

Abstract
Environmental exposures increase the risk for severe lung disease, but specific drivers of persistent epithelial injury and immune dysfunction remain unclear. Here we identify a feedback circuit triggered by chitin, a common component of airborne particles, that impacts lung health after epithelial injury. In mice, epithelial damage disrupts lung chitinase activity, leading to environmental chitin accumulation, impaired epithelial renewal, and group 2 innate lymphoid cell (ILC2) activation. ILC2s, in turn, restore homeostasis by inducing acidic mammalian chitinase (AMCase) in regenerating epithelial cells, promoting chitin degradation, epithelial differentiation, and inflammatory resolution. Mice lacking AMCase or ILC2s fail to clear chitin and exhibit increased mortality and impaired epithelial regeneration following injury. These effects are ameliorated by chitinase replacement therapy, demonstrating that chitin degradation is crucial for recovery after various forms of lung perturbation. Thus, the ILC2–chitinase response circuit may serve as a target for alleviating persistent post-injury lung epithelial and immune dysfunction.
One-Sentence Summary:
Environmental chitin accumulates after lung injury, triggering an ILC2-mediated epithelial regenerative circuit to restore homeostasis.
INTRODUCTION
Lung epithelial cells regulate fluid balance, gas exchange, and the clearance of continuously inhaled airborne particles (e.g., dust, smoke, bacteria, and fungal spores) from the airways. These homeostatic functions are impaired by epithelial cell injury after respiratory viral infection or other lung perturbations. Acute epithelial injury after viral infection is typically followed by a regenerative response that restores homeostasis. However, pathological outcomes including chronic inflammation, fibrosis, organ failure, and death can also occur, with morbidity and mortality risk linked to high concentrations of ambient particulate matter (1–5). Although mechanisms underlying these associations are unclear, a prominent organic constituent of environmental particles is chitin, an insoluble polysaccharide derived from a variety of sources including dust mites, cockroaches, and molds that are associated with poor air quality and housing conditions (6–8).
Chitin is present in standard specific pathogen–free animal housing conditions and accumulates in the lungs of AMCase-deficient mice over time, causing age-related pulmonary fibrosis (9). In the respiratory and gastrointestinal tracts, chitin activates group 2 innate lymphoid cells (ILC2s) via interleukin (IL)-25, IL-33, and thymic stromal lymphopoietin (TSLP) to promote lung eosinophil accumulation and alternative macrophage activation (10–13). These hallmarks of type 2 immune activation have also been linked with fibrosis, influenza infection, and severe SARS-CoV– and SARS-CoV-2–induced lung disease (14–16). However, the environmental drivers of type 2 triggering in such settings are undefined. Environmentally derived chitin accumulates in the airways of humans and mice with age-related pulmonary fibrosis (9), but can be degraded by mammalian chitinases including AMCase to lessen inflammatory and fibrotic pathology (7, 9, 17), suggesting that chitin and chitinases shape immune responses and disease persistence after injury. Here, we employed mouse models of respiratory viral infection and lung epithelial injury to investigate the contribution of chitin, AMCase, and ILC2s in restoration of health and epithelial homeostasis after perturbation. Unexpectedly, environmental chitin accumulated in the airways of mice following severe epithelial damage, in disparate settings including influenza A and SARS-CoV-2 infection, bleomycin-induced fibrosis, and after alveolar type 2 (AT2) epithelial cell depletion, concordant with loss of homeostatic AMCase activity. Chitin accumulation altered epithelial regeneration and ILC2 responses following injury, but restoration of chitinase activity was able to counteract these effects, revealing a host–environmental response circuit that contributes to lung homeostasis after epithelial perturbation.
RESULTS
Injury to AMCase-expressing epithelial cells causes accumulation of environmental chitin
We and others have previously shown that Chia1 (encoding AMCase) is highly expressed by mature secretory epithelial cells in the lung, particularly club cells and AT2s, consistent with scRNAseq data and studies demonstrating that Chia1 is induced upon AT2 differentiation (fig. S1, A to C) (9, 18–21). Using a method that enriches for alveolar epithelial cells (22), we compared distal epithelial cells from wild-type (WT) mice with AMCase-reporter (ChiaRed (CR))–expressing epithelial cells from heterozygous CR mice (9) by scRNAseq to further verify the cellular identity of AMCase-expressing cells. In the steady-state, EpCAM+ cells from WT mice primarily comprised mature AT2 (93%) and Tm4sf1-expressing alveolar epithelial progenitor cells (AEPs) (~6%) (23, 24). AMCase-expressing CR+ cells matched the transcriptional profile of mature AT2s, as expected based on prior studies (Fig. 1A and fig. S1, D and E) (9, 18, 19). In contrast to AT2s, AEPs were enriched for transcripts marking transitional alveolar epithelial cell states (Krt8, Krt19, Cldn4, and Cdkn1a) (fig. S1D), which expand in pathological settings such as severe SARS-CoV-2 infection and pulmonary fibrosis (25–30). In addition, AEPs and AT2s differentially expressed Epcam, Cdh1, and H2-Ab1, encoding cell surface markers EpCAM, E-cadherin, and MHC-II, respectively, which were used to distinguish these populations by flow cytometry (figs. S1, D and E, and S2A).
Fig. 1. Injury to AMCase-expressing cells causes accumulation of environmental chitin.

(A) Gating scheme and t-distributed stochastic neighbor embedding (tSNE) plots representing scRNAseq analysis of wild-type (WT) (3334 cells) or ChiaRed+ (CR+) (4489 cells) epithelial cells from lungs of WT or CR-heterozygous reporter mice (K-means clustering of populations comprising >5% of total). (B to J) Mice were intranasally mock-infected with PBS or inoculated with 250 plaque-forming units (PFU) of influenza A virus (IAV) and analyzed at indicated days post infection (dpi). (B) Total EpCAM+MHCII+ AT2 cells pooled from each experiment (n = 9–20 mice, N = 3–5 experiments per timepoint), (C) CR+ lung cells (n = 6–14 mice, N = 3–5 experiments per timepoint), (D) AMCase protein in BAL (n = 10–20 mice, N = 3 experiments), and (E) chitin in bronchoalveolar lavage (BAL) fluid (n = 15–24 mice, N > 5 experiments per timepoint) of IAV or mock-infected WT or CR mice at indicated timepoints after infection. R.U.: relative units. (F) Mouse breeding scheme to generate AMCase lineage-tracer (ChiaRed × R26(LSL)-zsGreen) (CR zsG) mice. (G) Quantification of AMCase lineage–traced zsG+ (green) cells in indicated lung areas and (H) representative lung sections from CR zsG mice after IAV or mock infection. Scale bar: 200 μm. Red boxes indicate higher magnification in (I) and the dotted line demarcates injured and uninjured areas. Representative images from n = 3 mice per group, N = 3 experiments. (I) Representative lung sections from CR zsG mice stained with keratin 8 (KRT8, red). Scale bar: 20 μm. Yellow arrowheads, CR zsG+KRT8+ cells. Yellow box indicates the subsection of the infected lung with individual channels represented on the right. (J) Quantification of KRT8 costaining within AMCase lineage–traced zsG+ (green) cells from IAV (14–21 dpi)- or mock-infected mice. Data represent individual biological replicates and are presented as mean±SE. P values were calculated using (B to E) one-way ANOVA with Dunnett’s multiple comparisons test, (G) Holm–Šídák method, or (J) unpaired t test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To study the turnover of Chia1-expressing cells after epithelial injury, we intranasally inoculated WT and CR mice with influenza A virus (IAV) (A/Puerto Rico/8/1934 (H1N1)), which causes acute epithelial cell loss followed by AEP-mediated alveolar regeneration (24, 31). AT2s and AMCase-expressing (CR+) AT2s were significantly reduced 8 days post infection (dpi) with IAV. This coincided with diminished AMCase protein in bronchoalveolar lavage (BAL) fluid, followed by recovery of steady-state AT2 numbers and restoration of BAL AMCase by 21 dpi (Fig. 1, B to D). AMCase was exclusively expressed by epithelial cells after IAV infection. CR reporter was not detected in other lung stromal and hematopoietic cells, including macrophage subsets and neutrophils, consistent with prior reports (fig. S2, A and B) (9, 21). The transient loss of AMCase preceded a rapid accumulation of chitin in the lungs of IAV-infected mice (Fig. 1E), suggesting that inhaled particles were abnormally retained in damaged lung tissue niches. Consistent with previous studies (7, 9), we detected chitin in food and bedding materials from two separate animal housing facilities used for these studies (fig. S3, A and B). Thus, this recalcitrant polysaccharide is broadly distributed in standard rodent housing conditions and accumulates in the lungs after epithelial damage.
To further examine the spatial distribution of AMCase-expressing lung epithelial cells, we crossed ChiaRed (encoding Cre) with Rosa26-CAG-LSL-zsGreen mice (CR zsG), enabling localization and fate-mapping of AMCase-expressing and zsGreen+ lineage–traced cells (Fig. 1F) (13). In mock-infected mice, AMCase lineage–traced CR zsG+ epithelial cells were uniformly distributed throughout proximal and distal lung regions, reflecting homeostatic AMCase expression in secretory club cells and AT2s (9). However, after injury, AMCase lineage–traced cells were absent in damaged lung areas, with a compensatory increase in the frequency of CR zsG-expressing cells in adjacent uninjured regions (Fig. 1, G and H, and fig. S4A). We verified these expression patterns using antibody staining for AMCase and surfactant protein C (SP-C) as an additional mature AT2 marker, which was coexpressed with the CR zsG reporter in a majority of cells in the steady-state and after injury (fig. S4, B to F). These data are consistent with prior reports demonstrating that healthy alveolar epithelial cells are replaced by Krt5-expressing dysplastic epithelial “pods” after severe viral injury (24, 31). Whereas AMCase lineage–traced cells were largely absent from these severely damaged regions early after injury, the frequency of CR zsG+ cells that coexpressed the AEP/transitional epithelial cell marker Krt8 was increased in regions adjacent to damage, where regenerative activity is enriched (Fig. 1, I and J) (24). In addition, rare CR zsG–expressing cell clusters within damaged lung areas increased in size by 21 dpi, consistent with de novo expansion of Chia1-expressing cells within regenerating epithelium (fig. S4, G and H). These data suggested that alveolar epithelial progenitor cells induce AMCase during the AT2 recovery phase after epithelial injury, resembling the developmental acquisition of mature AT2 markers during alveologenesis (18, 19, 24). Indeed, the progenitor:AT2 cell ratio was increased and CR reporter expression was significantly induced among AEPs at 14 dpi. By contrast, AT2s maintained steady-state CR expression during both injury and repair phases (fig. S4, I to L). Thus, lung AMCase expression appears to be restored by AEP-driven regeneration after respiratory viral injury.
AMCase is required for restoration of lung health after epithelial injury
The induction of AMCase in AEPs after viral damage suggested that re-establishing chitinase activity may be a key lung tissue adaptation to environmental chitin after severe epithelial injury. To evaluate this hypothesis, we examined IAV responses in AMCase-deficient (CC) mice, which lack airway chitinase activity (9). Compared to controls, CC mice exhibited increased morbidity and mortality after inoculation with two different doses of IAV (250 and 500 plaque-forming units (PFUs)) (Fig. 2A and fig. S5A). The lower 250 PFU dose was primarily employed in subsequent analyses to avoid potential survivor bias. Death among CC mice mostly occurred during the recovery phase following viral clearance and viral titers were similar between WT and CC mice (fig. S5B), suggesting that differences in disease severity were a result of epithelial injury and environmental chitin accumulation. Indeed, chitinase activity was absent (Fig. 2B) and chitin was significantly increased in the BAL fluid of surviving CC mice, compared to controls, for several months following infection (Fig. 2C and fig. S5C). Increased chitin was accompanied by exacerbated lung inflammatory cell infiltration and excessive protein accumulation in the BAL fluid (fig. S5D and Fig. 2D), which contained increased amounts of serum albumin as well as elevated lactate dehydrogenase (LDH) activity (Fig. 2E and fig. S5E), consistent with a sustained loss of barrier integrity (32, 33). Compared to controls, CC mice also exhibited enlarged areas of patchy consolidation by histology (Fig. 2, F to H). In addition, total lung epithelial cells, AT2s, and AEPs were all reduced in CC mice compared to controls (Fig. 2, I to K), indicative of a diminished capacity to restore epithelial barrier function after damage. By contrast, inoculation with low viral doses (e.g., 25 PFU) did not result in mortality, lung epithelial cell loss, or chitin accumulation in WT or CC mice (fig. S5, F to I). Thus, the chitin–chitinase axis was engaged during regenerative responses to severe epithelial injury.
Fig. 2. AMCase is required for restoration of lung health after epithelial injury.

(A) Survival of WT and CC mice inoculated with 500 PFU IAV. (B to G) WT and CC mice were inoculated with 250 PFU IAV and analyzed at indicated days post infection (dpi). (B) Chitinase activity, (C) chitin (21 dpi; n = 12–17 mice, N = 3 experiments), (D) total BAL protein (21 dpi; n = 6–8 mice, N = 3 experiments) and (E) LDH activity (14 dpi; n = 3–6 mice, N = 2 experiments) in BAL from indicated mice. (F) Representative H&E-stained lung sections at 14 and 21 dpi (n = 4–5 mice per group) and (G and H) quantification of inflamed areas at indicated timepoints at indicated timepoints. (I) Total lung epithelial cells, (J) AT2s, (K) AEPs (n = 3–23 mice, N ≥ 3 experiments), (L) ILC2s, and (M) eosinophils (n = 3–9 mice, N ≥ 3 experiments) at indicated timepoints. (N) Dosing regimen and of CC mice that received intranasal CHIT1 or heat-inactivated (hi)-CHIT1 after IAV inoculation. (O) BAL chitinase activity, (P) MFI of ChiaRed reporter among CR+ epithelial cells, (Q) survival, and (R) ILC2 numbers in CC mice treated with CHIT1 or hi-CHIT1 21 days after IAV inoculation (n = 7–9 mice, N = 3 experiments). (S) Lung ILC2s in WT mice that received intranasal AMCase or hi-AMCase after IAV inoculation (21 dpi; dosing regimen as in (N)). Dotted lines in (F) indicate inflamed areas and red boxes indicate insets for higher magnification. Scale bars: 1 mm (upper) and 200 μm (lower). Data represent individual biological replicates and are presented as mean±SE. Survival rates in (A) (WT IAV, n = 85 mice; CC IAV, n = 89 mice, N > 5 experiments) and (Q) (CHIT1, n = 13 mice; hi-CHIT1, n = 10 mice) were compared by log-rank (Mantel–Cox) test. P values were calculated using two-way ANOVA with multiple comparisons using (I to M) Holm–Šídák method. All other P values were calculated by unpaired t test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The epithelial defects in IAV-infected CC mice were accompanied by alterations in lung inflammatory infiltrates, which varied in composition at various timepoints after infection (fig. S6, A to F). Although alveolar macrophages contribute to particle clearance and tissue repair after IAV infection (34, 35), their numbers were similar in the lungs of infected WT and CC mice (fig. S6B). By contrast, γδ T cells and neutrophils were reduced in CC mice as compared to controls (fig. S6, E and F), consistent with enhanced activation of type 2 immune pathways during the repair phase after severe IAV infection. Accordingly, ILC2s and eosinophils, which are induced in the lungs by chitin exposure (10–12) and have been associated with epithelial repair after IAV (36, 37), were increased in the lungs of CC mice compared to controls (Fig. 2, L and M). We used mice expressing reporter alleles for ILC2 signature genes arginase-1 (Yarg; Arg1YFP), IL-5 (R5; Il5tdTomato), or IL-13 (Smart13; Il13hCD4) (38) to confirm the expansion of type 2 cytokine–expressing ILC2s during the recovery phase after IAV infection, as previously noted (36, 37) (fig. S7, A to E). Lung ILC2s remained elevated throughout the recovery phase (Fig. 2L). However, IL-13–producing ILC2s and BAL IL-5 were significantly greater in the lungs of CC mice (fig. S7, F and G). Rare IL-13–producing T helper 2 (Th2) cells were also detected during the recovery phase, although their numbers were similar in the lungs of WT and CC mice (fig. S7H).
The ILC2-activating cytokine IL-33, as well as amphiregulin (AREG)—an EGFR-binding growth factor previously implicated in ILC2-mediated lung epithelial restoration after injury (36)—were also elevated in the BAL of CC mice compared to controls (fig. S7, I and J). This suggested that IL33- and ILC2-mediated type 2 immune activation was sustained by chitin after viral injury. In uninfected WT mice, the activation of lung ILC2s with exogenous TSLP and IL-33—signals previously implicated in chitin responses (10–13)—markedly increased AMCase production and BAL chitinase activity, whereas chitinase activity remained absent in CC mice (fig. S7, K and L). Furthermore, ILC2 activation, IL-13 production, and eosinophil accumulation were significantly enhanced in CC versus WT mice after acute TSLP and IL-33 administration (fig. S7, M to P). Thus, steady-state chitin exposure in the absence of AMCase appears to differentially condition both epithelial and immune compartments in the lungs, preceding severe outcomes after infectious injury and later development of age-related fibrotic lung disease (9).
These results indicated that the inability to restore chitinase activity after infection in CC mice led to increased chitin accumulation, ILC2 activation, dysregulated epithelial repair, and exacerbated lung disease. To test whether exogenous chitinase replacement therapy could rescue these effects, we intranasally administered recombinant chitinases to CC mice during the recovery phase after IAV infection (Fig. 2N). To avoid potential immunogenicity in AMCase-deficient CC mice and to test the sufficiency of chitinase enzymatic activity, we treated with chitotriosidase (CHIT1), another active mammalian endochitinase similar to AMCase. CHIT1 administration increased BAL chitinase activity and recovery of mature AT2 cells, identified by epithelial AMCase reporter expression (Fig. 2, O and P). Treatment with CHIT1—but not heat-inactivated CHIT1 (hi-CHIT1)—also prevented IAV-induced mortality in CC mice (Fig. 2Q). Administration of either recombinant CHIT1 or AMCase was also sufficient to reduce lung ILC2 expansion in CC and WT mice, respectively, after severe IAV infection (Fig. 2, R and S), suggesting that ILC2s are triggered by environmental chitin after severe epithelial injury. Thus, chitinase enzyme replacement therapy demonstrates efficacy in restoring chitinase activity and aiding recovery after epithelial injury.
To extend these findings to other settings of respiratory virus–induced epithelial injury, we examined chitin and AMCase in mouse models of SARS-CoV-2 infection. Consistent with IAV infection, we observed chitin accumulation in the airways of mice that had been transiently transduced with an adenoviral vector expressing human ACE2 (AdV-hACE2) (39) and inoculated with SARS-CoV-2 (strain WA1/2020) (fig. S8, A and B). At the same time, AMCase expression was lost within the inflamed regions of the lung, demonstrating that SARS-CoV-2 infection also impairs epithelial AMCase in a spatially restricted manner (fig. S8, C and D), akin to IAV infection. AdV-hACE2-transduced CC mice exhibited exacerbated inflammatory lung pathology and prolonged weight loss compared to WT controls after SARS-CoV-2 infection (fig. S8, E to G). Moreover, environmental chitin accumulated in the lungs of mice infected with B.1.351 SARS-CoV-2, a strain naturally adapted to mice. This viral strain also caused more severe lung pathology in CC versus WT mice, while viral titers remained similar (fig. S8, H to K). Thus, respiratory viruses appear to cause epithelial damage, which results in the accumulation of environmental chitin. This chitin can then be degraded by AMCase to promote the resolution of inflammation during the post-acute recovery phase.
To circumvent antiviral immune responses and test whether the genetically targeted ablation of AT2 cells was sufficient to trigger chitin accumulation and ILC2 activation, we transiently depleted AT2s using mice expressing an inducible surfactant protein C-driven diphtheria toxin A allele (Sftpctm1(cre/ERT2,rtTA)Hap × Gt(Rosa)26Sortm1(DTA)Lky/J; AT2-deleter) (23, 40, 41) (fig. S9A). We verified the loss of AT2 cells following tamoxifen administration in AT2-deleter mice (fig. S9B), which exhibited lung edema and increased BAL protein accumulation compared to Sftpccre/ERT2-negative controls (fig. S9, C and D), consistent with AT2 cell deletion and loss of alveolar integrity. AT2 deletion also resulted in reduced BAL AMCase protein and chitinase activity, whereas BAL chitin, lung ILC2s, eosinophils, and AREG were all significantly elevated compared to controls (fig. S9, E to K). Other lung-resident immune cells, such as alveolar macrophages, were unaltered by AT2 cell deletion (fig. S9L). Thus, the loss of AT2 cells specifically impacts AMCase expression, chitin accumulation, and ILC2 activation.
AMCase prevents severe progressive fibrotic lung disease and mortality
Persistent AT2 cell injury and epithelial regenerative dysfunction are prominent in fibrotic lung diseases (4). We therefore tested the involvement of chitin and chitinase in a mouse model of bleomycin-induced pulmonary fibrosis. Bleomycin administration resulted in sustained loss of mature AT2s, accompanied by reduced BAL chitinase activity and accumulation of environmental chitin, which was significantly increased in the CC mice compared to WT controls (Fig. 3, A to C). CC mice also exhibited increased mortality during the later repair/resolution phase following bleomycin (Fig. 3D), in agreement with the viral infection models and supporting a crucial role for AMCase in promoting resolution of fibrotic lung disease and survival after injury. Indeed, by 42 days after bleomycin administration, WT mice had largely resolved fibrosis. However, the lungs from surviving CC mice were marked by prominent interstitial fibrotic lesions (Fig. 3, E and F) and persistently elevated collagen content, which was rescued in CC mice by expression of a lung-specific AMCase transgene (CC × SPAM Tg mice) (Fig. 3G) (9). Moreover, therapeutic administration of recombinant AMCase after bleomycin injury reduced BAL chitin accumulation, improved body weights, and attenuated the severity of lung fibrosis in WT mice (Fig. 3, H to J). Thus, AMCase counteracts the exacerbating effects of environmental chitin in the setting of pulmonary fibrosis, suggesting broad involvement of chitin and AMCase in the repair and regeneration of damaged lung epithelium.
Fig. 3. AMCase prevents severe progressive fibrotic lung disease and mortality.
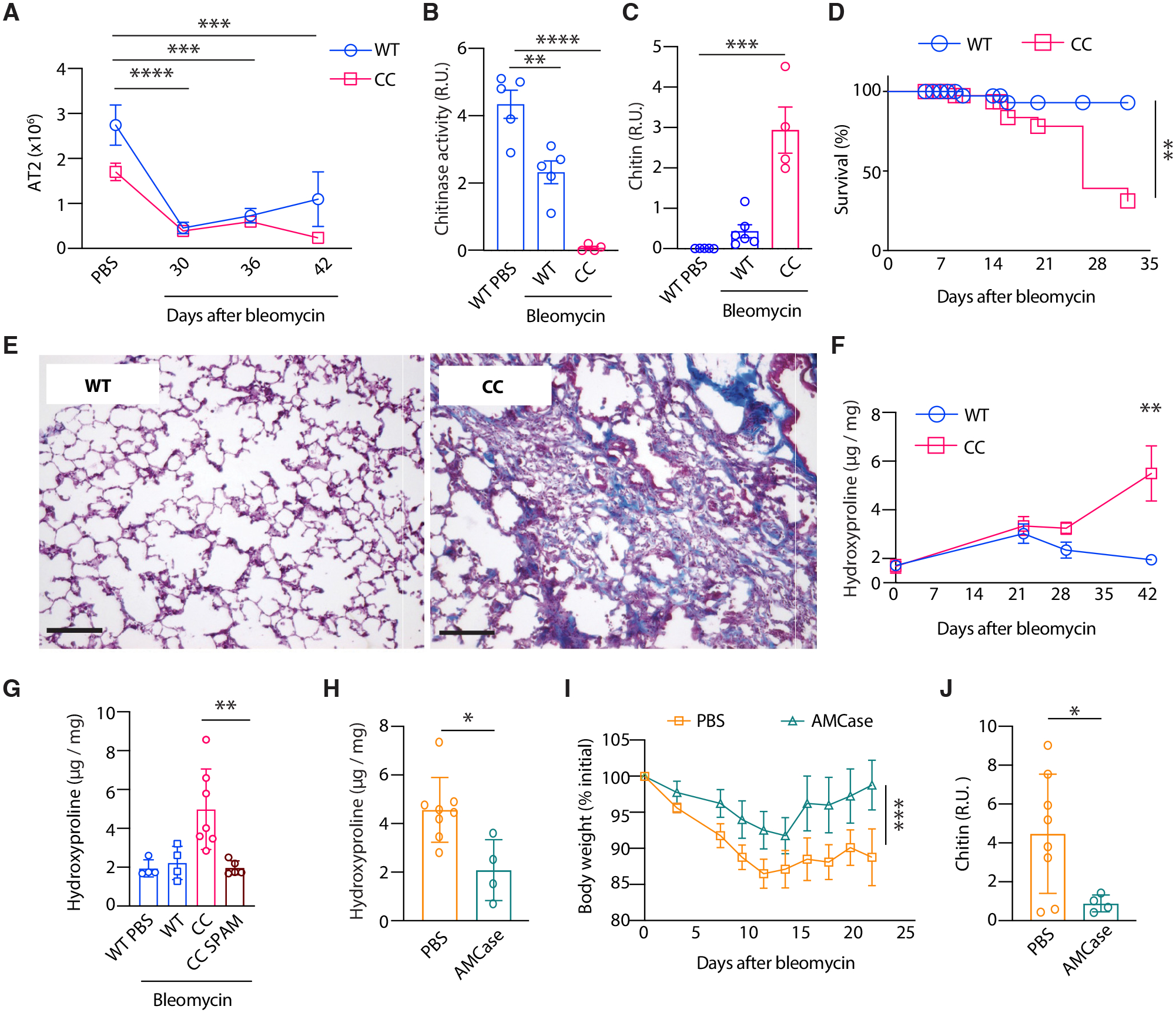
(A) Total lung AT2s (n = 4–7 mice, N = 3–5 experiments per timepoint), (B) BAL chitinase activity, (C) BAL chitin (n = 4–6 mice, N > 2 experiments), and (D) survival of WT and CC mice 21 days after bleomycin challenge (WT, n = 58 mice; CC, n = 63 mice; N ≥ 3 experiments). R.U.: relative units. (E) Representative Masson’s trichrome-stained lung sections from WT and CC mice 42 days after bleomycin administration (n > 5 mice per genotype; N > 3 experiments). Scale bar: 20 μm. (F) Lung hydroxyproline content in bleomycin-challenged WT and CC mice and (G) WT, CC, and CC SPAM mice 42 days after bleomycin administration (n = 4–7 mice per genotype, N > 2 experiments). (H) Lung hydroxyproline, (I) body weights, and (J) BAL chitin among 21 days post bleomycin-challenged WT mice receiving intranasal PBS or recombinant AMCase. Data represent individual biological replicates. P values were calculated by two-way ANOVA with correction for multiple comparisons using (A) Dunnett’s method or (F) Holm–Šídák method, (B, C, G) one-way ANOVA with multiple comparisons using Dunnett’s method, (H and J) unpaired t test, (I) paired t test, and (D) log-rank (Mantel–Cox) test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Environmental chitin and chitinase activity control ILC2s and recovery after IAV infection
Our results implicated environmental chitin as a driver of lung disease severity and progression after epithelial injury. We therefore tested whether reducing chitin in the mouse cage environment could impact immune and epithelial recovery after IAV. Because standard mouse housing conditions contain natural sources of chitin (e.g., food and bedding) (fig. S3) (9) and are subject to airborne particulate contamination, we reduced environmental chitin by (i) housing mice in sealed positive pressure cages that received double HEPA-filtered air, (ii) using a low dust–generating cellulose-based bedding substrate, and (iii) placing moistened food into feeders located on the cage floor, thereby minimizing inhalation of food dust particles. WT and CC mice were placed in either standard or “low-chitin” caging conditions, acclimated for 1 week, then intranasally inoculated with IAV (Fig. 4A). Both WT and CC mice housed in chitin-reduced caging conditions maintained body weight (Fig. 4B–C) and exhibited less airway chitin accumulation (Fig. 4, D and E), and improved epithelial recovery after IAV inoculation compared to mice housed in standard conditions (Fig. 4, F and G). Furthermore, the enhanced lung ILC2 and eosinophil accumulation observed in IAV-infected CC mice housed in standard conditions was ameliorated by reducing environmental chitin (Fig. 4, H and I), implicating chitin as a common environmental driver of type 2 innate immune triggering after respiratory viral infection.
Fig. 4. Environmental chitin and chitinase activity control ILC2 responses and recovery after IAV infection.

(A) WT and CC mice were acclimated to standard or low-chitin housing conditions prior to intranasal inoculation with 250 PFU IAV followed by analysis at 14 days post-infection (dpi). [Part of the illustration was created with Biorender.com]. (B) Body weights of IAV-infected WT or (C) CC mice housed in standard or low chitin conditions. (D) Representative chitin blot and (E) quantification of BAL chitin, (F) total AT2s, (G) AEPs, (H) lung ILC2s, and (I) eosinophils in WT and CC mice housed as indicated (low chitin: n > 9 mice per group; standard: n = 3–15 mice per group). (J) Representative image and (K) quantification of AMCase (protein immunoblot) and total protein (Coomassie blue stain) in BAL from WT and SPAM Tg mice at 14 dpi (n = 3–6 mice per group). (L) BAL chitinase activity, (M) BAL chitin, (N) body weights (n = 3–6 mice per group), (O) survival, (P) BAL protein, and (Q) total AT2s in WT and SPAM transgenic (Tg) mice at 14 dpi. (K to M and P) n = 4–8 mice, N = 3 experiments; Q, n = 9–18, 3 experiments). R.U.: relative units. P values were calculated by unpaired t test (K-M, P-Q), two-way ANOVA with multiple comparisons using Holm–Šídák method (B and C, E-I), or (O) log-rank (Mantel–Cox) test (WT IAV, n = 32 mice; Spam Tg IAV, n = 26 mice, >5 experiments). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Since AMCase-expressing cells were lost and chitin accumulated in the lungs of IAV-infected WT mice housed in standard conditions, we also tested whether enhancing lung chitinase activity by transgene-mediated overexpression or chitinase therapy could improve epithelial regenerative responses in standard settings. We examined responses to IAV infection in WT mice expressing a SP-C promotor–driven AMCase transgene (SPAM Tg) (17). Compared to WT controls, SPAM Tg mice maintained higher BAL AMCase expression (Fig. 4, J and K) and chitinase activity during the recovery phase post-IAV infection (Fig. 4L), concordant with reduced BAL chitin accumulation (Fig. 4M) and improved weight gain during the epithelial recovery phase (Fig. 4N). Transgenic AMCase expression also improved survival (Fig. 4O) and lung epithelial function during the recovery phase after injury, as SPAM Tg mice displayed reduced BAL protein accumulation (Fig. 4P) and increased lung AT2 numbers compared to WT controls (Fig. 4Q). In addition, therapeutic administration of recombinant AMCase to WT mice was sufficient to boost BAL chitinase activity (fig. S10A), reduce chitin accumulation (fig. S10B), and improve AT2 recovery compared with heat-inactivated control AMCase (fig. S10C). Thus, enhancing chitin degradation promotes epithelial regeneration and barrier restoration after injury.
Epithelial regeneration is modulated by chitin and chitinase activity
Epithelial cells initiate immune triggering in response to chitin (11, 12, 42), and engage cellular stress pathways in the homeostatic absence of AMCase (9), suggesting that injury-related chitin accumulation might also influence epithelial regeneration and repair. To assess this, we performed RNAseq on lung epithelial cells isolated from CC and WT mice in the steady state and during the recovery phase (14 dpi) after IAV infection. AEPs from WT mice induced several genes (FDR-adjusted P < 0.05) and gene pathways involved in epithelial proliferation and differentiation after infection (Fig. 5A and fig. S11A), consistent with the capacity of AEPs to expand and regenerate mature alveolar cells during the recovery phase post-IAV infection (24). Regenerating WT AEPs also induced expression of mature alveolar type 1 (AT1) and AT2 signature genes. By contrast however, AEPs from infected CC mice failed to induce epithelial maturation markers and proliferation and differentiation gene pathways were suppressed compared to WT controls (Fig. 5, B and C), consistent with reduced epithelial cell numbers in CC mice (Fig. 2, I to K). Thus, epithelial regeneration is impaired in the absence of AMCase by environmental chitin.
Fig. 5. Epithelial regeneration is modulated by chitin and chitinase activity.

(A) RNA-seq analysis comparing differentially expressed genes (log2FC P < 0.05) in AEPs sorted from the lungs of naïve versus IAV-infected (14 dpi) WT mice and (B) downregulated genes in AEPs from IAV-infected CC versus WT mice. (C) Relative expression of AT1 and AT2 marker genes in WT and CC AEPs, shown as log2FC of IAV-infected (14 dpi) as compared to genotype-matched AEPs from naïve mice. (D) Experimental approach for AEP sorting after chitin challenge. (E) AT1 and AT2 marker gene expression and (F) downregulated gene ontology (GO) pathways in AEPs from chitin- versus PBS-treated WT mice. (G) Average expression of Egfr mRNA and (H) EGFR+ AT2s and AEPs in the lungs of naïve WT mice. (I) Average expression of Egfr in sorted AEPs from WT and CC mice following intranasal PBS or chitin administration. (J) EGFR-regulated genes in CC versus WT AEPs after chitin challenge. (K) Downregulated GO pathways in naïve AEPs sorted from the lungs of CC versus WT mice. RNA-seq expression data represent n ≥ 3 mice per group and were filtered with Benjamini–Hochberg false-discovery rate adjusted P values ≤ 0.05. P values were calculated by unpaired t test (H), *P < 0.05; **P < 0.01.
To determine whether chitin directly alters epithelial transcriptional programs, we performed RNAseq analysis on AEPs isolated from the lungs of WT and CC mice 12 hours after intranasal instillation of PBS or chitin (Fig. 5D) (11, 12). In contrast to IAV-induced injury, chitin did not alter lung epithelial cell numbers or the ratio of AEP to AT2 (fig. S11, B to D). However, proliferation and differentiation gene pathways, along with the expression of AT1 and AT2 marker genes, were suppressed in AEPs from chitin-treated mice versus PBS-treated controls (Fig. 5, E and F), indicating that chitin exerts direct effects on regenerative gene expression in AEPs, even in the absence of infectious injury. Indeed, AEP gene pathways affected by chitin overlapped with proliferation and differentiation programs that were blunted in CC versus WT AEPs after IAV injury (fig. S11E).
Lung epithelial proliferation and differentiation can be promoted by epidermal growth factor receptor (EGFR) signaling after injury (43–45), and AEPs were relatively enriched in Egfr expression compared to AT2s (Fig. 5, G and H), suggesting enhanced responsiveness to the EGFR ligand AREG. Furthermore, Egfr mRNA was significantly lower among CC AEPs compared to controls in the steady-state and was further diminished by chitin exposure (Fig. 5I). Accordingly, EGFR-regulated genes (46) were reduced in CC versus WT AEPs in the steady-state and after challenge with chitin (Fig. 5J and fig. S11F), along with alterations in proliferative pathways such as DNA replication and mitotic cell cycle processes (Fig. 5K). Compared to WT, AEPs in CC mice are therefore relatively unresponsive to AREG signaling. This suggests the presence of a negative feedback loop that may prevent uncontrolled proliferation and may account for the failure of AEPs to differentiate in the presence of exacerbated ILC2 responses in CC mice (Fig. 2 and fig. S7). Thus, AMCase plays a role in establishing the lung immune and epithelial set-point upon environmental chitin conditioning, which is crucial for healthy recovery after injury.
ILC2s promote epithelial regeneration by restoring AT2 AMCase expression
These data prompted us to further test the involvement of ILC2s on AT2 restoration after injury. Although IL-13 is produced by ILC2s in response to IAV injury, mice lacking IL-4Rα, the common receptor chain for IL-4 and IL-13, showed no differences in morbidity or mortality, chitinase activity, or chitin accumulation (fig. S12, A to D) compared to WT mice after IAV infection. Total epithelial cell recovery and the AEP:AT2 ratio in Il4ra−/− mice were also unaltered compared to controls (fig. S11, E to G), indicating that IL-4 and IL-13 signaling is dispensable for chitinase restoration and epithelial recovery after IAV-induced injury. We then further explored the contribution of AREG, which is also produced by ILC2s to influence epithelial recovery after viral injury (36). Consistent with this, recombinant AREG administration during the recovery phase induced AEP AMCase expression, increased BAL chitinase activity, reduced chitin accumulation and tissue inflammation, and enhanced AT2 recovery in the lungs of IAV-infected WT mice (Fig. 6, A to H). By contrast, AEP AMCase expression, AT2 maturation, and mouse weight recovery after IAV infection were reduced and lung inflammation was enhanced by the administration of AREG-blocking antibodies (Fig. 6, I to M). Moreover, administration of the ILC2-activating cytokines TSLP and IL-33 during the recovery phase post-IAV significantly increased ILC2s numbers, AREG production, AEP AMCase expression, BAL chitinase activity, and AT2 numbers (Fig. 6, N to R). Thus, the modulation of ILC2s and ILC2-derived AREG appears to influence epithelial regeneration and AMCase restoration after viral injury.
Fig. 6. Amphiregulin promotes AT2 maturation and chitinase expression following injury.

(A) Recombinant amphiregulin (AREG) was administered intranasally to IAV-infected mice every other day from 10 to 21 dpi. (B) ChiaRed (CR)-expressing AEPs and CR MFI after PBS or AREG administration to mock- or IAV-infected (21 dpi) mice (n = 3–7 mice per group, N = 3 experiments). (C) BAL chitinase activity, (D) BAL chitin, (E) AT2s, (F) AEP:AT2 frequency (as a percentage of total EpCAM+ lung epithelial cells; n = 3–7 mice per group, N = 3 experiments), (G) representative H&E-stained lung sections, and (H) quantification of inflamed area in the lungs of IAV-infected mice treated with PBS or AREG at 21 dpi (n = 3–5 mice per group, N = 2 experiments). (I to M) IAV-infected WT mice received intranasal anti-AREG or isotype control antibodies every other day from 10 to 21 dpi. (I) MFI of CR-expressing AEPs, (J) AT2s, (K) mouse body weights, (L) representative H&E-stained lung sections, and (M) quantification of the inflamed area after anti-AREG or isotype administration (n = 3–6 mice per group, N = 3 experiments, 21 dpi). (N) Lung ILC2s, (O) BAL AREG, (P) CR MFI among AEPs, (Q) BAL chitinase activity, and (R) AT2s in IAV-infected mice that were treated with PBS or IL33+TSLP between 10 and 16 dpi (n = 5–9 mice per group, N = 2 experiments). Bold yellow lines in (G and L) indicate inflamed areas. Scale bars: 1 mm. R.U.: relative units. Data represent individual biological replicates and are presented as mean±SE. P values were calculated by (K) paired t test and (B and O) one-way ANOVA with Sidak’s multiple comparison test. All other P values were calculated by unpaired t test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To determine the contribution of ILC2s to the restoration of chitinase activity after epithelial injury, we examined responses to IAV infection in ILC2-deleter (Il5tm1.1(icre)Lky × Gt(Rosa)26Sortm1(DTA)Lky/J) (38) and control R5-Cre (Il5tm1.1(icre)Lky) mice. We verified that R5-Cre–mediated ILC2 deletion eliminated lung ILC2s before and after IAV infection (fig. S13, A and B). Compared to R5-Cre controls, IAV-infected ILC2-deleter mice exhibited increased morbidity and mortality, particularly during the epithelial recovery phase (Fig. 7A), consistent with prior observations employing different ILC2 depletion strategies (36, 37) and confirming a crucial role for ILC2s in restoring lung health after viral infection. Notably, BAL from surviving ILC2-deleter mice showed elevated chitin amounts and exacerbated protein accumulation compared to controls (Fig. 7B and fig. S13C), along with reduced AREG (fig. S13D), corroborating previous findings demonstrating diminished AREG in ILC2-depleted mice after injury (36). The AEP-to-AT2 transition was concomitantly blunted in the lungs of ILC2-deleter mice, along with skewed AEP:AT2 ratios (Fig. 7, C and D). ILC2-deleter mice also exhibited markedly reduced BAL AMCase and chitinase activity compared to control mice, and chitinase activity was additionally reduced in triple knockout (TKO) mice that lack the major ILC2-activating components IL-25, IL-33R, and TSLPR (11, 38) (Fig. 7E and fig. S13E). Moreover, AEPs from ILC2-deleter mice failed to induce Chia1 transcript (Fig. 7F), and total AT2 recovery was significantly diminished in comparison with controls during the recovery phase after infection (Fig. 7G), suggesting that ILC2 deficiency results in an inability to clear chitin due to impaired AT2 maturation and AMCase restoration after injury.
Fig. 7. ILC2s control epithelial regeneration by restoring AT2 AMCase expression.

(A to F) Mice were intranasally infected with 250 PFU of IAV and analyzed for (A) survival (WT IAV, n = 13 mice; ILC2-deleter IAV, n = 19 mice), or (B to F) euthanized and analyzed at 14 days post infection (dpi). (B) BAL protein (n = 6 mice per group, N = 3 experiments), (C) representative flow cytometry and (D) frequency of AEP and AT2 cells (n = 8–15 mice, N = 3 experiments). R.U.: relative units. (E) BAL chitinase activity in R5, ILC2-deleter mice and TKO mice after IAV infection (n = 4–8 mice, N > 2 experiments). (F) Quantitative real-time PCR (qPCR) analysis of Chia1 transcript abundance in AEPs after IAV infection (n = 3–5 mice per group). (G) Total AT2 cells (n = 4–6 mice, representative of N = 3 experiments in R5 and ILC2-deleter mice after 21 dpi. (H) Dosing regimen, (I) body weights, (J) survival (ILC2-deleter IAV-AMCase, n = 22 mice; ILC2-deleter IAV-hiAMCase, n = 12 mice, N > 3 experiments) (K) BAL chitinase activity (n = 4–7 mice, N = 3 experiments) (L), BAL chitin (n = 9–10 mice, N = 3 experiments), (M) BAL total protein (n = 6–8 mice, N = 3 experiments), and (N) AT2 numbers (n = 7–10, N = 3 experiments) in ILC2-deleter mice at 21 dpi or as indicated after IAV infection and intranasal administration of active AMCase or hiAMCase. Arrows in (I and J) represent active AMCase or hi-AMCase intranasal doses. Data represent individual biological replicates and are presented as mean±SE. P values were calculated by (A and J) log-rank (Mantel–Cox) test, (B, D, F, G, I, and K to N) unpaired t test, or (E) one-way ANOVA with Holm–Šídák method. *P < 0.05; **P < 0.01; ***P < 0.001.
These results suggested that the induction of AMCase crucially mediates the ability of ILC2s to restore epithelial function and lung health after viral injury. In support of this idea, the requirement for ILC2s in AMCase restoration could be bypassed via exogenous administration of AREG (fig. S13F), which was sufficient to restore AEP Chia1 expression and BAL chitinase activity in IAV-infected ILC2-deleter mice (fig. S13, G and H). Furthermore, mice administered active AMCase, compared with a heat-inactivated AMCase control, improved body weight recovery (Fig. 7H) as well as survival among IAV-infected ILC2-deficient mice (Fig. 7, H to J). These improvements were accompanied by increased BAL chitinase activity, reduced BAL chitin and protein accumulation, and significantly improved AT2 cell restoration during the recovery phase in AMCase-treated ILC2-deleter mice compared to controls (Fig. 7, K to N). Thus, chitinase replacement therapy compensates for the loss of ILC2s to promote lung epithelial regeneration after injury, indicating that ILC2s are an essential component of the homeostatic circuit that is engaged to restore chitinase activity after perturbation.
DISCUSSION
Our data demonstrate that a key determinant of lung repair and regeneration after epithelial injury is the ubiquitous environmental polysaccharide chitin, which accumulates in AMCase-depleted lung niches after various forms of epithelial damage. We show that chitin is prevalent in standard, specific-pathogen-free mouse housing conditions and influences the severity and persistence of lung disease in commonly employed animal models of respiratory viral infection and pulmonary fibrosis. Following acute damage, chitin triggers innate immune and epithelial responses that facilitate its own enzymatic digestion by an endogenous glycosyl hydrolase, AMCase. This sequence comprises a host–environmental circuit that drives divergent lung disease outcomes in a chitin- and chitinase-dependent manner. This adaptation to chitin is coordinated by ILC2s, a class of sentinel tissue-resident cells that maintain barrier integrity in several tissues including lung (36), gastrointestinal tract (13, 43, 47), and skin (48), suggesting that an essential homeostatic function of ILC2s is to impart mucosal barrier cells with specificity for a widespread environmental substrate.
We found that homeostatic chitinase function is compromised by severe respiratory viral infection and fibrotic injury, which impacts chitin-mediated inflammatory triggering and epithelial regeneration. Although we show that transient AT2 depletion is sufficient to cause loss of AMCase activity and chitin accumulation, other routine particle clearance mechanisms including mucociliary function and alveolar macrophage phagocytosis are also impaired in disease settings (34, 35, 49, 50), implying that a lack of chitinase and possibly other AT2-derived proteins (e.g., surfactant proteins) is compounded by an overall reduced capacity to maintain the integrity of the airspaces. In humans, high concentrations of airborne particulate matter increase the risk for severe respiratory viral infection, pulmonary disease, and mortality, including COVID-19–related lung epithelial alterations that resemble age-related fibrosis (1, 2, 5, 28, 29). Severe COVID-19 lung pathology is marked by cell types and cytokines that are induced by chitin exposure (10, 11, 14, 16, 17, 30, 51). In addition, we previously showed that chitin accumulates in the airways of humans with pulmonary fibrosis, as well as in aging mice that genetically lack AMCase (9). These findings implicate chitin as a common environmental driver of lung disease severity even among younger, WT mice, induced by both infectious and non-infectious stimuli that cause AT2 cell injury, which impairs critical epithelial-derived chitinase effector function. As we show, AMCase-deficient mice that are unable to restore chitinase activity exhibit severe lung disease and mortality following AT2 injury.
Our results further identify chitin as an environmental trigger of lung ILC2 activation following respiratory viral infection. Prior studies have established that ILC2s contribute to epithelial repair after IAV (36). However, the identity of specific environmental factors that contribute to sustained type 2 immune triggering in post-viral lung disease has remained unclear. We show that environmental chitin and lung chitinase activity influence both the lung immune and epithelial cellular landscape in the context of infectious disease, revealing a host–environmental interaction that determines respiratory disease outcomes. After lung epithelial injury, chitin triggers ILC2s to restore homeostasis by inducing AMCase in regenerating epithelial cells, thereby equipping mature AT2s with chitin-degrading capacity required for tissue restoration and resilience. We further demonstrate that this circuit is partially mediated by AREG after IAV. However, this growth factor likely synergizes with a complex network that integrates interstitial macrophages and fibroblasts—among other cellular and growth factor components—to ultimately resolve tissue injury.
The lack of AMCase led to the development of a non-resolving, progressive lung fibrosis after bleomycin-induced injury, suggesting that chitin accumulation overrides normal mechanisms of epithelial restitution in disparate contexts. Both AEP and ILC2 compartments are consequently altered in the absence of AMCase, both in the steady-state and after IAV infection, consistent with low-level homeostatic “conditioning” of ILC2s and AEPs by environmental chitin that subsequently accumulates and impairs recovery following injury. Accordingly, although lung ILC2s from CC mice exhibit increased responsiveness and AREG production in response to stimuli, these signals are coupled with AEPs that express diminished EGFR and associated growth factor components and are therefore relatively unresponsive to differentiation signals due to the suppressive effects of environmental chitin. This conditioning perhaps reflects a negative feedback loop that prevents uncontrolled AEP proliferation in the presence of exacerbated ILC2 responses. Nevertheless, in the absence of AMCase in the setting of epithelial injury, chitin provokes chronic ILC2 triggering and impairment of epithelial regeneration, resembling epithelial alterations that occur in chronic lung disease, pulmonary fibrosis, rhinosinusitis, and ILC2–stem cell interactions during hair follicle cycling (4, 24, 52, 53). Our results suggest this ILC2–AMCase circuit is broadly involved in shaping responses to epithelial dysfunction and inflammation. Furthermore, it may represent a general feature of insoluble particle–driven stromal alterations and recurrent injury, which may ultimately produce long-lasting alterations in lung architecture and function.
Both ILC2 triggering and lung epithelial impairments can be ameliorated by reducing environmental chitin or by boosting airway chitinase activity, implicating the ILC2–chitinase circuit as a critical determinant of lung health post-injury. Additionally, we demonstrate that the requirement for ILC2s in restoring epithelial health after IAV infection can be bypassed by exogenous chitinase therapy, showing that a primary function of ILC2s in mucosal barrier restoration is to coordinate chitinase responses to chitin, analogous to their essential role in driving dietary chitin adaptations in the gastrointestinal tract (13). In humans, CHIA gene variants encoding AMCase enzyme isoforms with increased chitinase activity show protective associations among asthma cohorts (54, 55), suggesting that robust enzymatic activity is crucial to degrading chitin-containing stimuli that contribute to chronic airway disease. Here, we show that both active mammalian endochitinases, AMCase and CHIT1, exhibit efficacy in therapeutic targeting of environmental chitin following lung epithelial injury. AMCase and CHIT1 differ with regard to expression patterns, substrate preferences, and pH-dependent activities (56–58) suggesting that engineered enzyme variants might be tailored to improve degradation of naturally occurring substrates in a tissue-specific manner. In conclusion, this work demonstrates that boosting chitinase activity to reduce airway chitin improves lung health following injury, suggesting additional therapeutic avenues for restoring barrier integrity in a wide range of diseases involving epithelial damage.
MATERIALS AND METHODS
Mice
For all experiments, age- and sex-matched mice on C57BL/6J backgrounds were used between 6–12 weeks of age and maintained under specific pathogen–free conditions. ChiaRed knockin/knockout reporter (CR; Chia1ChiaRed) mice (9) were bred with Rosa26-flox-stop-zsGreen (Gt(Rosa)26Sortm6(CAG-ZsGreen1)Hze/J; 007906; The Jackson Laboratory) mice to generate AMCase-zsGreen (CR-zsG) reporter / lineage tracer mice, and additionally crossed with Smart13 (Il13hCD4) (59) reporter mice as indicated. WT SPAM Tg, CC SPAM Tg, TKO (IL-25−/−, IL-33R−/−, and TSLPR−/−), Red5 (Il5Red5), Yarg (Arg1Yarg), and ILC2-deleter (Il5Red5 × Gt(Rosa)26Sortm1(DTA)Lky/J) mice were bred and maintained as previously described (9, 11, 38). Sftpctm1(cre/ERT2,rtTA)Hap mice, kindly provided by Dr. H. Chapman (UCSF), were bred with Gt(Rosa)26Sortm1(DTA)Lky/J (009669; Jackson) mice to generate AT2-deleter mice. Whole-body Il4ra−/− mice were generated by crossing CMV-cre (006054; Jackson) with Il4rafl/fl mice (60), kindly provided by Dr. F. Brombacher (University of Cape Town). All procedures were approved by the Washington University School of Medicine in St. Louis Institutional Animal Care and Use Committee (Assurance #D16–00245).
In vivo treatments
Mice were anesthetized with isoflurane and intranasally inoculated with IAV [A/Puerto Rico/8/1934(H1N1)], which was kindly provided by Dr. A. Boon, Washington University School of Medicine), or mock-infected with phosphate buffered saline (PBS) (30 μl). Transient transduction of mice with AdV-hACE2 and inoculation with 105 FFU of SARS-CoV-2 strain 2019 n-CoV/USA_WA1/2020 was performed as described (39). Where indicated, mice were intranasally inoculated with 106 FFU of SARS-CoV-2 WA1/2020 D614G, B.1.351 in 50 μl PBS as described (61). IAV titers were determined by plaque assay on Madin–Darby canine kidney cells. After all challenges, mice were monitored and weighed daily, and humanely euthanized if 30% loss of initial weight occurred. Recombinant mouse AMCase (generated as previously described (62)), chitotriosidase (CHIT1; R&D Systems), or matched control aliquots that were heat-inactivated (5 min, 95°C), were intranasally administered at a dose of 2 μg in 50 μl PBS every 2 days between 10–21 dpi after IAV inoculation. Recombinant mouse TSLP (10 ng) and IL-33 (500 ng; R&D Systems) were administered intranasally to mice in 50 μl of PBS for 3 consecutive days prior to euthanasia and tissue collection. Recombinant mouse amphiregulin (5 μg) (R&D Systems), anti-amphiregulin (2 μg) (R&D Systems), and IgG isotype control (2 μg) (R&D Systems) were administered intranasally to mice in 50 μl PBS for every 2 days between 10–21 dpi after IAV inoculation prior to euthanasia and tissue collection. Bleomycin (2 mg per kilogram of body weight) (Cayman) was resuspended in PBS and administered to mice intranasally once at start of experiment prior to euthanasia and tissue collection. Tamoxifen (2 μg) (Sigma-Aldrich) was resuspended in 1 ml of peanut oil and administered to mice intraperitoneally twice a week, every 3 days prior to euthanasia and tissue collection. Serum was collected from supernatants of non-heparinized blood that was allowed to clot at room temperature for 1 hour. For bronchoalveolar lavage (BAL), sterile PBS (1 ml) was intratracheally instilled and centrifuged for 10 min at 3000g before collecting supernatants for biochemical assays. BAL pellets (BALp) were boiled for 5 min to lyse cellular material and denature proteins before assaying for chitin content as described (9).
Standard and reduced chitin housing conditions
For standard conditions, mice were housed in filter-top cages receiving room air 1 week prior to placement in negative pressure cages that received double HEPA–filtered air (PNC IVC rack system; Allentown) for IAV infections. Standard cages contained autoclaved corn cob bedding (Andersons) with irradiated PicoLab 5053 Rodent Diet (LabDiet) placed in hoppers located at the top of the cage (as shown in Fig. 2). For low-chitin conditions, mice were housed in sealed positive pressure cages that received double HEPA–filtered air (Sentry SPP rack system; Allentown) and lined with ALPHA-dri PLUS bedding (Shepherd Specialty Papers), a low dust–generating bedding substrate comprised of virgin paper pulp cellulose squares. In addition, PicoLab 5053 Rodent Diet (identical to that used in standard conditions) was moistened and placed into feeders located on the floor of the cage to minimize the inhalation of food dust particles generated during feeding.
Tissue preparation and flow cytometry
Mice were euthanized, and lung immune cells were enumerated by flow cytometry as described (11). Lung epithelial cells were prepared as described (9), with minor modifications. Briefly, lungs were perfused with 10 ml of PBS through the right ventricle of the heart before intratracheal instillation of 2 ml of dispase II (5 mg/ml) (ThermoFisher) and 1 ml of 0.1% low-melting point agarose, followed by cooling with ice in situ for 1 min. Lung lobes were then dissected and incubated in 2 ml of dispase II (5 mg/ml) (ThermoFisher) for 45 min at room temperature, transferred to C tubes (Miltenyi) containing 5 ml of DNase I (50 μg/ml) (Sigma), and dispersed into single-cell suspensions using a gentleMACS automated tissue dissociator (Miltenyi). Cells were passed through 100-μm nylon filters, washed, incubated with red blood cell lysis buffer (BioLegend), and resuspended in PBS with 2% fetal bovine serum prior to Fc blocking with anti-mouse CD16/CD32 (BioXCell) and cell staining.
After Fc blocking, cells were stained using antibodies listed in table S1. Live cells were identified by exclusion of DAPI (4′,6-diamidine-2′-phenylindole dihydrochloride) (Roche) and enumerated with Precision Count Beads (BioLegend). Flow cytometry data acquisition and analysis was performed with a BD FACSymphony A3 flow cytometer and FlowJo software (BD Biosciences), and cells were sorted using a MoFlo XDP (Beckman Coulter).
Biochemical and protein assays
Chitinase activity was measured in BAL supernatants using 4-methylumbelliferyl-N,N′-diacetyl-β-D-chitobioside (Sigma) substrate as described (9) for 30 min at 37°C. Chitin was quantified by dot-blot assay as described (9), with minor modifications. Briefly, 1 μl of BALp and sonicated chitin (Sigma) controls were pipetted onto 0.2-μm nitrocellulose membranes, dried overnight, rinsed in Tris-buffered saline containing 0.05% Tween 20 (TBST), blocked for 30 min with 5% bovine serum albumin (BSA) in TBST, and incubated with enhanced GFP-labeled chitin binding probe (eGFP-CBP) (48) in 1% BSA/TBST overnight at 4° C with gentle agitation. After washing with TBST, the membranes were incubated with biotinylated anti-GFP antibody (13-6498-82), followed by streptavidin–HRP (RABHRP-3) (Invitrogen) (Table S1) for 45 min at room temperature, washed, and developed with ECL substrate (170–5060; BioLegend), before image acquisition on a ChemiDoc MP (BioRad). For protein and AMCase quantification, equal volumes of BAL supernatant were diluted in 4X Laemmli SDS-PAGE buffer containing 2-mercaptoethanol (Bio-Rad) and incubated at 95°C for 5 min. Samples were then electrophoresed on 4 to 15% TGX protein gels (Bio-Rad), transferred to 0.2-μm nitrocellulose membranes, blocked with 5% milk, and probed with anti-AMCase (207169; Abcam) followed by goat anti-rabbit IgG (H+L)-HRP (4049–05; SouthernBiotech) (table S1). Total BAL protein quantities were expressed as relative units minus AMCase signal, unless specified otherwise. IL-13 (88-7137-22; ThermoFisher), IL-33 (DY362605; R&D), and IL-5 (431204; BioLegend) were quantified by ELISA. Hydroxyproline was determined as previously described (9), by homogenizing snap-frozen whole lung tissue prior to hydrolysis in concentrated hydrochloric acid for 3 hours at 120°C, followed by drying and incubation with 4-(dimethylamino)benzaldehyde as recommended by the manufacturer (Sigma). Absorbance was measured at 560 nm using a Synergy H1 Plate Reader (BioTek).
Histology and immunofluorescence microscopy
Formaldehyde-fixed, paraffin-embedded mouse lung tissues sections (5 μm) were stained with hematoxylin and eosin (H&E) or Masson’s trichrome by standard methods. Cryosections (7 μm) were prepared from paraformaldehyde-fixed, OCT (Sakura)-embedded frozen lung tissue from indicated fluorescent reporter mice. For immunofluorescence imaging, OCT-embedded lung tissue sections were washed three times in PBS at room temperature for at least 5 min, blocked with blocking solution (1:100 diluted goat serum (ThermoFisher; 50197Z) and 2% BSA in PBS) for 2 hours at room temperature, followed by incubation for 24 hours at 4°C with primary antibody as listed in table S1, in blocking solution. After washing three times in PBS, samples were stained for 1 hour at room temperature with a secondary antibody diluted in blocking solution. Nuclei were stained with DAPI and mounted with Fluoromount-G™ Mounting Medium (ThermoFisher; 00-4958-02) before acquiring images with an AxioImager Z2 microscope or Axio Scan Z1 slide scanner (Carl Zeiss) at the Washington University Center for Cellular Imaging. An Orca Flash sCMOS camera (Hamamatsu) with LED illumination source and filter sets for DAPI, GFP, and RFP imaging were used to acquire fluorescence images at 20X magnification. A color CCD camera (Carl Zeiss) was used to acquire H&E images at 20X magnification. Image analysis of H&E and immunofluorescence slides was performed using QuPath software (version 0.2.3) (63). Briefly, tissue was detected by thresholding a downsampled and smoothed image of the core and individual cells were identified by separating stains using color deconvolution and identifying peaks in each channel (or sum of the hematoxylin channels) after smoothing and assigning these as positive or negative cells (64). The percentage of positive cells per area was calculated and results were exported along with markup images showing the detected cells, for visual verification.
RNA sequencing
Lung alveolar epithelial type 2 cells (AT2s) (DAPI−CD45−EpCAM+MHCIIhi) and alveolar epithelial progenitors (AEPs) (DAPI−CD45−EpCAM+MHCIIlo) were sorted as described above and prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovoSeq 6000 (Illumina). Gene counts were imported into the R/Bioconductor package EdgeR (65), normalized for library size after calculating trimmed mean of M-values (TMM). Ribosomal genes and genes not expressed in the smallest group size minus one samples greater than one count per million were excluded, TMM size factors and count matrices were imported into Limma (66), and weighted likelihoods based on mean-variance relationships were calculated with voomWithQualityWeights (67). Differential expression results were filtered for Benjamini–Hochberg false-discovery rate–adjusted P values of 0.05 or less. For each contrast identified using Limma, global perturbations in established Gene Ontology (GO) terms and KEGG pathways were identified using the GAGE package from R/Bioconductor (68). This package tested for expression changes in log2-fold changes reported by Limma in each term, compared to the background log2-fold changes of genes not included in that term. Heatmaps for each GO term with a Benjamini–Hochberg false-discovery rate–adjusted P value of 0.05 or lower were generated using the R/Bioconductor package heatmap3 (69) to visualize differences across groups of samples.
Single-cell RNA sequencing (scRNAseq) was performed as described (70) on WT (DAPI−CD45−EpCAM+) or CR+ (DAPI−CD45−EpCAM+CR+) epithelial cells, sorted into ice-cold 0.5% BSA in PBS and processed through the Chromium Single Cell 3′ v2 Library Kit (10X Genomics) per the manufacturer’s protocol. Single-cell libraries from 10,000 cells per sample were sequenced with standard Illumina sequencing primers on an Illumina HiSeq 4000, using paired-end sequencing with single indexing, in which read 1 comprised 26 cycles and read 2 comprised 98 cycles. The resulting bcl files were demultiplexed using bcl2fastq2.1.7v, and the resultant paired-end fastq files were aligned to the mm10 transcriptome (71, 72) using STAR aligner in the Cellranger toolkit (10X Genomics). Lung cell plots shown in fig. S1, A to C were generated from scRNAseq data (GEO accession number: GSE149563) (21) accessed via the LungMAP Consortium and Data Coordinating Center (73).
Quantitative RT-PCR
Sorted AEPs (CD45−EpCAM+MHCIIlo) were lysed in RLT Plus. Total RNA was then eluted from RNeasy Plus Micro columns (Qiagen). Total RNA was reverse-transcribed with SuperScript IV VILO, then quantitative real-time PCR using Power SYBR Green (Thermo) was run on a CFX Connect Real-Time System (BioRad). Transcripts were normalized to 18S ribosomal gene, and relative gene expression was determined by comparative ΔCT (2−ΔΔCT) method. Primer pair sequences (listed 5′ to 3′): Chia1: (F) TACCAGACAGGCTGGGTTCT; (R) GGAGTAGTCACTGGCTCGGA. 18S: (F) TTACAGGGCCTCGAAAAGAGTC; (R) AACTTTGGCATTGTGGAAGG.
Statistical analysis
Data represent mean ± SE, and results from independent experiments were pooled unless otherwise indicated. P values were calculated by unpaired two-tailed Student’s t test, multiple t test, or one-way or two-way ANOVA as indicated using Prism software (GraphPad).
Supplementary Material
Acknowledgements:
We thank A. Boon and M. Holtzman for advice and reagents, E. Penrose for technical assistance, E. Wan at the UCSF Institute for Human Genetics for assistance with RNAseq, and K. Ravichandran for comments on the manuscript. We thank the Pulmonary Morphology Core for lung histology, and the Genome Technology Access Center at the McDonnell Genome Institute at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1TR002345 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH.
Funding:
National Institutes of Health grant R01HL148033 (S.J.V.D.)
National Institutes of Health grant R01AI176660 (S.J.V.D.)
National Institutes of Health grant R21AI163640 (S.J.V.D.)
National Institutes of Health grant R01AI026918 (R.M.L.)
Bursky Center for Human Immunology and Immunotherapy Programs, Center for Cellular Imaging, Rheumatic Diseases Research Resource-based Center (NIH P30 AR073752) at Washington University in St. Louis
Howard Hughes Medical Institute (R.M.L.)
Competing interests:
S.J.V.D. and R.M.L. are listed as inventors on a patent for the use of chitinases to treat fibrotic lung disease and S.J.V.D, R.M.L., and J.A.F. are listed as inventors on a patent for mutant chitinases with enhanced expression and activity. M.S.D. is a consultant for Inbios, Vir Biotechnology, Ocugen, Topspin, Moderna, and Immunome. The Diamond laboratory has received unrelated funding support in sponsored research agreements from Vir Biotechnology, Emergent BioSolutions, and Moderna.
Data and materials availability:
Bulk RNA sequencing data generated in this study are deposited in Gene Expression Omnibus (GEO) under accession codes GSE275075 and GSE275319. Single-cell RNA sequencing data are available in Dryad (74). Tabulated data underlying Figs. 1 to 7; figs. S1, S3, and S4 to S13 are archived in data S1. Immunoblot scans are archived in data S2. All other data are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.Di Q, Wang Y, Zanobetti A, Wang Y, Koutrakis P, Choirat C, Dominici F, Schwartz JD, Air Pollution and Mortality in the Medicare Population. N. Engl. J. Med 376, 2513–2522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, Balakrishnan K, Brunekreef B, Dandona L, Dandona R, Feigin V, Freedman G, Hubbell B, Jobling A, Kan H, Knibbs L, Liu Y, Martin R, Morawska L, Pope CA 3rd, Shin H, Straif K, Shaddick G, Thomas M, van Dingenen R, van Donkelaar A, Vos T, Murray CJL, Forouzanfar MH, Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 389, 1907–1918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horne BD, Joy EA, Hofmann MG, Gesteland PH, Cannon JB, Lefler JS, Blagev DP, Korgenski EK, Torosyan N, Hansen GI, Kartchner D, Pope CA 3rd, Short-Term Elevation of Fine Particulate Matter Air Pollution and Acute Lower Respiratory Infection. Am. J. Respir. Crit. Care Med 198, 759–766 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, Windmueller R, Ysasi AB, Zacharias WJ, Chapman HA, Kotton DN, Rock JR, Snoeck H-W, Vunjak-Novakovic G, Whitsett JA, Morrisey EE, The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present, and Future. Cell Stem Cell 26, 482–502 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao Y, Pan J, Liu Z, Meng X, Wang W, Kan H, Wang W, Temporal association between particulate matter pollution and case fatality rate of COVID-19 in Wuhan. Environ. Res 189, 109941 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevenson LA, Gergen PJ, Hoover DR, Rosenstreich D, Mannino DM, Matte TD, Sociodemographic correlates of indoor allergen sensitivity among United States children. J. Allergy Clin. Immunol 108, 747–752 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Van Dyken SJ, Garcia D, Porter P, Huang X, Quinlan PJ, Blanc PD, Corry DB, Locksley RM, Fungal chitin from asthma-associated home environments induces eosinophilic lung infiltration. J. Immunol 187, 2261–2267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salo PM, Arbes SJ Jr, Jaramillo R, Calatroni A, Weir CH, Sever ML, Hoppin JA, Rose KM, Liu AH, Gergen PJ, Mitchell HE, Zeldin DC, Prevalence of allergic sensitization in the United States: results from the National Health and Nutrition Examination Survey (NHANES) 2005–2006. J. Allergy Clin. Immunol 134, 350–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Dyken SJ, Liang H-E, Naikawadi RP, Woodruff PG, Wolters PJ, Erle DJ, Locksley RM, Spontaneous Chitin Accumulation in Airways and Age-Related Fibrotic Lung Disease. Cell 169, 497–509.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, Taki Y, Futatsugi-Yumikura S, Tsutsui H, Ishii KJ, Yoshimoto T, Akira S, Nakanishi K, Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. U. S. A 109, 3451–3456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, McKenzie ANJ, Krummel MF, Liang H-E, Locksley RM, Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and γδ T cells. Immunity 40, 414–424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang H-E, Locksley RM, Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol 9, 275–286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim D-H, Wang Y, Jung H, Field RL, Zhang X, Liu T-C, Ma C, Fraser JS, Brestoff JR, Van Dyken SJ, A type 2 immune circuit in the stomach controls mammalian adaptation to dietary chitin. Science 381, 1092–1098 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, Ellingson MK, Mao T, Oh JE, Israelow B, Takahashi T, Tokuyama M, Lu P, Venkataraman A, Park A, Mohanty S, Wang H, Wyllie AL, Vogels CBF, Earnest R, Lapidus S, Ott IM, Moore AJ, Muenker MC, Fournier JB, Campbell M, Odio CD, Casanovas-Massana A, Yale IMPACT Team, Herbst R, Shaw AC, Medzhitov R, Schulz WL, Grubaugh ND, Dela Cruz C, Farhadian S, Ko AI, Omer SB, Iwasaki A, Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Page C, Goicochea L, Matthews K, Zhang Y, Klover P, Holtzman MJ, Hennighausen L, Frieman M, Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J. Virol 86, 13334–13349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B, Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reese TA, Liang H-E, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM, Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature 447, 92–96 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR, Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacob A, Morley M, Hawkins F, McCauley KB, Jean JC, Heins H, Na C-L, Weaver TE, Vedaie M, Hurley K, Hinds A, Russo SJ, Kook S, Zacharias W, Ochs M, Traber K, Quinton LJ, Crane A, Davis BR, White FV, Wambach J, Whitsett JA, Cole FS, Morrisey EE, Guttentag SH, Beers MF, Kotton DN, Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 21, 472–488.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, Huang D, Xu Y, Huang W, Jiang M, Jiang X, Mao J, Chen Y, Lu C, Xie J, Fang Q, Wang Y, Yue R, Li T, Huang H, Orkin SH, Yuan G-C, Chen M, Guo G, Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Zepp JA, Morley MP, Loebel C, Kremp MM, Chaudhry FN, Basil MC, Leach JP, Liberti DC, Niethamer TK, Ying Y, Jayachandran S, Babu A, Zhou S, Frank DB, Burdick JA, Morrisey EE, Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 371, eabc3172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corti M, Brody AR, Harrison JH, Isolation and primary culture of murine alveolar type II cells. Am. J. Respir. Cell Mol. Biol 14, 309–315 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH, Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice. J. Clin. Invest 121, 2855–2862 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, Morrisey EE, Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, Ogar P, Sengupta A, Kukhtevich I, Schneider R, Zhao Z, Voss C, Stoeger T, Neumann JHL, Hilgendorff A, Behr J, O’Reilly M, Lehmann M, Burgstaller G, Königshoff M, Chapman HA, Theis FJ, Schiller HB, Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun 11, 3559 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi Y, Tata A, Konkimalla A, Katsura H, Lee RF, Ou J, Banovich NE, Kropski JA, Tata PR, Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol 22, 934–946 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi J, Park J-E, Tsagkogeorga G, Yanagita M, Koo B-K, Han N, Lee J-H, Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 27, 366–382.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, Abbondanza D, Fleming SJ, Subramanian A, Montoro DT, Jagadeesh KA, Dey KK, Sen P, Slyper M, Pita-Juárez YH, Phillips D, Biermann J, Bloom-Ackermann Z, Barkas N, Ganna A, Gomez J, Melms JC, Katsyv I, Normandin E, Naderi P, Popov YV, Raju SS, Niezen S, Tsai LT-Y, Siddle KJ, Sud M, Tran VM, Vellarikkal SK, Wang Y, Amir-Zilberstein L, Atri DS, Beechem J, Brook OR, Chen J, Divakar P, Dorceus P, Engreitz JM, Essene A, Fitzgerald DM, Fropf R, Gazal S, Gould J, Grzyb J, Harvey T, Hecht J, Hether T, Jané-Valbuena J, Leney-Greene M, Ma H, McCabe C, McLoughlin DE, Miller EM, Muus C, Niemi M, Padera R, Pan L, Pant D, Pe’er C, Pfiffner-Borges J, Pinto CJ, Plaisted J, Reeves J, Ross M, Rudy M, Rueckert EH, Siciliano M, Sturm A, Todres E, Waghray A, Warren S, Zhang S, Zollinger DR, Cosimi L, Gupta RM, Hacohen N, Hibshoosh H, Hide W, Price AL, Rajagopal J, Tata PR, Riedel S, Szabo G, Tickle TL, Ellinor PT, Hung D, Sabeti PC, Novak R, Rogers R, Ingber DE, Jiang ZG, Juric D, Babadi M, Farhi SL, Izar B, Stone JR, Vlachos IS, Solomon IH, Ashenberg O, Porter CBM, Li B, Shalek AK, Villani A-C, Rozenblatt-Rosen O, Regev A, COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595, 107–113 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, Katsyv I, Rendeiro AF, Amin AD, Schapiro D, Frangieh CJ, Luoma AM, Filliol A, Fang Y, Ravichandran H, Clausi MG, Alba GA, Rogava M, Chen SW, Ho P, Montoro DT, Kornberg AE, Han AS, Bakhoum MF, Anandasabapathy N, Suárez-Fariñas M, Bakhoum SF, Bram Y, Borczuk A, Guo XV, Lefkowitch JH, Marboe C, Lagana SM, Del Portillo A, Tsai EJ, Zorn E, Markowitz GS, Schwabe RF, Schwartz RE, Elemento O, Saqi A, Hibshoosh H, Que J, Izar B, A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dinnon KH 3rd, Leist SR, Okuda K, Dang H, Fritch EJ, Gully KL, De la Cruz G, Evangelista D, Asakura T, Gilmore RC, Hawkins P, Nakano S, West A, Schäfer A, Gralinski LE, Everman JL, Sajuthi SP, Zweigart MR, Dong S, McBride J, Cooley MR, Hines JB, Love MK, Groshong SD, VanSchoiack A, Phelan SJ, Liang Y, Hether T, Leon M, Zumwalt RE, Barton LM, Duval EJ, Mukhopadhyay S, Stroberg E, Borczuk A, Thorne LB, Sakthivel MK, Lee YZ, Hagood JS, Mock JR, Seibold MA, O’Neal WK, Montgomery SA, Boucher RC, Baric RS, SARS-CoV-2 infection produces chronic pulmonary epithelial and immune cell dysfunction with fibrosis in mice. Sci. Transl. Med 14, eabo5070 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Planer JD, Morrisey EE, After the Storm: Regeneration, Repair, and Reestablishment of Homeostasis Between the Alveolar Epithelium and Innate Immune System Following Viral Lung Injury. Annu. Rev. Pathol 18, 337–359 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia Y, Chen K, Lin P, Lieber G, Nishi M, Yan R, Wang Z, Yao Y, Li Y, Whitson BA, Duann P, Li H, Zhou X, Zhu H, Takeshima H, Hunter JC, McLeod RL, Weisleder N, Zeng C, Ma J, Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun 5, 4387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikami Y, Grubb BR, Rogers TD, Dang H, Asakura T, Kota P, Gilmore RC, Okuda K, Morton LC, Sun L, Chen G, Wykoff JA, Ehre C, Vilar J, van Heusden C, Livraghi-Butrico A, Gentzsch M, Button B, Stutts MJ, Randell SH, O’Neal WK, Boucher RC, Chronic airway epithelial hypoxia exacerbates injury in muco-obstructive lung disease through mucus hyperconcentration. Sci. Transl. Med 15, eabo7728 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghoneim HE, Thomas PG, McCullers JA, Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol 191, 1250–1259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M, Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog 10, e1004053 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CGK, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Others, Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol 12, 1045–1054 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, Metzger DW, IFN-γ increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol 11, 209–219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang H-E, Pollack JL, Gate RE, Haliburton GE, Ye CJ, Marson A, Erle DJ, Locksley RM, A tissue checkpoint regulates type 2 immunity. Nat. Immunol 17, 1381–1387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hassan AO, Case JB, Winkler ES, Thackray LB, Kafai NM, Bailey AL, McCune BT, Fox JM, Chen RE, Alsoussi WB, Turner JS, Schmitz AJ, Lei T, Shrihari S, Keeler SP, Fremont DH, Greco S, McCray PB Jr, Perlman S, Holtzman MJ, Ellebedy AH, Diamond MS, A SARS-CoV-2 Infection Model in Mice Demonstrates Protection by Neutralizing Antibodies. Cell 182, 744–753.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, Thannickal VJ, Moore BB, Christensen PJ, Simon RH, Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am. J. Respir. Crit. Care Med 181, 254–263 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, Noble PW, Stripp BR, Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 203, 707–717 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roy RM, Wüthrich M, Klein BS, Chitin elicits CCL2 from airway epithelial cells and induces CCR2-dependent innate allergic inflammation in the lung. J. Immunol 189, 2545–2552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D, IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. U. S. A 112, 10762–10767 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JS, McKinnis VS, Nawrocki A, White SR, Stimulation of migration and wound repair of guinea-pig airway epithelial cells in response to epidermal growth factor. Am. J. Respir. Cell Mol. Biol 18, 66–74 (1998). [DOI] [PubMed] [Google Scholar]
- 45.Kheradmand F, Folkesson HG, Shum L, Derynk R, Pytela R, Matthay MA, Transforming growth factor-alpha enhances alveolar epithelial cell repair in a new in vitro model. Am. J. Physiol 267, L728–38 (1994). [DOI] [PubMed] [Google Scholar]
- 46.Nava M, Dutta P, Zemke NR, Farias-Eisner R, Vadgama JV, Wu Y, Transcriptomic and ChIP-sequence interrogation of EGFR signaling in HER2+ breast cancer cells reveals a dynamic chromatin landscape and S100 genes as targets. BMC Med. Genomics 12, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsou AM, Yano H, Parkhurst CN, Mahlakõiv T, Chu C, Zhang W, He Z, Jarick KJ, Zhong C, Putzel GG, Hatazaki M, JRI IBD Live Cell Bank Consortium, Lorenz IC, Andrew D, Balderes P, Klose CSN, Lira SA, Artis D, Neuropeptide regulation of non-redundant ILC2 responses at barrier surfaces. Nature 611, 787–793 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ricardo-Gonzalez RR, Kotas ME, O’Leary CE, Singh K, Damsky W, Liao C, Arouge E, Tenvooren I, Marquez DM, Schroeder AW, Cohen JN, Fassett MS, Lee J, Daniel SG, Bittinger K, Díaz RE, Fraser JS, Ali N, Ansel KM, Spitzer MH, Liang H-E, Locksley RM, Innate type 2 immunity controls hair follicle commensalism by Demodex mites. Immunity 55, 1891–1908.e12 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pittet LA, Hall-Stoodley L, Rutkowski MR, Harmsen AG, Influenza virus infection decreases tracheal mucociliary velocity and clearance of Streptococcus pneumoniae. Am. J. Respir. Cell Mol. Biol 42, 450–460 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ural BB, Caron DP, Dogra P, Wells SB, Szabo PA, Granot T, Senda T, Poon MML, Lam N, Thapa P, Lee YS, Kubota M, Matsumoto R, Farber DL, Inhaled particulate accumulation with age impairs immune function and architecture in human lung lymph nodes. Nat. Med 28, 2622–2632 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, Debnath O, Thürmann L, Kurth F, Völker MT, Kazmierski J, Timmermann B, Twardziok S, Schneider S, Machleidt F, Müller-Redetzky H, Maier M, Krannich A, Schmidt S, Balzer F, Liebig J, Loske J, Suttorp N, Eils J, Ishaque N, Liebert UG, von Kalle C, Hocke A, Witzenrath M, Goffinet C, Drosten C, Laudi S, Lehmann I, Conrad C, Sander L-E, Eils R, COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol 38, 970–979 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, Wadsworth MH 2nd, Hughes TK, Kazer SW, Yoshimoto E, Cahill KN, Bhattacharyya N, Katz HR, Berger B, Laidlaw TM, Boyce JA, Barrett NA, Shalek AK, Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 560, 649–654 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ricardo-Gonzalez RR, Kotas ME, Tenvooren I, Marquez DM, Fassett MS, Lee J, Daniel SG, Bittinger K, Díaz RE, Fraser JS, Mark Ansel K, Spitzer MH, Liang H-E, Locksley RM, Type 2 innate immunity regulates hair follicle homeostasis to control Demodex pathosymbionts, bioRxiv (2021)p. 2021.03.29.437438. [Google Scholar]
- 54.Seibold MA, Reese TA, Choudhry S, Salam MT, Beckman K, Eng C, Atakilit A, Meade K, Lenoir M, Watson HG, Thyne S, Kumar R, Weiss KB, Grammer LC, Avila P, Schleimer RP, Fahy JV, Rodriguez-Santana J, Rodriguez-Cintron W, Boot RG, Sheppard D, Gilliland FD, Locksley RM, Burchard EG, Differential enzymatic activity of common haplotypic versions of the human acidic Mammalian chitinase protein. J. Biol. Chem 284, 19650–19658 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okawa K, Ohno M, Kashimura A, Kimura M, Kobayashi Y, Sakaguchi M, Sugahara Y, Kamaya M, Kino Y, Bauer PO, Oyama F, Loss and Gain of Human Acidic Mammalian Chitinase Activity by Nonsynonymous SNPs. Mol. Biol. Evol 33, 3183–3193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sutherland TE, Andersen OA, Betou M, Eggleston IM, Maizels RM, van Aalten D, Allen JE, Analyzing airway inflammation with chemical biology: dissection of acidic mammalian chitinase function with a selective drug-like inhibitor. Chem. Biol 18, 569–579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kimura M, Umeyama T, Wakita S, Okawa K, Sakaguchi M, Matoska V, Bauer PO, Oyama F, Direct comparison of chitinolytic properties and determination of combinatory effects of mouse chitotriosidase and acidic mammalian chitinase. Int. J. Biol. Macromol 134, 882–890 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Barad BA, Liu L, Diaz RE, Basilio R, Van Dyken SJ, Locksley RM, Fraser JS, Differences in the chitinolytic activity of mammalian chitinases on soluble and insoluble substrates. Protein Sci 29, 966–977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang H-E, Reinhardt RL, Bando JK, Sullivan BM, Ho I-C, Locksley RM, Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat. Immunol 13, 58–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Herbert DR, Hölscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, Kirsch R, Hall P, Mossmann H, Claussen B, Förster I, Brombacher F, Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity 20, 623–635 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Chong Z, Karl CE, Halfmann PJ, Kawaoka Y, Winkler ES, Keeler SP, Holtzman MJ, Yu J, Diamond MS, Nasally delivered interferon-λ protects mice against infection by SARS-CoV-2 variants including Omicron. Cell Rep 39, 110799 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Díaz RE, Ecker AK, Correy GJ, Asthana P, Young ID, Faust B, Thompson MC, Seiple IB, Van Dyken S, Locksley RM, Fraser JS, Structural characterization of ligand binding and pH-specific enzymatic activity of mouse Acidic Mammalian Chitinase. Elife 12 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qupath software. https://qupath.github.io/.
- 64.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, James JA, Salto-Tellez M, Hamilton PW, QuPath: Open source software for digital pathology image analysis. Sci. Rep 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin-Labat M-L, Smyth GK, Ritchie ME, Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 43, e97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ, GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao S, Guo Y, Sheng Q, Shyr Y, Advanced heat map and clustering analysis using heatmap3. Biomed Res. Int 2014, 986048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang H-E, Vaka D, Eckalbar WL, Molofsky AB, Erle DJ, Locksley RM, Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol 19, 1093–1099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.mm10 transcriptome_1 ftp://ftp.ensembl.org/pub/release-84/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.primary_assembly.fa.gz.
- 72.mm10 transcriptome_2 ftp://ftp.ensembl.org/pub/release-84/gtf/mus_musculus/Mus_musculus.GRCm38.84.gtf.gz.
- 73.LungMAP Consortium. www.lungmap.net.
- 74.Jung H, Van Dyken S, Lung distal epithelium_ChiaRed and WT epithelial cells [Dataset], (2024); 10.5061/dryad.47d7wm3pc. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk RNA sequencing data generated in this study are deposited in Gene Expression Omnibus (GEO) under accession codes GSE275075 and GSE275319. Single-cell RNA sequencing data are available in Dryad (74). Tabulated data underlying Figs. 1 to 7; figs. S1, S3, and S4 to S13 are archived in data S1. Immunoblot scans are archived in data S2. All other data are available in the main text or the supplementary materials.