vector in early passage primary prostate tumor cells isolated from the RP mouse (RP-DKO-PrCa cells) (Fig. 4a). Doxycycline was used to induce Cks1 KD (Fig. 4h), and as expected, there was increased p27 protein accumulation (Fig. 4i) and a dramatic decrease in cell proliferation (Fig. 4j). Consistent with the effect of Cks1-KD, cell proliferation of Cks1N45R RP-DKO-MEFs was significantly reduced compared to Cks1-WT RP-DKO-MEFs (Fig. 4k).
The cell cycle distributions of the Cks1-WT and Cks1N45R RP-DKO-MEFs were analyzed by flow cytometry. The population of G1 and S phase cells increased in Cks1N45R RP-DKO-MEFs to 48% and 44%, respectively, from 30% and 28%, compared to Cks1-WT RP-DKO-MEFs (Fig. 5a, b). The G2/M population decreased from 41% to 6.05%. These data indicate the reduced cell proliferation was due to the arrest of the Cks1N45R RP-DKO-MEFs in G1 and S phases, consistent with the known role of p27 in regulating the cell cycle. In addition to inhibiting G1-S transition, p27 also inhibits the S-G2 transition, and consequently, cells synthesize DNA but don’t divide. In line with this finding, immunofluorescence showed that Cks1N45R RP-DKO-MEFs and Skp2-KO RP-DKO-MEFs have enlarged nuclei, compared to control RP-DKO MEFs (Fig. 5d). An additional fraction of cells were undergoing apoptosis, as evidenced by a small, but distinct, sub-G1 seen in the Cks1N45R RP-DKO-MEFs (Fig. 5b) and the induction of active caspase 3 in the cells (Fig. 5c). The induction of apoptosis was p27-dependent, since p27 KO in Cks1N45R cells reduced active caspase 3 to the same level in Cks1-WT RP-DKO-MEFs (Fig. 5c).
Fig. 5. Cks1N45R induces apoptosis and causes cell cycle arrest in G1 and S phases, in a p27-dependent manner.

a, b Cell cycle distribution and quantitation of Cks1-WT (a) and Cks1N45R (b) in RP-DKO-MEFs as analyzed by flow cytometry (PI, propidium iodide; EdU, 5-ethynyl-2´-deoxyuridine). Distribution of cells in sub-G1, G1, S, and G2-M are indicated. c Western blot of active caspase 3 levels in RP-DKO MEFs with Cks1-WT, Cks1N45R, and combined Cks1N45R with p27 KO. d Immunofluorescence of nuclei of RP-DKO-MEFs stained with DAPI. e Western blot of Skp2 in RP-DKO-MEFs of indicated genotypes with and without MG132. f Quantification of Skp2 protein levels of RP-DKO-MEFs as shown in (e) (Mean ± SEM, n = 6 biologically independent western blots). g Corresponding Skp2 mRNA in RP-DKO-MEFs. ns, not significant. Mean ± SEM, n = 3 biologically independent samples. h, i Skp2 protein stability in RP-DKO-MEFs was determined and quantified by treating cells with cycloheximide (CHX) for the indicated times. j Skp2 and p27 protein in RP-DKO-PrCa cells with Cks1 knock down and cycloheximide.
Elevated p27 protein promotes Skp2 degradation
The N-terminus of Skp2 is an intrinsically disordered region that comprises several characteristic sequences on a stretch of 100 residues, including a cyclin-binding motif, an Rb1-binding motif, a nuclear-localization signal, and APCCdh1-binding motifs (Fig. S3)44. We hypothesized that these features of Skp2 may have evolved as feedback loops; however, the contribution of p27 and Cks1 in the context of SCFSkp2 stability has not been investigated. Importantly, western blot quantitation showed a 40% reduction of Skp2 in RP-DKO-MEFs expressing p27T187A and a 76% reduction in Cks1N45R RP-DKO-MEFs, compared to control RP-DKO-MEFs (Fig. 5e, f), in all cases, without significant changes in Skp2 mRNA levels (Fig. 5g). The decrease was due to a large decrease in the estimated half-life of the Skp2 protein (1.9 hours) in the Cks1N45R RP-DKO-MEFS, compared to 16 hours in the WT RP-DKO-MEFs; it was 3.8 hours in p27T187A RP-DKO-MEFs (Fig. 5h, i). We also confirmed these results in RP-DKO-PrCa cells, in which the inducible knockdown of Cks1 produced a comparable effect promoting faster Skp2 degradation over time (Fig. 5j).
Having observed that the increased degradation of Skp2 coincided with large increases in p27 protein levels, we speculated that p27 itself regulated Skp2 stability, either directly or indirectly. To test this, p27 was KD in both Cks1N45R RP-DKO-MEFs and RP-DKO-PrCa cells (Fig. 6a, b), which caused a marked elevation in Skp2 in both cell types (Fig. 6c, d) (note that p27 levels were only decreased by approximately 50%). We confirmed that this was due to an increase in Skp2 protein stability, as measured by half-life, after either p27 KD or KO, an effect observed in both RP-DKO-MEFs and RP-DKO-PrCa cells (Fig. 6e–h). These results showed that differences in p27 abundance proportionally account for Skp2 protein stability alterations. Since p27 is degraded by Skp2-mediated ubiquitination, these data suggest that the two proteins form a mutually inhibitory (double-negative) feedback loop.
Fig. 6. Knockdown of p27 causes increased Skp2 protein due to a reduction in Skp2 proteasomal degradation.

a, b p27 was knocked down in RP-DKO-Cks1N45R MEFs (a) and RP-DKO-PrCa cells (b), and p27 mRNA levels determined by RT-qPCR. Mean ± SEM, n = 3 biologically independent samples. c, d Western blots of Skp2 and p27 from cells in (a, b). e, f Cells treated with cycloheximide (CHX) to determine the degradation rates of Skp2. Effect of p27 knockdown in Cks1N45R RP-DKO-MEFs (e) and RP-DKO-PrCa (f). g Skp2 protein levels in Cks1N45R RP-DKO-MEFs with and without p27 knockout, and with CHX. h Quantification of Skp2 degradation from (e–g). i, j Effect of Cks1 knock down (i) and Skp2 knock down (j) in RP-DKO-PrCa cells on mRNA levels of the indicated E2F1-regulated apoptotic genes, relative to GAPDH mRNA. Mean of two biologically independent samples.
The effect of Cks1 versus p27 knockdown on the transcription of E2F1-regulated genes supported the mutually inhibitory hypothesis (Fig. 6i, j). Our previous work has shown that in Rb1-null cells, the E2F transcription factors are chronically uninhibited, but the pro-apoptotic transcriptional activity of E2F1 remains in check by cyclin A-E2F1 complexes, and by SCFSkp2 mediated degradation of E2F114. Therefore, by inhibiting the SCFSkp2 protein-protein interaction with Cks1N45R, the resulting elevated levels of p27 are poised to out-compete the homologous cyclin-binding motif of E2F1 from Cdk2-cyclinA complexes. We corroborated that this ultimate pro-apoptotic fate of Rb1-null cells is also in effect in RP-DKO-PrCa cells, by showing that Cks1 knockdown caused significantly elevated mRNA transcription of four E2F1-regulated apoptotic genes. Conversely, decreased expression of the pro-apoptotic genes was observed with the knockdown of p27 in the RP-DKO-PrCa cells (Fig. 6i, j).
p27 accumulation increases Skp2 binding to APC/CCdh1 and promotes Skp2 degradation
Finally, we sought to investigate the underlying mechanism by which p27 abundance regulates Skp2 stability. Skp2 activity during the cell cycle is regulated, in part, at the protein level by APC/CCdh1 (Cdh1), which is the primary ubiquitin ligase that targets Skp2 for degradation in G1 phase45,46. We previously reported that the intrinsically disordered N-terminus region of Skp2 contains a cyclin-binding motif that interacts with the same cyclin A binding sites as p2744,47. This tandem arrangement of cyclin- and Cdh1-binding motifs raises the possibility that mutually exclusive binding events control the APC/CCdh1-mediated ubiquitination of Skp2 via its N-terminus (Fig. S3)45,46.
To test if elevated p27 could outcompete cyclin A and render Skp2 more accessible to Cdh1 for degradation, we expressed p27 in 293 T cells (which do not normally express p27) and determined its effect on the association of Skp2 with cyclin A and Cdh1 in coimmunoprecipitation experiments. HA-tagged Cdh1 and Flag-tagged Skp2 were overexpressed, and Skp2 was immunoprecipitated by anti-Flag antibody. In the absence of p27, there was a strong association of Skp2 with cyclin A, and little association with Cdh1 (Fig. 7a, lane 3). The opposite was observed in the presence of p27, where less cyclin A and more HA-Cdh1 coimmunoprecipitated with Flag-Skp2 (Fig. 7a, lane 6). These data are consistent with our hypothesis that high levels of p27 compete with Skp2 binding to cyclin A and expose the N-terminus tail of Skp2 to Cdh1-dependent ubiquitination and degradation. We directly confirmed the ability of Cdh1 to ubiquitinate Skp2 in 293 T cells in cells expressing Flag-Skp2, HA-Cdh1, and Myc-p27 (Fig. 7b). Adding MG132 to cells, especially those expressing Cdh1, increased the levels of ubiquitinated Skp2 (lanes 2,4,6). Expressing p27 (lanes 5-6) further increased the levels of ubiquitinated Skp2 (panel c) and reduced the levels of non-ubiquitinated Skp2 (panel d), compared to cells without this construct (lanes 3-4). These data are consistent with the hypothesis that increased p27 is accelerating Skp2 ubiquitination and degradation via Cdh1. We could not test the effect of CKS1N45R in this assay because the 293 T cells lack endogenous p27, which would be required to mediate the effect of an altered Cks1.
Fig. 7. Increased p27 promotes Skp2 protein binding to APCCDH1 and APCCDH1-dependent Skp2 ubiquitination and proteasomal degradation.
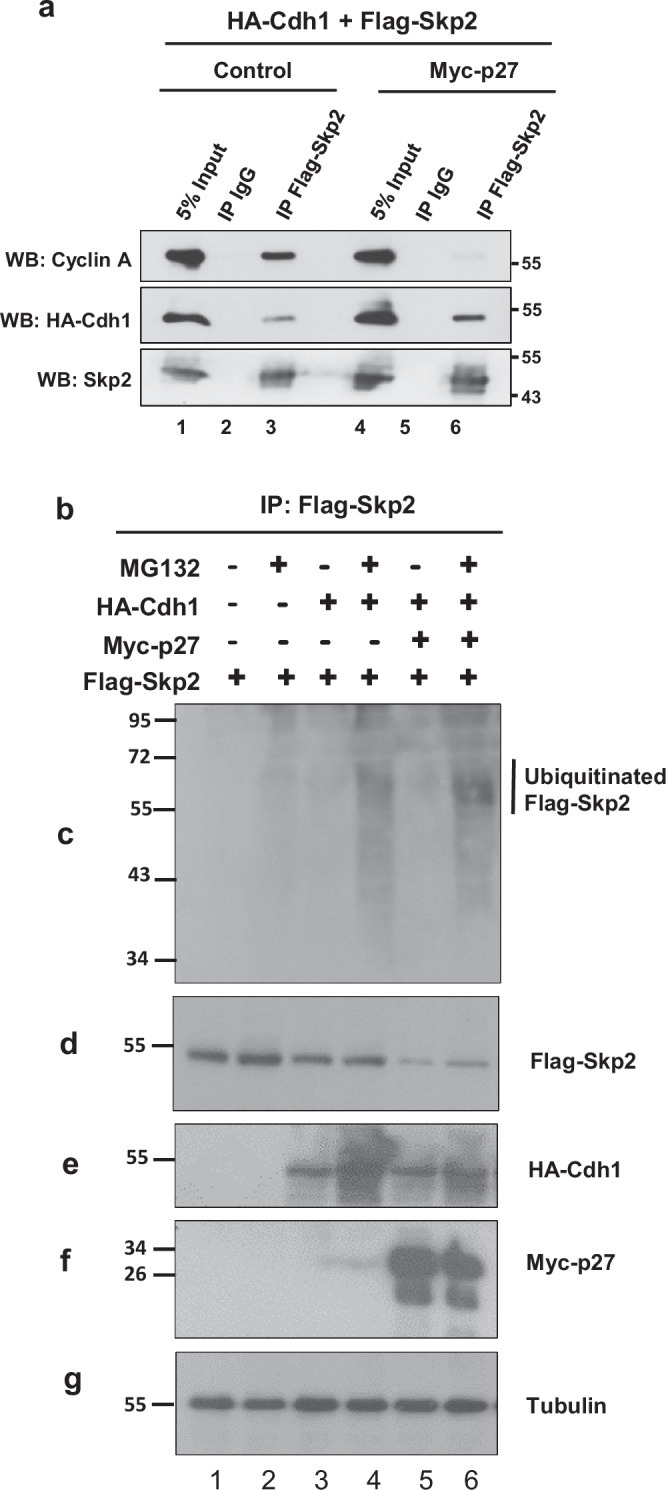
a Coimmunoprecipitation of cyclin A or HA-Cdh1 with Flag-Skp2 in 293 T cells overexpressing Flag-Skp2, with (lanes 4–6) or without (lanes 1–3) Myc-p27 overexpression. b–g 293 T cells were transfected with Flag-Skp2 (lanes 1–6), HA-Cdh1 (lanes 3–6), or Myc-p27 (lanes 5, 6), as indicated. All groups were treated with and without MG132. After IP of Skp2 with an anti-mouse Flag antibody, its ubiquitinated forms were detected with an anti-rabbit ubiquitin antibody (c). Ubiquitinated Flag-tagged Skp2 has an estimated molecular weight of 58-63 kDa. Total cell extracts were probed with an anti-mouse Flag (d), anti-rabbit HA (e), anti-mouse Myc (f), or anti-mouse tubulin antibodies (g).
Discussion
The mutation and loss of function of the tumor suppressor RB1 is often associated with tumor progression, including in highly aggressive small cell lung cancers, metastatic prostate cancers, non-small cell lung cancers that have become resistant to EGFR-directed chemotherapy, and neuroendocrine tumors in multiple organs18. In normal cells, pRb represses Skp2 mRNA expression, directly inhibits SCFSkp2 ubiquitin ligase activity, and associates with Cdh1, facilitating APC/CCdh1-mediated Skp2 degradation15,22,45,46. Hence the functional loss of pRb enhances SCFSkp2 function through multiple mechanisms. The objective of this study was to shed light on the non-equivalence of different approaches to inhibit SCFSkp2 ligase activity in Rb1-null tumors, using a combination of structural analyses, mechanistic biochemical approaches, and gene-modified mouse tumor models. These studies were primarily done in cells derived from mice with double-deficient Rb1 and Trp53 prostate tumors, which correspond to the most highly aggressive and drug-resistant prostate tumors seen clinically.
Herein, we demonstrated that this tumorigenesis model unambiguously discriminated among distinct genetic perturbations to SCFSkp2 (Fig. 2a–d), each of which is informative towards defining a putative pharmacological approach to inhibit this E3 ligase. Specifically, we sought to test gene-edited mice carrying the single-residue mutation (Cks1N45R) that is structurally predicted to mimic the inhibition of the protein-protein interaction between Skp2 and Cks1 (Fig. 2d). In the presence of Cks1N45R, the E3 ligase SCFSkp2 is unperturbed except for its ability to assemble into the higher-order complex SCFSkp2-Cks1 that ubiquitinates a distinct repertoire of phosphodegron-substrates such as p27, in addition to supporting the association with Cdk2-cyclin A that provides an additional binding site for p27 (Fig. 2a, Table S1)31,39,40. Importantly, the presence of Cks1N45R only disrupts its ability to associate with Skp2, and does not perturb other Cks1-dependent functions, suggesting that its effects are on-target41.
The most striking finding in this study was the dramatic anti-tumorigenic protection observed in mice expressing Cks1N45R, which completely blocked the development of Rb1/Trp53 double-KO metastatic prostate cancers and also prevented Rb1 loss-of-heterozygosity pituitary tumors. Importantly, the Cks1N45R mutation, which only abolishes the Skp2-Cks1 interaction, protected mice from tumorigenesis to the same extent as Skp2-KO mice that lack the SCFSkp2 E3 ligase altogether. These findings bode well for the development of protein-protein inhibitors targeting the Skp2-Cks1 interface, for which the Cks1N45R mutation is an accurate model. However, contrary to expectation, attempting to force the accumulation of p27 by KI of the non-phosphorylatable p27T187A mutation, which is predicted to avoid SCFSkp2-mediated ubiquitination, only weakly and temporarily protected the mice from developing Rb1/Trp53 double-KO prostate cancer21. In the presence of p27T187A, the complex of SCFSkp2-Cks1 is predicted to be stalled in a higher-order complex with Cdk2-cyclin A and p27T187A. Our findings suggest this complex can engage in ubiquitin transfer, albeit only as an inefficient substrate. This hypothesis was supported by experiments confirming the binding of p27T187A to cullin1, and the slow degradation of p27 in p27T187A cells.
There are caveats to these conclusions. While we have documented that p27T187A binds to the SCFSkp2/Cks1/Cdk2/CyclinA complex, and that the cellular levels of p27 protein in p27T187A cells differ from those in both Skp2-WT and Skp2-KO cells, we have not directly shown that the changes in p27 levels are due to partial SCFSkp2-mediated degradation of p27T187A. In addition, the results were obtained in the presence of genetic manipulations using knocked-out, overexpressed, mutated, or tagged proteins. These could activate compensatory mechanisms or cause conformational changes in key proteins. Hence these experiments may not fully represent the physiologic condition.
Through our studies seeking to understand the mechanistic differences between Cks1N45R and p27T187A, we observed that the fates of Skp2 and p27 are mutually interconnected. While it is well established that increases in Skp2 lead to decreases in p27, we are now proposing an additional aspect to this regulatory process, in which increases in p27 lead to decreases in Skp2. Skp2’s interactions with different protein complexes within the cell are complex, multifaceted, and not fully understood. In addition to the SCF complex, Skp2 binds to CDK2, cyclin A, and the E3 ligase APC/CCdh1, the latter of which promotes its degradation45,46. Detailed analysis of the structure-function of Skp2 has defined the nature of some of these interactions. For example, the cyclin A binding site on Skp2 has been mapped to four residues within the N-terminal of Skp2, while Cks1 binds to the C-terminal leucine-rich repeat domain of Skp247,48. Of note for our studies, the association of the N-terminus of Skp2 with Cdk2-cyclin A is mutually exclusive with p27 (Fig. S3), suggesting that their binding sites on cyclin A likely overlap, as demonstrated with reconstituted proteins and peptides49. Our studies in intact cells support this hypothesis; presumably, the partial or complete displacement of Skp2 would make it available for association with APC/CCdh1. The proposed mutually inhibitory feedback loop between Skp2 and p27 is consistent with the switch-like behavior required by pathways at critical regulatory junctions. Under normal conditions, cells need to rapidly degrade key proteins to allow for cell cycle progression, and to just as quickly restore the same proteins to reengage cell cycle checkpoint control.
Our study strengthens the rationale and provides a biological basis for targeting the Skp2-Cks1 interface as a promising therapeutic strategy for mCRPC and other cancers with RB1 deficiency. Our findings with Cks1N45R suggest that efforts to inhibit this protein-protein interaction are well warranted, even if it has previously been challenging to discover potent and selective small-molecule SCFSkp2 inhibitors (Table S1)28. On the other hand, targeting the phosphodegron binding site of SCFSkp2 could be bypassed in favor of additional sites on higher-order p27 complexes with Cdk2/cyclinA. These studies highlight the importance of conducting animal tumor studies using specific mutations in addition to gene KOs as targeted perturbations to validate distinct modes of inhibition prior to engaging in pharmacological screening. Importantly, this and other studies continue to support the concept that SCFSkp2 E3 ligase activity is non-essential for quiescent and normally replicating cells in animals44. Therefore, inhibitors targeting the Skp2-Cks1 interface could be both efficacious and well-tolerated as a therapeutic strategy. More broadly, the approach using a gene-edited subunit of a multi-protein complex to mimic a drug-surrogate mutation may be useful as a blueprint to validate the therapeutic potential of other multi-protein biomedical targets. While several small molecule SCFSkp2 inhibitors have been identified to date, none have been shown to have the requisite combination of potency, specificity, and drug-like properties needed to advance to clinical trials28,50.
Materials and methods
Mice
POMC-Cre mice51 PB-Cre4 mice52, Rb1lox/lox mice53, Trp53lox/lox mice54, p27T187A/T187A mice20, and p27−/− mice55 have been described previously. Mice used were FVB, C57BL6J and 129 Sv hybrid background; male mice were used for prostate cancer experiments, male and female for pituitary cancer experiments. Mice were genotyped as previously described15,16. Mice were followed from birth to up to 20 months of age.
Cks1N45R/N45R mice were generated by CRISPR/Cas9-mediated genome engineering with the help of the Albert Einstein Gene Modification Facility56. gRNA targeting to the Exon 2 of mouse CKS1 gene was designed by an online tool (http://crispr.mit.edu/) and generated by in vitro transcription. Cas9 protein was purchased from PNA Bio Inc. The Cks1 N45R Homologous Recombination Donor (HRD) 5’-CCA AGG ACA TAG CCA AGC TGG TCC CGA AAA CCC ATC TGA TGT CTG AAT CTG AAT GGA GGC GTC TCG GCG TTC AGC AGA GTC AGG GAT GGG TCC ACT ATA TGA TCC ATG AAC CAG GTC AGT-3’ containing the around 60nt homologous arms at each side and the surrounded N45R mutation was synthesized chemically from Integrated DNA Technologies Corp (IDT).
Superovulated female C57BL6 mice (3–4 weeks old) were mated to C57BL6 males, and fertilized embryos were collected from oviducts. An in vitro transcribed guide RNA (gRNA) 5’- CCT GAC TCT GCT GAA CGC CGA GG -3’ targeting to the Exon 2 of mouse CKS1 gene, Cas9 protein and Cks1N45R HRD were mixed and microinjected into the pronuclear of fertilized eggs. The injected zygotes were transferred into pseudopregnant CD1 females, and the resulting pups were obtained and genotyped by genomic sequencing. The F0 Cks1N45R/WT founder mice were bred with C57BL6 mice to get F1 offspring. F1 and next generations mice were genotyped by PCR-restriction digestion strategy using primers Cks1-Forward, 5’-CGC AGG ATA GCC TGA AAC TC-3’, Cks1-Reverse, 5’-CAG TGC AGT GCT ACG GAC TC-3’. PCR products were digested by restriction enzyme BsmBI ordered from New England Biolabs (NEB). Cks1N45R PCR products can be cut into two fragments, 237 bp, and 287 bp, whereas the Cks1 wild type 524 bp PCR products cannot.
All animal experiments were approved by the Einstein Institutional Animal Care and Use Committee. We have complied with all relevant ethical regulations for animal use. Novel genetic mice are available for research purposes upon completion of a Materials Transfer Agreement.
Cell culture, cell proliferation measurement, and infection
MEFs and primary prostate tumor cells were prepared and cultured as previously described21. Measurement of cell proliferation was performed by plating 2 × 105 cells in 6 cm plates in triplicate, and cell numbers were counted every 2 days.
Vector containing small hairpin RNA targeting p27, which is complementary to p27 mRNA 5’- CGC AAG TGG AAT TTC GAC TTT-3’, was obtained from Einstein shRNA core facility. Knockdown was carried out by lentiviral transduction. Helper vectors pMDLg/p-RRE, pRSV-REV, and pMD2-VSVG were transfected into 293 T cells to generate lentiviral stocks. Successful lentiviral transduction was selected by puromycin (400-128p, GeminiBio-Products), followed by mRNA (RT–qPCR) and protein (western blots) measurements. Small hairpin RNA targeting Cks1, which is complimentary to Cks1 mRNA 5’-CGTGACCATGTTGCTTTCTTAT-3’, was digested from a pGipZ lentiviral vector obtained from the Einstein shRNA core facility and cloned into a pTripZ lentiviral vector obtained from Dharmacon. Lentiviral helper constructs psPAX2 and pMD2.G were gifts from Didier Trono (Addgene plasmid #12260 and #12259). Doxycycline (2 mg/mL; Sigma-Aldrich) was used to induce Cks1 knockdown in cultured cells.
Structural models
The Protein Data Bank (PDB) entry 6TTU comprising Neddylated-CUL1, RBX1, E2~Ub, and entry 7B5R comprising CUL1, SKP1, SKP2(95–418), CKS1, CDK12, CyclinA2 were downloaded in Schrödinger Maestro and submitted to standard protein preparation33,34. The global coordinates of 6TTU and 7B5R were aligned across the shared CUL1 secondary structure. The CryoEM and crystallographically resolved regions of the p27 N-terminus (25–93) and C-terminus (181–190), respectively matching the high-resolution X-ray structures of PDB entries 2AST and 1JSU, were joined with intrinsically disordered sections predicted by the p27 AlphaFold database entry AF-P46527-F1 and represented as structureless loops30,33. The model of the intrinsically disordered N-terminal tail of SKP2 shown in Fig. S4 was extracted from the AlphaFold entry AF-Q13309-F133,34.
Flow cytometry
Procedures for preparing cells for cell cycle and sub-G1 population analysis were same as previously described18. Cells were analyzed by using BD LSR II Flow Cytometer at Einstein FACS core facility. Data were analyzed by software FlowJo. The gating strategy for the flow cytometry is shown in Fig. S4.
Western blots, and coimmunoprecipitation assay
Cells or tumor masses were lysed in RIPA buffer (#89901, Pierce). Protein concentrations were determined by Bradford Protein Assay Kit (#18-442, Genesee Scientific) using SmartSpec 3000 Spectrophotometer for equal loading by protein content onto SDS-PAGE gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membrane (IPVH00010, Millipore) and probed with the following antibodies: Skp2 (15010-1-AP, Proteintech), α-tubulin (#sc-8035, Santa Cruz Biotechnology), GAPDH (#2118, Cell Signaling Technology), p27 (#610242, BD Biosciences), cyclin A (#sc-596, Santa Cruz Biotechnology), cleaved caspase 3 (#9664S, Cell Signaling Technology), caspase 3 (#9662, Cell Signaling Technology).
For coimmunoprecipitation assays, cells were lysed in Nonidet P-40 buffer [50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 5 mmol/L EDTA, 0.5% Nonidet P-40, and 10% glycerol] containing protease inhibitors. Precleared extracts were incubated with mouse anti-Myc antibody (#sc-40, Santa Cruz Biotechnology), rabbit anti-HA antibody (#3724, Cell signaling Technology), rabbit anti-Myc antibody (#9402, Cell Signaling Technology), or mouse anti-Flag antibody (F1804, Sigma), as shown in the figures. Immunocomplexes were recovered using protein G-agarose (#22851, Pierce) or protein A-agarose (P1406, Millipore Sigma), separated on SDS-polyacrylamide gels, and transferred to PVDF membrane. For Western blotting, the primary antibodies used were rabbit Skp2 (15010-1-AP, Proteintech), horseradish peroxidase (HRP)-conjugated mouse anti-Cks1/2 antibody (#sc-376663, Santa Cruz Biotechnology), rabbit anti-HA antibody (#3724S, Cell Signaling Technology), mouse anti-Myc antibody (#sc-40, Santa Cruz Biotechnology), p27 (#610242, BD Biosciences), rabbit anti-Myc antibody ((#9402, Cell Signaling Technology), Cdk2 antibody (#2546, Cell Signaling Technology), and cyclin A (#sc-596, Santa Cruz Biotechnology). The secondary antibodies were HRP-conjugated mouse anti-rabbit IgG (#5127S, Cell Signaling Technology) or HRP-conjugated anti-mouse IgG (NEF822001EA, Revvity). Proteins of interest were detected using chemiluminescence (Revvity).
For Flag-Skp2 ubiquitination determination, transfected cells were lysed in NP-40 buffer, and precleared cell lysates were incubated with anti-mouse Flag and protein G-agarose beads overnight to pull down Flag-Skp2. Ubiquitinated Flag-Skp2 was detected using an anti-rabbit ubiquitin antibody (#91112, Cell Signaling Technology) by western blot assay.
Cycloheximide chase and protein degradation analysis
For protein stability analysis, cells were plated into 60-mm dishes at 70%–80% confluence. Cycloheximide (CHX, #239764; Calbiochem) was added at 50 µg/mL. At the indicated time points, cell extracts were prepared for western blot. For protein degradation determination, MG132 (Cayman Chemical Company) was added into medium at 20 µM for 6 hours. Cells were harvested and indicated proteins were separated by western blot, with untreated cells served as control.
Reverse transcription and real-time quantitative PCR
RNA was extracted by Trizol reagent (Invitrogen). Oligo-dT and SuperScript II (Invitrogen) were used for the synthesis of the first-strand cDNA at 42 °C for 60 mins. SYBR green PCR mixture (4309155, ABI) and the standard program of ABI 7500 Fast real-time PCR were used. GAPDH was used as an internal control. The qPCR reactions were performed in a final volume of 10 μl. qPCR data were analyzed using the ΔΔCt analysis method. All were done in triplicates and performed three separate times.
Plasmids
Myc-p27, Myc-p27T187A, Myc-Cks1, and Myc-Cks1N45R were generated and cloned into pLenti-CMV vector by Albert Einstein Gene Modification Facility. Other plasmids were described previously12,47. All the coding sequences of the plasmids above are derived from mouse gene. Plasmids were transfected into 293 T cells by Lipofectamine 2000 (11668030, Invitrogen).
Immunohistochemistry and immunofluorescence staining
Tissue sections were prepared, and immunohistochemistry was performed as previously described55. The following antibodies were used: Ki67 (ab16667, Abcam), active caspase-3 (#9664S, Cell Signaling Technology), p27 (ab92741, Abcam). For immunofluorescence of tissue sections, Alexa Fluor 594 Tyramide SuperBoost Kit (goat anti-rabbit IgG, B40944, Life Technologies Corporation) was used to enhance the signal of Caspase 3. For BrdU labeling in vitro, cells were incubated with 10 uM BrdU (B5002; Sigma-Aldrich) for 1 h at 37 °C in a CO2 incubator. Cells were incubated in 2 M HCL for 30 mins at room temperature for DNA hydrolysis. Mouse anti-BrdU (Ab-3, NA61, Calbiochem) and goat anti-mouse IgG Secondary Antibody (Alexa Fluor 594, #A-11020, Life Technologies Corporation) were used for immunofluorescence. Tissue sections or glass coverslips were mounted by Vectashield antifade mounting medium with DAPI (H-1500, Vector Laboratories).
MRI
Magnetic resonance imaging for mouse prostate was performed by Einstein Gruss Magnetic Resonance Research Center. MRI at 9.4 Tesla (T) was used to serially image the mouse prostate and obtain three dimensional images. Images were analyzed by NIH’s software MIPAV.
Statistical and reproducibility
Kaplan-Meier survival curves were analyzed using a log-rank test with GraphPad Prism 9 software. P-values for differences between indicated samples were analyzed by Student’s t test. All statistical analyses are two-sided and p < 0.05 was considered as statistically significant. Replicates (n) are indicated in each figure legend and refer to biologically independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by NIH RO1CA201458. We thank Dr. Richard Stanley, Dr. Antonio Di Cristofano, and Dr. Streamson Chua for providing valuable advice on the research plan and feedback on the experimental results. Technical assistance was provided by the flow cytometry, histopathology, and analytical imaging core facilities of the Albert Einstein College of Medicine, supported by NIH P30 CA013330. The P250 high-capacity slide scanner from the analytical imaging facility was supported by 1S10OD026852-01. Flow cytometry cell cycle analysis was performed using BD LSR-II with the help of Fnu Aodengtuya.
Author contributions
Conception and design: Y. X., H.Z., L.Z., and E.L.S. Acquisition of data: Y.X., H.Z., and S.K. Analysis and interpretation of data: J.P.M., Y.X., H.Z., J.L., C.B., J.Z., B.H., and E.L.S. Writing, review, and/or revision of the manuscript: Y.X., J.P.M., H.Z., and E.L.S.
Peer review
Peer review information
Communications Biology thanks Leslie Gold and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Johannes Stortz.
Data availability
All data supporting the findings of this study are included in the article and its Supplementary information. Numerical source data for the graphs in the manuscript are available in Supplementary Data. Uncropped and unedited western blot images are found in supplemental information. Other data that support the findings of this study are available from the corresponding author upon request.
Competing interests
The authors declare the following competing interests: E.S. is a consultant for Sandoz Pharmaceuticals. J.P.M. is a co-founder of Exo Therapeutics, a company focused on substrate-binding sites. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Juan Pablo Maianti, Email: juanpablo.maianti@einsteinmed.edu.
Hongling Zhao, Email: hongling.zhao@einsteinmed.edu.
Edward L. Schwartz, Email: edward.schwartz@einsteinmed.edu
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-025-07662-3.
References
- 1.Armenia, J. et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet.50, 645–651 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nava Rodrigues, D. et al. RB1 heterogeneity in advanced metastatic castration-resistant prostate cancer. Clin. Cancer Res.25, 687–697 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Mateo, J. et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Invest.130, 1743–1751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou, Z. et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res.66, 7889–7898 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Zhao, H. et al. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell24, 645–659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nyquist, M. D. et al. Combined TP53 and RB1 loss promotes prostate cancer resistance to a spectrum of therapeutics and confers vulnerability to replication stress. Cell Rep.31, 107669 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mu, P. et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science355, 84–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabari, J. K., Lok, B. H., Laird, J. H., Poirier, J. T. & Rudin, C. M. Unravelling the biology of SCLC: implications for therapy. Nat. Rev. Cancer14, 549–561 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Musgrove, E. A. & Sutherland, R. L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer9, 631–643 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Wang, J. et al. Targeted inhibition of SCFSKP2 confers anti-tumor activities resulting in a survival benefit in osteosarcoma. Oncogene43, 962–975 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyson, N. J. RB1: a prototype tumor suppressor and an enigma. Genes Dev.30, 1492–1502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji, P. et al. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell16, 47–58 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Zhang, L. & Wang, C. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene25, 2615–2627 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu, Z. et al. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat. Commun.5, 3463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skaar, J. R., Pagan, J. K. & Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov.13, 889–903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang, H. et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/- mice. Nat. Genet.42, 83–88 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao, H. et al. Deletions of retinoblastoma 1 (Rb1) and its repressing target S phase kinase-associated protein 2 (Skp2) are synthetic lethal in mouse embryogenesis. J. Biol. Chem.291, 10201–10209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta, P., Zhou, H., Hoang, B. & Schwartz, E. L. Targeting the untargetable: RB1-deficient tumors are vulnerable to Skp2 ubiquitin ligase inhibition. Br. J. Cancer127, 969–975 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacks, T. et al. Effects of an Rb mutation in the mouse. Nature359, 295–300 (1992). [DOI] [PubMed] [Google Scholar]
- 20.Ruan, D. et al. Skp2 deficiency restricts the progression and stem cell features of castration-resistant prostate cancer by destabilizing Twist. Oncogene36, 4299–4310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao, H. et al. p27T187A knockin identifies Skp2/Cks1 pocket inhibitors for advanced prostate cancer. Oncogene36, 60–70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, H. K. et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature464, 374–379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frecas, D. & Pagano, M. Deregulated proteolysis by the F-box proteins Skp2 and ß-TrCP: tipping the scales of cancer. Nat. Rev. Cancer8, 438–449 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrano, A. C., Eytan, E., Hershko, A. & Pagano, M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol.1, 193–199 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Sheaff, R. J., Groudine, M., Gordon, M., Roberts, J. M. & Clurman, B. E. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev.11, 1464–1478 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Timmerbeul, I, et al. Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc. Natl. Acad. Sci. USA103, 14009–14014 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malek, N. P, et al. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature413, 323–327 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Lough, L. et al. Chemical probes of Skp2-mediated p27 ubiquitylation and degradation. MedChemComm9, 1093–1104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganoth, D, et al. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat. Cell Biol.3, 321–324 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Spruck, C, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol. Cell7, 639–650 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Hao, B. et al. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol. Cell20, 9–19 (2005). [DOI] [PubMed]
- 32.Hoellein, A, et al. Cks1 promotion of S phase entry and proliferation is independent of p27Kip1 suppression. Mol. Cell Biol.32, 2416–2427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garriga, J. et al. CDK9 is constitutively expressed throughout the cell cycle, and its steady-state expression is independent of SKP2. Mol. Cell Biol.23, 5165–5173 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hao, B., Oehlmann, S., Sowa, M. E., Harper, J. W. & Pavletich, N. P. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol. Cell26, 131–143 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Zhang, H., Kobayashi, R., Galaktionov, K. & Beach, D. P19skpl and P45skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell22, 915–925 (1995). [DOI] [PubMed] [Google Scholar]
- 36.Rowland, R. J. et al. Cryo-EM structure of SKP1-SKP2-CKS1 in complex with CDK2-cyclin A-P27KIP1. Sci. Rep.13, 10718 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baek, K. et al. NEDD8 nucleates a multivalent cullin–RING–UBE2D ubiquitin ligation assembly. Nature578, 461–466 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horn-Ghetko, D. et al. Ubiquitin ligation to F-box protein targets by SCF–RBR E3–E3 super-assembly. Nature590, 671–676 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeffrey, P. D. et al. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature376, 313–320 (1995). [DOI] [PubMed] [Google Scholar]
- 40.Schulman, B. A., Lindstrom, D. L. & Harlow, E. Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc. Natl. Acad. Sci. USA95, 10453–10458 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sitry, D. et al. Three different binding sites of Cks1 are required for p27-ubiquitin ligation. J. Biol. Chem.277, 42233–42240 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Bornstein, G, et al. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J. Biol. Chem.278, 25752–25757 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Kamura, T, et al. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc. Natl. Acad. Sci. USA100, 10231–10236 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakayama, K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J.19, 2069–2081 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bashir, T, Dorrello, N. V, Amador, V, Guardavaccaro, D & Pagano, M Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature428, 190–193 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Wei, W, et al. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature428, 194–198 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Ji, P, et al. Skp2 contains a novel cyclin A binding domain that directly protects cyclin A from inhibition by p27Kip1. J. Biol. Chem.281, 24058–24069 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Kelso, S. et al. Bipartite binding of the N terminus of Skp2 to cyclin A. Structure29, 975–988 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salamina, M. et al. Discriminative SKP2 interactions with CDK-cyclin complexes support a cyclin A-specific role in p27KIP1 degradation. J. Mol. Biol.433, 166795 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee, Y. & Lim, H. S. Skp2 inhibitors: novel anticancer strategies. Curr. Med. Chem.23, 2363–2379 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Balthasar, N. et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron42, 983–991 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Wu, X, et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech. Dev.101, 61–69 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Sage, J, Miller, A. L, Pérez-Mancera, P. A, Wysocki, J. M & Jacks, T Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature424, 223–228 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Jonkers, J, et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet.29, 418–425 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Fero, M. L. et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell85, 733–744 (1996). [DOI] [PubMed] [Google Scholar]
- 56.Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
All data supporting the findings of this study are included in the article and its Supplementary information. Numerical source data for the graphs in the manuscript are available in Supplementary Data. Uncropped and unedited western blot images are found in supplemental information. Other data that support the findings of this study are available from the corresponding author upon request.