

Abstract
Pheochromocytoma is a rare form of adrenal hypertension. This study aimed to investigate the clinical characteristics and associated genetic mutations in patients with pheochromocytoma and primary aldosteronism. We retrospectively analyzed data from 23 patients with pheochromocytoma diagnosed and treated between 2011 and 2022. Three cases were complicated by primary aldosteronism. Compared to 15 other patients without primary aldosteronism complications, these three patients had a greater suppression of plasma renin activity (0.2 vs. 2.3 ng/mL/h, p < 0.01) and a higher aldosterone-to-renin ratio (p < 0.01). No significant differences were found in blood pressure, serum potassium levels, or plasma aldosterone concentrations between the two groups. In genetic analysis, among the three patients with pheochromocytoma and primary aldosteronism, two had a KCNJ5 (G151R) mutation in the pheochromocytoma tumor tissues. However, no CYP11B2- or CYP11B1-positive cells were detected via immunostaining in the pheochromocytoma tissues of these three patients. To our knowledge, this is the first study to reveal the presence of the KCNJ5 mutation, commonly considered specific to primary aldosteronism, in pheochromocytoma cases clinically complicated by primary aldosteronism. The findings suggest that patients with pheochromocytoma and suppressed plasma renin activity should be assessed for primary aldosteronism.
Keywords: KCNJ5, Next-generation sequencing, Pheochromocytoma, Primary aldosteronism
Graphical Abstract

Introduction
Hypertension is a significant cause of cerebrovascular, cardiac, renal, and macrovascular diseases. Secondary hypertension has a specific cause that is distinct from essential hypertension and has a different pathogenesis and treatment strategy. The frequency of secondary hypertension is higher than that previously reported, and it accounts for at least 10% of all patients with hypertension [1]. Secondary hypertension can be caused by various factors. Moreover, hypertension caused by excess hormones is known as endocrine hypertension, with the most common causative organ being the adrenal gland, as observed in pheochromocytoma (PHEO).
PHEO is a neuroendocrine tumor originating from chromaffin cells in the adrenal medulla that presents with a series of catecholamine-excess symptoms, such as paroxysmal hypertension, headache, palpitations, and sweating [2]. The prevalence of PHEO in patients with hypertension is reported to be relatively rare, at 0.3%–0.6% [3, 4]. However, a systematic review examining PHEO incidence over the past seven decades showed an increase in mean incidence from 0.19/100,000 annually before 2000 to 0.58/100,000 annually after 2010, a rise attributed to advancements in imaging methods [5]. Consequently, the actual prevalence of PHEO among patients with hypertension may have been previously underestimated. Moreover, recent studies have highlighted a significant frequency of hereditary PHEO. In their meta-analysis, Lenders et al. [6] reported a hereditary frequency of 33.8%, representing 1,250 cases out of 3,694. Multiple causative genes have been identified in this context.
In adrenal hypertension, conditions of adrenocortical origin, such as primary aldosteronism (PA), are more prevalent than PHEO. PA has been reported to account for 5%–10% of hypertension cases [4]. However, according to a recent review on the evolution of PA [7], the prevalence of PA might range from 6%–14% in normotensive individuals and from 15.7%–24% in those with resistant hypertension. PA is characterized by renin-independent autonomous production of aldosterone and low renin activity and typically presents with hypokalemia. In addition to its high prevalence, cerebrovascular disease, coronary artery disease, and arrhythmias such as atrial fibrillation are reported to be 3–5 times more common in patients with essential hypertension than in those without [8]. Similarly, a large longitudinal study showed that compared with patients with primary hypertension, patients with PA had a 1.3-fold higher mortality rate and approximately 2-fold higher incidence of cardiovascular events [9]. Recent advances in genetic analysis techniques have led to the identification of causative genes that are frequently mutated in PA [10, 11].
Over the past few decades, an increasing coexistence of PHEO and PA has been reported [12]. Generally, compared with individual or single disease, coexisting disease cases show more severe symptoms, such as a higher rate of electrolyte disturbance [12]. Moreover, a case report of the coexistence of PHEO and PA indicated that multiple aldosterone-producing micronodules may be responsible for tumorigenesis [13].
Although these instances of PHEO coexisting with primary aldosteronism have offered some clinical evidence or possible insights, the underlying reasons are still unclear. Specifically, information on the relevant gene mutations is lacking. Here, we conducted a retrospective study of our case series of PHEO to examine the prevalence and clinical features of cases of PHEO coexisting with primary aldosteronism and to determine the types of associated gene mutations.
Materials and Methods
Study design
This retrospective study was conducted in Kanazawa University Hospital. The study was approved by the ethics committee of Kanazawa University (no. 2015121). The genetic experiment was approved by the ethics committee of Kanazawa University (nos. 2012013 and 2012019).
Patients
At Kanazawa University Hospital, surgical treatment was performed on 48 patients with PHEO from 2011 to 2022. Of the 48 patients, 23 were included in the study because consent for the study was obtained and sufficient amounts of PHEO and surrounding tissues were available for clinical and genetic analysis. For laboratory values, we adopted values obtained during screening tests for hypertension and adrenal tumors, as well as during tests for definitive diagnosis. Given the association of PHEO with severe hypertension and the risks inherent in adjusting antihypertensive medications, unadjusted values were also utilized for screening purposes. Conversely, for the captopril challenge test (CCT) used in diagnosing primary aldosteronism, antihypertensive drugs were adjusted in accordance with Japanese guidelines. Adrenalectomies were performed in all 23 patients. Postoperatively, the endocrinologists divided the resected tissues into two parts for separate pathological and genetic sequencing analyses. Diagnosis of PHEO was confirmed after the histopathological analysis.
Data collection
The patients were divided into three groups according to whether they had been diagnosed with aldosterone excess or presented with comorbid characteristics without a confirmatory test. The variables recorded for comparing the two groups included sex, age at diagnosis, systolic blood pressure (SBP), diastolic blood pressure (DBP), and biochemical measurements of PA with serum potassium concentration, plasma aldosterone concentration (PAC), plasma renin activity (PRA), and aldosterone-to-renin ratio (ARR). Additionally, biochemical measurements for PHEO included plasma adrenaline (AD), plasma noradrenaline (NAD), urine metanephrine (MN), and urine normetanephrine (NMN).
Hypertension was defined as SBP ≥140 mmHg, and/or DBP ≥90 mmHg, and/or receiving antihypertensive treatment. BP assessment was conducted following the Japanese Society of Hypertension (JSH) guidelines [1], and the BP values on the morning of the second day of hospitalization were documented. Hypokalemia was defined as a serum potassium concentration of <3.5 mEq/L. The imaging findings included tumor size and lateralization. The pathological analysis included Ki-67 staining in tumor cells.
Next-generation sequencing
The DNA of resected PHEO tissues was extracted using the DNeasy Blood and Tissue Kit (QIAGEN, Hilden, Germany). DNA extraction was also conducted on those cases with enough adjacent adrenal glands and/or surrounding fat tissue. Extracted DNA was subjected to library preparation according to the protocol provided with the KAPA HyperPlus Kit (KAPA Biosystems, Wilmington, MA, USA). For target gene sequencing, the custom xGen NGS Hybridization capture system was used for enrichment, followed by sequencing on NextSeq 2000 (Illumina, San Diego, CA, USA) or MiSeq systems (Illumina, San Diego, CA, USA). In the sequence data analysis, the best practices of GATK (https://gatk.broadinstitute.org/) were utilized to call variants in both germline and somatic mutations. ANNOVAR (http://annovar.openbioinformatics.org/) was used for variant annotation. The gnomAD database (https://gnomad.broadinstitute.org/) was used to determine the population frequencies of variants. Pathogenicity of the variants was assessed using ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). Variants were classified into four categories based on their clinical significance: pathogenic, likely pathogenic, uncertain significance, and others. Overall, this comprehensive variant analysis pipeline facilitates the identification and annotation of genetic variants with potential clinical significance and provides valuable insights into the underlying genetic causes of diseases. In this study, we evaluated several genetic mutations known to be associated with various adrenal disorders. For PHEO, in addition to the well-established mutations in RET, MEN1, NF1, and VHL, we also assessed mutations in KCNJ5, ATP1A1, ATP2B3, CACNA1D, and CACNA1H, which are recognized in PA. Additionally, mutations in PRKACA, PRKAR1A, GNAS, CTNNB1, and ARMC5, known to be involved in CS, were included in our analysis. Other genes studied were CYP11B1, a steroid synthase; HSD3B2 and PDE1A, associated with congenital adrenocortical hyperplasia; and APC, implicated in the development of adrenal tumors.
Immunohistochemistry
All pathological specimens were prepared according to the General Rule for Clinical and Pathological Studies on Adrenal Tumor in Japan. PHEO tumor tissues and adjacent adrenal glands from three PHEO + PA cases were paraffin-embedded and stained with hematoxylin and eosin (HE). Immunohistochemical staining was performed using anti-CYP11B1 and -CYP11B2 antibodies (kindly gifted by Dr. Celso E. Gomez-Sanchez in the Endocrinology Section, G.V. [Sonny] Montgomery VA Medical Center and University of Mississippi Medical Center, Jackson, MS 39216, USA), using a ChemMate ENVISION kit (DAKO, Glostrup, Denmark). The specificities of anti-CYP11B1 and -CYP11B2 antibodies were established in a previous study [14]. We defined CYP11B2-positive areas as lesions responsible for aldosterone overproduction.
Statistical analysis
Statistical analyses were performed using the PRISM software (OMS, Tokyo, Japan). A nonparametric two-tailed Mann-Whitney U test was used for multiple comparisons between the PHEO, PHEO with high ARR, and PHEO + PA groups. Data are expressed as medians (minimum-maximum). Differences between groups were considered significant at p < 0.017, following the Bonferroni correction method.
Results
Clinical characteristics
This study included 23 patients with PHEO (9 men and 14 women), with a median age of 63 years (range, 18–84). Clinical characteristics and laboratory results of the patients are shown in Tables 1 and 2, respectively. Hypertension was initially recorded in all 23 patients, and 78% (18/23) of them received antihypertensive treatment. Fourteen patients (61%) were treated with a calcium channel blocker or an α-blocker alone, and up to 9% of patients received combination therapy (cases 17 and 23). The proportion of hypokalemia was 13% (case 8, case 19, and case 20). Nine PHEO cases (39%) showed suppressed PRA (<1 ng/mL/h), with a median of 0.2 (0.1–0.9) ng/mL/h. Among them, three patients were diagnosed as PA based on positive results of the captopril challenge test. Imaging findings indicated a median tumor size of 42 (15–120) mm, with an approximately threefold higher incidence of tumors on the left adrenal side.
Table 1. Clinical characteristics and laboratory results of the PHEO with normal ARR, PHEO with high ARR, and PHEO with PA groups.
| PHEO with normal ARR group (n = 15) (A) |
PHEO with high ARR group (n = 5) (B) |
PHEO + PA group (n = 3) (C) |
Total (n = 23) |
p value | |||
|---|---|---|---|---|---|---|---|
| A vs. B | B vs. C | A vs. C | |||||
| Male/Female | 5/10 | 2/3 | 2/1 | 9/14 | |||
| Age (years) | 57 (18–84) |
64 (48–75) |
53 (52–77) |
63 (18–84) |
0.62 | >0.99 | 0.65 |
| SBP (mmHg) | 127 (94–223) |
147 (127–163) |
154 (152–174) |
133 (94–223) |
0.07 | 0.14 | 0.03 |
| DBP (mmHg) | 72 (66–107) |
92 (71–100) |
88 (86–89) |
80 (66–107) |
0.19 | 0.79 | 0.41 |
| Serum K (mEq/L) | 4.2 (3.1–5.1) |
3.8 (3.3–4.3) |
4.1 (4.0–4.2) |
4.1 (3.1–5.1) |
0.31 | 0.64 | 0.89 |
| PAC (pg/mL) | 89.0 (28.6–339.0) |
104.0 (74.0–153.0) |
97.9 (73.9–159.0) |
97.9 (28.6–339.0) |
0.61 | >0.99 | 0.91 |
| PRA (ng/mL/h) | 2.3 (0.5–15.2) |
0.2 (0.2–0.6) |
0.2 (0.1–0.9) |
1.4 (0.1–15.2) |
0.0003 | 0.77 | 0.005 |
| ARR | 45.1 (5.8–86.2) |
370.0 (255.0–520.0) |
369.5 (108.8–1,590.0) |
62.0 (5.8–1,590.0) |
0.0001 | >0.99 | 0.003 |
| AD (pg/mL) | 27 (8–600)1 |
70 (25–446) |
80 (50–110) |
45 (8–600)2 |
0.23 | >0.99 | 0.29 |
| NAD (pg/mL) | 1,427 (124–36,000)1 |
840 (232–2,839) |
1,100 (280–1,300) |
1,250 (124–36,000)2 |
0.62 | >0.99 | 0.30 |
| Urine MN (mg/d) | 0.26 (0.06–9.00) |
0.48 (0.22–5.50) |
0.24 (0.11–0.67) |
0.26 (0.06–9.00) |
0.46 | 0.63 | 0.75 |
| Urine NMN (mg/d) | 4.90 (0.30–22.00)1 |
1.18 (0.80–2.10) |
1.18 (0.80–2.10) |
1.79 (0.30–22.0)2 |
0.34 | 0.79 | 0.25 |
| Tumor size (mm) | 42 (15–85) |
52 (22–120) |
30 (26–43) |
42 (15–120) |
0.69 | 0.57 | 0.34 |
| Lateralization (Left/Right) | 10/5 | 5/0 | 2/1 | 17/6 | |||
| Ki-67 cells | |||||||
| <1% | 7 | 3 | 1 | 11 | |||
| ≥1% | 6 | 2 | 2 | 10 | |||
Data are expressed as median (minimum-maximum) unless otherwise noted.
PHEO, pheochromocytoma; PA, primary aldosteronism; SBP, systolic blood pressure; DBP, diastolic blood pressure; K, potassium; PAC, plasma aldosterone concentration; PRA, plasma renin activity; ARR, aldosterone to renin ratio; AD, adrenaline; NAD, noradrenaline; MN, metanephrine; NMN, normetanephrine.
1 n = 14
2 n = 22
Table 2. Characteristics and laboratory results associated with PA in 23 patients with pheochromocytoma.
| Case | Sex/Age (years) | Blood pressure (mmHg) | Serum K (mEq/L) | PAC (pg/mL) | PRA (ng/mL/h) | ARR | Measurement method | Medication | CCT | Gene mutation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/52 | 154/86 | 4.2 | 159 | 0.1 | 1,590 | RIA | none | positive | none |
| 2 | M/77 | 174/89 | 4.1 | 98 | 0.9 | 109 | RIA | α-blocker | positive | KCNJ5 |
| 3 | F/53 | 152/88 | 4.0 | 74 | 0.2 | 370 | RIA | none | positive | KCNJ5 |
| 4 | F/57 | 110/66 | 3.7 | 31 | 0.5 | 62 | CLEIA | α-blocker | — | none |
| 5 | M/64 | 163/100 | 4.2 | 153 | 0.6 | 255 | RIA | CCB | — | GNAS |
| 6 | M/48 | 148/98 | 3.8 | 137 | 0.4 | 343 | RIA | none | — | none |
| 7 | F/63 | 138/92 | 4.3 | 104 | 0.2 | 520 | RIA | none | — | none |
| 8 | F/75 | 127/71 | 3.3 | 80 | 0.2 | 400 | RIA | α-blocker | — | NF1 |
| 9 | F/67 | 147/80 | 3.7 | 74 | 0.2 | 370 | RIA | α-blocker | — | PRKACA |
| 10 | F/47 | 94/66 | 4.3 | 78 | 1.7 | 46 | RIA | α-blocker | — | none |
| 11 | F/74 | 130/70 | 3.9 | 46 | 4.7 | 10 | RIA | α-blocker | — | CYP11B1 |
| 12 | F/40 | 127/72 | 3.9 | 198 | 2.3 | 86 | RIA | α-blocker | — | RET |
| 13 | M/70 | 165/101 | 4.3 | 29 | 4.9 | 6 | CLEIA | α-blocker | — | none |
| 14 | F/29 | 124/72 | 4.3 | 134 | 9.3 | 14 | RIA | α-blocker | — | RET |
| 15 | F/23 | 114/66 | 5.1 | 219 | 4.8 | 46 | RIA | α-blocker | — | VHL |
| 16 | F/48 | 141/94 | 3.8 | 71 | 1.1 | 65 | RIA | α-blocker | — | VHL |
| 17 | M/84 | 124/69 | 4.5 | 79 | 1.3 | 61 | RIA | α-blocker | — | PRKACA |
| CCB | ||||||||||
| 18 | M/76 | 132/90 | 4.3 | 339 | 15.2 | 22 | RIA | α-blocker | — | APC |
| 19 | F/18 | 117/93 | 3.2 | 89 | 1.4 | 64 | RIA | none | — | PRKACA |
| 20 | M/43 | 223/107 | 3.1 | 100 | 2.3 | 43.5 | RIA | α-blocker | — | ATP2B3 |
| 21 | F/83 | 133/78 | 4.2 | 48.3 | 1.4 | 34.5 | CLEIA | ARB | — | none |
| 22 | F/72 | 110/68 | 5 | 176 | 3.9 | 45.1 | RIA | MRA | — | RET |
| 23 | M/67 | 137/77 | 4 | 99 | 10.6 | 9.3 | RIA | ARB | — | MEN1 |
| CCB | ||||||||||
| α-blocker |
PA, primary aldosteronism; K, potassium; PAC, plasma aldosterone concentration; PRA, plasma renin activity; ARR, aldosterone to renin ratio; CCT, captopril challenge test; RIA, radioimmunoassay; CLEIA, chemiluminescence immune assay; CCB, calcium channel blocker; ARB, angiotensin receptor blocker; MRA, mineralocorticoid receptor antagonist.
Baseline characteristics of PHEO, PHEO with high ARR, and PHEO with PA groups
According to the JSH guidelines [1], three patients were diagnosed with PA based on positive results of the CCT. The 23 patients with PHEO were divided into three groups: PHEO (n = 15), PHEO with high ARR (n = 5), and PHEO + PA (n = 3). Since the results of CCT were lacking, the five PHEO cases with ARR >200 were analyzed separately to reduce the potential confounding effects of coexisting PHEO and PA. The baseline characteristics of the PHEO, PHEO with high ARR, and PHEO + PA groups are shown in Table 1. For the three patients (13%, 3/23) diagnosed with PHEO with PA (two men, one woman), the median age was 61 years (52–77). All three PHEO + PA patients had hypertension. PRA was lower in the PHEO + PA group than in the PHEO group (0.2 [0.1–0.9] vs. 2.3 [0.5–15.2], p < 0.01). Compared to the PHEO group, ARR in the PHEO + PA group was higher, with a median of 369.5 (108.8–1,590.0) (p < 0.01). The median PAC in the PHEO + PA group was 73.9 pg/mL (64.0–159.0). No statistically significant differences were observed in SBP, DBP, and PAC between the two groups. No hypokalemia was observed in the PHEO + PA group, with a median value of 4.1 mEq/L (4.0–4.2). The laboratory results associated with PHEO, including AD, NAD, urine MN, and urine NMN, were similar between the PHEO and PHEO + PA groups. Imaging findings revealed a median tumor size of 30 mm (26–43) in the PHEO + PA group, and all tumors were unilateral (two left-side, one right-side).
Postoperative biochemical outcomes of PA in the PHEO with PA group
The preoperative and postoperative outcomes of PA in the PHEO with PA group are shown in Table 3. Among the three PHEO + PA patients, adrenal vein sampling (AVS) was conducted solely on Case 1 before the adrenalectomy, revealing a bilateral PA condition. Postoperative CCT was performed on Case 1 and Case 3, with a positive result and negative result, respectively. Three months after the left adrenalectomy to treat PHEO, the values of PAC, PRA, and ARR in Case 1 were 102 pg/mL, 0.3 ng/mL/h, and 340, respectively. Case 2 exhibited persistent hypertension after the left adrenalectomy, and angiotensin receptor blocker (ARB) was adopted as a drug treatment. With an ARB therapy, a 3-month postoperative assessment of Case 2 revealed a PAC of 114 pg/mL, PRA of 1.0 ng/mL/h, and an ARR of 114. Case 3 underwent right adrenalectomy, with a postoperative biochemical evaluation at the 3-month mark indicating a PAC of 65 pg/mL, PRA of 0.7 ng/mL/h, and an ARR of 93.
Table 3. Preoperative and postoperative outcomes of PA in PHEO with PA group.
| Case | Pre-operation | Post-operation | ||||||
|---|---|---|---|---|---|---|---|---|
| PAC (pg/mL) |
PRA (ng/mL/h) |
ARR | CCT | PAC (pg/mL) |
PRA (ng/mL/h) |
ARR | CCT | |
| 1 | 159 | 0.1 | 1,590 | positive | 102 | 0.3 | 340 | positive |
| 2 | 98 | 0.9 | 109 | positive | 114 | 1.0 | 114 | — |
| 3 | 74 | 0.2 | 370 | positive | 65 | 0.7 | 93 | negative |
PA, primary aldosteronism; PHEO, pheochromocytoma; PAC, plasma aldosterone concentration; PRA, plasma renin activity; ARR, aldosterone to renin ratio; CCT, captopril challenge test.
Gene mutation analysis with next-generation sequencing
Genetic analysis of the 23 patients with PHEO is presented in Fig. 1 and Table 2. Gene mutations defined as uncertain significance (22%, 5/23) included APC p.E1645K, APC p.E1663K, GNAS p.A436D, MEN1 p.T456A, VHL p.T124K, and RET p.V292M. Gene mutations defined as pathogenic or likely pathogenic (26%, 6/23) included RET p.M918T, RET p.C634G, KCNJ5 p.G151R, NF1 p.S1063X, and VHL p.N78S. Gene mutations defined as other (22%, 5/23) included CYP11B1 p.R374R, PRKACA p.P62P, and ATP2B3 p.G491G. The proportion of non-mutated cases was 30% (7/23).
Fig. 1. Genetic analysis of the 23 patients with pheochromocytoma.
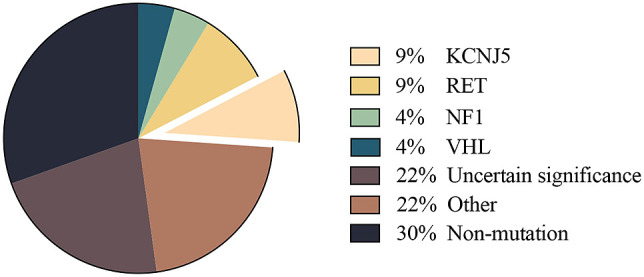
Gene mutations, defined as those of uncertain significance, included APC p.E1645K, APC p.E1663K, GNAS p.A436D, MEN1 p.T456A, VHL p.T124K, and RET p.V292M. Gene mutations defined as pathogenic or likely pathogenic included RET p.M918T, RET p.C634G, KCNJ5 p.G151R, NF1 p.S1063X, and VHL p.N78S. Gene mutations defined as other included CYP11B1 p.R374R, PRKACA p.P62P, and ATP2B3 p.G491G.
Notably, next-generation sequencing performed on the PHEO tumor tissue, adjacent adrenal gland, and fat tissue derived from Case 2 showed that the KCNJ5 mutation was present only in the PHEO tumor tissue. Case 3 was solely analyzed using the PHEO tumor tissue. The Sanger sequencing technique was used to confirm the presence of a KCNJ5 mutation. Case 3 exhibited a KCNJ5 (G151R) mutation in the PHEO tumor tissue (Fig. 2), while the amount of PHEO tumor tissue from Case 2 was inadequate for this analysis.
Fig. 2. Radiological, histopathological, and genetic findings in Case 3.

A, abdominal computed tomography; B, I-123 metaiodobenzylguanidine (MIBG); C, hematoxylin and eosin staining (HE); D, gene analysis of KCNJ5 using the Sanger DNA sequencing technique; the yellow arrow in A indicates a tumor with a diameter of 26 mm in the right adrenal gland; the red arrow in B indicates an uptake of MIBG in the right adrenal gland, which is not inconsistent with pheochromocytoma; a mutation was detected in KCNJ5, and the change in the corresponding amino acid was confirmed (c.451C).
Immunohistochemical analysis
The PHEO tumor tissues and adjacent adrenal glands from the three PHEO + PA cases were stained with HE, CYP11B1 immunohistochemistry, and CYP11B2 immunohistochemistry (Fig. 3, Supplementary Fig. 1). Case 1 (Fig. 3A–C) was a PHEO + PA case without KCNJ5 mutation, while cases 2 (Fig. 3D–F) and 3 (Fig. 3G–I) were PHEO + PA cases with KCNJ5 mutations. The results of CYP11B1 staining were negative in the PHEO tissues of cases 1 (Fig. 3B), 2 (Fig. 3E), and 3 (Fig. 3H). No CYP11B2-positive cells or aldosterone-producing cell clusters (APCCs) were detected in the PHEO tissues of cases 1 (Fig. 3C), 2 (Fig. 3F), or 3 (Fig. 3I). In the adjacent adrenal glands, APCCs were observed in Case 1 (Supplementary Fig.1C, black arrow) and Case 3 (Fig. 3I, Supplementary Fig.1I, black arrow).
Fig. 3. Immunohistochemistry findings in the three cases of PHEO coexistence with PA.

Case 1: A–C; Case 2: D–F; Case 3: G–I. Cases 2 and 3 involved PHEO with a KCNJ5 mutation. A, D, and G, hematoxylin and eosin staining (HE); B, E, and H, immunostaining for CYP11B1; C, F, and I, immunostaining for CYP11B2; t, tumor; a, adjacent adrenal gland; APCC, black arrow in I. PHEO, pheochromocytoma; PA, primary aldosteronism; APCC, aldosterone-producing cell cluster.
Association between steroid hormone excess and gene mutation in PHEO
Of the three patients clinically diagnosed with PHEO and PA, KCNJ5 (G151R) mutations were found in two. This site of KCNJ5 mutation is frequently observed in aldosterone-producing adenomas (APAs). In the other 20 patients with PHEO without PA, no specific genetic mutations in APA, including the KCNJ5, were found.
Discussion
In the present study, 39% of PHEO cases (9/23) exhibited a suppressed level of PRA, at <1 ng/mL/h. This cut-off value in patients with PA indicated a nearly threefold higher risk of cardiovascular events and a significantly higher risk of death compared to patients with unsuppressed PRA (≥1 ng/mL/h) [15]. Generally, PRA levels are elevated in patients with PHEO due to the overactivation of β-adrenergic receptors [16], with levels 1.9- to 2.3-fold higher than those in patients with essential hypertension [17]. Similarly, Yamada et al. reported an unsuppressed PRA level in patients with PHEO (n = 12), averaging 2.1 (1.3–2.8) ng/mL/h [18]. To comprehend the potential reasons for PRA suppression in some patients with PHEO, the negative feedback loops of renin secretion should be considered. Angiotensin II (Ang II) regulates renin synthesis and secretion both in vivo and in vitro [19, 20]. Regarding the relationship between Ang II and PHEO, the density of Ang II binding sites is markedly low in PHEO tissues, occasionally undetectable, contrasting with their common presence in tissues of normal human, rat, and bovine adrenal glands [21, 22]. Nonetheless, few studies addressed PRA levels in patients with PHEO. This is partially evidenced in a case report where a patient with PHEO exhibited a 50% reduction in Ang II post-laparoscopic adrenalectomy [23]. While the exact mechanism behind PRA suppression in PHEO is yet to be elucidated, increased plasma Ang II levels in patients with PHEO can be possibly attributed to diminished Ang II binding sites, contributing to renin suppression.
Among these nine patients with suppressed PRA levels, three of them were diagnosed as PA based on the positive results of CCT; therefore, they were included in the PHEO + PA group, while the remaining patients were included in the PHEO group (n = 15) and the PHEO with high ARR group (n = 5). Significant differences were observed between the PHEO and PHEO + PA groups in the PRA and ARR (p < 0.001), indicating that PHEO + PA cases may aggravate blood pressure through the renin-angiotensin-aldosterone system. However, no significant differences were observed in serum potassium levels or PAC. Collectively, these outcomes of the coexistence of PHEO with PA were not completely consistent with the findings of the study by Mao et al. [12], which comprised 15 patients with concomitant PHEO and PA before 2020, of whom 87% (13/15) initially showed hypokalemia. Furthermore, the PAC in our study was significantly lower than that in Mao’s study (97.9 [73.9–159.0] vs. 287.5 [110.0–1,171.0] pg/mL, p < 0.01), while the PRA level was markedly similar (0.2 [0.1–0.9] vs. 0.255 [0.004–1.2] ng/mL/h, p = 0.98). A recent case report also supported our findings by demonstrating normal serum potassium levels and a high PAC of 4.7 mmol/L and 139 pg/mL, respectively [13], indicating that the prevalence of hypokalemia occurring in concomitant PHEO and PA cases may not be as high as previously described and that the PAC in these cases could be normal. None of the cases with coexistence in our study initially showed catecholamine excess symptoms, whereas this proportion in Mao’s study was 40% [12].
For the lateralization of PHEO and PA, nine cases (9/15) in Mao’s study, assessed through AVS, revealed that three cases (33%) displayed bilateral manifestation of both PHEO and PA. Additionally, two cases (22%) were diagnosed with bilateral PHEO and unilateral PA. Of the remaining four cases with unilateral PA, two (22%) were on the same side as PHEO, and the other two (22%) were contralateral to PHEO [12]. Another PHEO + PA patient exhibited persistent PA after unilateral adrenalectomy, indicating a condition of unilateral PHEO with bilateral PA [13]. In our study, only one case (Case 1) was performed with AVS before adrenalectomy, revealing a concomitant unilateral PHEO and bilateral PA. After left-sided adrenalectomy, Case 1 continued to exhibit signs of persistent PA, with ARR >200 and a positive result of CCT, although the value of ARR had decreased by nearly 80%, from 1,590 to 340. Nevertheless, persistent hypertension was observed in Case 2 following unilateral adrenalectomy targeting the PHEO, suggesting the possibility of bilateral PA cannot be excluded, although an ARR value of 114 was recorded during treatment with an ARB. In Case 3, the resolution of PA was confirmed by ARR <200 and a negative CCT, indicating an ipsilateral PA to PHEO. Gathering together, these findings indicated that PA, when occurring alongside PHEO, may manifest as either unilateral or bilateral without a predisposition for either presentation, independent of the laterality of the PHEO. Hence, we recommend considering the performance of confirmatory tests for PA in patients with PHEO, particularly in cases with suppressed PRA, as potential coexistence with PA warrants consideration for administering both α-blocker and mineralocorticoid receptor antagonists for adrenalectomy.
In order to clarify the mechanism of the coexistence of PHEO with aldosterone excess, genetic analysis via next-generation sequencing was performed in all 23 cases (Fig. 1). Tissue samples of PHEO derived from two out of three patients with PHEO and PA had KCNJ5 (G151R) mutations. Nevertheless, the gene expression profiles in the medulla from these two cases remain unknown, as the medulla tissues were not extracted during the adrenalectomy. The subsequent Sanger sequencing also identified a mutation responding to a change in the amino acid at position 451 of the KCNJ5 gene in the PHEO tumor tissue derived from Case 3 (Fig. 2D). However, the signal intensity of c.451C observed in the Sanger sequencing result was somewhat insufficient. This did not exclude the possibility of contamination by APCCs presented in the adjacent adrenal in this case. In Fig. 3 and Supplementary Fig. 1, APCCs were also found surrounding the PHEOs. On the other hand, the next-generation sequencing analysis had confirmed the same genetic mutation. Further research is required to address the underlying mechanisms of KCNJ5 mutations in PHEO tumorigenesis and the potential molecular characterization with pathogenetic significance.
KCNJ5 is a well-known gene mutation in APAs. Since its first report in 2011 [24], the high prevalence of KCNJ5 in APA has been extensively studied, with a prevalence of 65%–77% in Asian populations [25-27] and 33%–38% in Western countries [28, 29]. The two patients with PHEO who exhibited KCNJ5 mutations were classified into the PHEO + PA group, as previously mentioned. However, both patients presented with hypertension, accompanied by normal serum potassium levels (4.0–4.1 mEq/L) and PAC (73.9–97.9 pg/mL). These laboratory results do not align with the manifestations of KCNJ5 (G151R) mutations in APAs, which typically increase aldosterone production by opening voltage-gated calcium channels [28]. Both patients exhibited suppressed PRA, measuring 0.2 and 0.9 ng/mL/h, respectively.
To date, the pathogenetically molecular characterization of numerous gene mutations in PHEO has become increasingly clear and can be primarily classified into two molecular clusters/subtypes: (1) pseudohypoxia, inducing excessive angiogenesis and uncontrolled tumor growth due to VHL or SDH mutations and (2) kinase signaling, leading to cell proliferation and inhibited apoptosis caused by RET, NF1, TMEM127, or MAX mutations [30]. Meanwhile, in PHEO, mutations such as VHL not only primarily affect hypoxia-inducible factor-1 but also involve proteins like matrix metalloproteinases, which are crucial for tumorigenesis [30]. Consequently, despite the limited number of PHEO cases with KCNJ5 mutations (n = 2), their significant prevalence (8.7%) warrants further investigations to potentially uncover additional mechanisms in PHEO tumorigenesis.
These findings demonstrate that: (1) KCNJ5 mutations in PHEO do not encompass all the typical symptoms observed in PA; (2) cases of PHEO coexisting with PA may exacerbate blood pressure through the renin-angiotensin-aldosterone system; and (3) given the distinct embryological origins of PHEO and PA—chromaffin cells of the adrenal medulla and adrenocortical cells, respectively—and the prominence of hereditary genetic mutations in PHEO compared to the higher prevalence of somatic mutations such as KCNJ5 in PA, the simultaneous occurrence of PHEO and PA may be incidental.
The immunohistochemistry results support our hypothesis. No CYP11B2-positive cells or APCCs were found in the tissue samples of PHEO with KCNJ5 mutations (Fig. 3F, I). However, in a study of 45 APAs with successful CYP11B2-staining and sequencing [31], KCNJ5 mutations exhibited a high prevalence (78%, 35/45). An increase in CYP11B2 expression induced by KCNJ5 mutations has been demonstrated to result in hypersecretion of aldosterone [24-28, 31]. The negative results of CYP11B1 staining in tissue samples of PHEO with KCNJ5 mutations (Fig. 3E, H) were not consistent with the finding of high expression of CYP11B1 in APAs harboring KCNJ5 [32]. Furthermore, we found that the manifestations of KCNJ5 mutations differed in PHEO. Therefore, understanding the underlying mechanisms that contribute to the aggravation of blood pressure in these cases requires further research.
This study has several limitations. First, being a retrospective study, our results were susceptible to selection and confounding biases. Second, the small sample number of the PHEO + PA group (n = 3) could be a reason for the occurrence of statistically significant differences between groups despite the rarity of coexistence cases. Third, due to the insufficient quantity of tumor tissue or absence of adjacent tissue samples, gene expression profile analyses associated with PHEO and PA were not feasible, leaving the impact of KCNJ5 mutations on catecholamine or aldosterone secretion by PHEO unclear. As the PHEO tumor was divided into two sections for pathological and genetic sequencing analyses, confirming identical genetic mutations and hormone production in these sections was challenging. Nonetheless, separating non-tumor tissue was straightforward since the endocrinologist had divided the tissue and the PHEO tumor was large and distinct. Still, it cannot be dismissed that some tissue might not have been entirely segregated from the adjacent adrenal gland and surrounding fat during division, potentially compromising the accuracy of KCNJ5 mutation detection in PHEO + PA cases. At last, not all patients, especially those with ARR >200, underwent CCT, potentially resulting in an oversight of coexistence cases.
Despite the limitations, the strength of this study lies in the analysis of PHEO cases by integrating genetic variations associated with PA through next-generation sequencing. Furthermore, to our knowledge, this was the first study to identify KCNJ5 mutations in patients with PHEO.
In conclusion, compared to patients with PHEO, those with concomitant PHEO and PA showed greater suppression of PRA and an upper level of ARR. We recommend that the patients with PHEO who exhibit both suppressed PRA and hypertension require increased attention because the potential for coexistence with PA in such situations should not be overlooked. A novel insight provided by this study was the observation of a KCNJ5 (G151R) mutation in PHEO cases, which is commonly considered specific to APA (Graphical Abstract). However, not all typical symptoms associated with PA were present in these cases of PHEO and PA coexistence, which could be partially explained by the negative CYP11B2 staining results in the two PHEO cases with KCNJ5 mutations. Furthermore, the underlying mechanisms of suppressed PRA in PHEO cases warrant further investigation.
Graphical Abstract.
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Contributions
Conceptualization, M.K., Sh.K., Y.T., and T.Y.; manuscript drafting and revision, X.M., M.K., K.A., and T.Y.; experiment performing, T.K. and K.H.; immunohistochemical analysis, K.N.; analysis and evaluation of genetic mutations, A.W.; data collection, M.K., D.A., Se.K., and Sh.K.; clinical data analysis, X.M., M.K., and Y.N. All authors have read and agreed to the published version of the manuscript.
Disclosure
None of the authors have any potential conflicts of interest associated with this research.
Ethics Approval
The study was approved by the ethics committee of Kanazawa University (no. 2015121). The genetic experiment was approved by the ethics committee of Kanazawa University (nos. 2012013 and 2012019).
Funding
This work was supported by the Japan Society for the Promotion of Science (Grant no. 19111212) and JST SPRING (Grant no. JPMJSP2135).
Data Availability
The data relevant to the present study are available from the corresponding author upon request.
Supplementary Material
Supplementary Fig. 1
Immunohistochemistry findings in the three cases of PHEO coexistence with PA
References
- 1.Umemura S, Arima H, Arima S, Asayama K, Dohi Y, et al. (2019) The Japanese Society of Hypertension guidelines for the management of hypertension (JSH 2019). Hypertens Res 42: 1235–1481. [DOI] [PubMed] [Google Scholar]
- 2.Lenders JWM, Eisenhofer G, Mannelli M, Pacak K (2005) Phaeochromocytoma. Lancet 366: 665–675. [DOI] [PubMed] [Google Scholar]
- 3.Anderson GH Jr, Blakeman N, Streeten DH (1994) The effect of age on prevalence of secondary forms of hypertension in 4,429 consecutively referred patients. J Hypertens 12: 609–615. [DOI] [PubMed] [Google Scholar]
- 4.Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T (2004) Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res 27: 193–202. [DOI] [PubMed] [Google Scholar]
- 5.Al Subhi AR, Boyle V, Elston MS (2022) Systematic review: incidence of pheochromocytoma and paraganglioma over 70 years. J Endocr Soc 6: bvac105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SKG, et al. (2014) Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 99: 1915–1942. [DOI] [PubMed] [Google Scholar]
- 7.Vaidya A, Carey RM (2020) Evolution of the primary aldosteronism syndrome: updating the approach. J Clin Endocrinol Metab 105: 3771–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohno Y, Sone M, Inagaki N, Yamasaki T, Ogawa O, et al. (2018) Prevalence of cardiovascular disease and its risk factors in primary aldosteronism: a multicenter study in Japan. Hypertension 71: 530–537. [DOI] [PubMed] [Google Scholar]
- 9.Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A (2018) Incidence of atrial fibrillation and mineralocorticoid receptor activity in patients with medically and surgically treated primary aldosteronism. JAMA Cardiol 3: 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scholl UI (2022) Genetics of primary aldosteronism. Hypertension 79: 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamilaris CDC, Stratakis CA, Hannah-Shmouni F (2021) Molecular genetic and genomic alterations in Cushing’s syndrome and primary aldosteronism. Front Endocrinol (Lausanne) 12: 632543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mao JJ, Baker JE, Rainey WE, Young WF Jr, Bancos I (2021) Concomitant pheochromocytoma and primary aldosteronism: a case series and literature review. J Endocr Soc 5: bvab107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ugi S, Yonishi M, Sato D, Nakaizumi N, Horikawa O, et al. (2023) Coexistence of pheochromocytoma and primary aldosteronism due to multiple aldosterone-producing micronodules in the ipsilateral adrenal gland. Intern Med 62: 2685–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, et al. (2014) Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol 383: 111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A (2018) Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. Lancet Diabetes Endocrinol 6: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canu L, Parenti G, De Filpo G, Mannelli M (2019) Pheochromocytomas and paragangliomas as causes of endocrine hypertension. Front Endocrinol (Lausanne) 10: 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haase M, Dringenberg T, Allelein S, Willenberg H, Schott M (2017) Excessive catecholamine secretion and the activation of the renin-angiotensin-aldosterone-system in patients with pheochromocytoma: a single center experience and overview of the literature. Horm Metab Res 49: 748–754. [DOI] [PubMed] [Google Scholar]
- 18.Yamada T, Fukuoka H, Hosokawa Y, Odake Y, Yoshida K, et al. (2020) Patients with pheochromocytoma exhibit low aldosterone renin ratio-preliminary reports. BMC Endocr Disord 20: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schunkert H, Ingelfinger JR, Jacob H, Jackson B, Bouyounes B, et al. (1992) Reciprocal feedback regulation of kidney angiotensinogen and renin mRNA expressions by angiotensin II. Am J Physiol 263: E863–E869. [DOI] [PubMed] [Google Scholar]
- 20.Müller MWH, Todorov V, Krämer BK, Kurtz A (2002) Angiotensin II inhibits renin gene transcription via the protein kinase C pathway. Pflugers Arch 444: 499–505. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Garcia C, Keiser HR (1990) Angiotensin II and angiotensin converting enzyme binding in human adrenal gland and pheochromocytomas. J Hypertens 8: 433–441. [DOI] [PubMed] [Google Scholar]
- 22.Opocher G, Rocco S, Cimolato M, Vianello B, Arnaldi G, et al. (1997) Angiotensin II receptors in cortical and medullary adrenal tumors. J Clin Endocrinol Metab 82: 865–869. [DOI] [PubMed] [Google Scholar]
- 23.Col V, de Cannière L, Messaoudi L, Michel L, Donckier J (1999) Heart failure induced by pheochromocytoma: laparoscopic treatment and intraoperative changes of several new cardiovascular hormones. Horm Res 51: 50–52. [DOI] [PubMed] [Google Scholar]
- 24.Choi M, Scholl UI, Yue P, Björklund P, Zhao B, et al. (2011) K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331: 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taguchi R, Yamada M, Nakajima Y, Satoh T, Hashimoto K, et al. (2012) Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J Clin Endocrinol Metab 97: 1311–1319. [DOI] [PubMed] [Google Scholar]
- 26.Zheng FF, Zhu LM, Nie AF, Li XY, Lin JR, et al. (2015) Clinical characteristics of somatic mutations in Chinese patients with aldosterone-producing adenoma. Hypertension 65: 622–628. [DOI] [PubMed] [Google Scholar]
- 27.Hong AR, Kim JH, Song YS, Lee KE, Seo SH, et al. (2016) Genetics of aldosterone-producing adenoma in Korean patients. PLoS One 11: e0147590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, et al. (2014) Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 64: 354–361. [DOI] [PubMed] [Google Scholar]
- 29.Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, et al. (2013) Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 45: 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galan SR, Kann PH (2013) Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma. Clin Endocrinol (Oxf) 78: 165–175. [DOI] [PubMed] [Google Scholar]
- 31.Nanba K, Baker JE, Blinder AR, Bick NR, Liu CJ, et al. (2022) Histopathology and genetic causes of primary aldosteronism in young adults. J Clin Endocrinol Metab 107: 2473–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Sousa K, Boulkroun S, Baron S, Nanba K, Wack M, et al. (2020) Genetic, cellular, and molecular heterogeneity in adrenals with aldosterone-producing adenoma. Hypertension 75: 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1
Immunohistochemistry findings in the three cases of PHEO coexistence with PA
Data Availability Statement
The data relevant to the present study are available from the corresponding author upon request.