

Abstract
Studying the thermodynamics of biochemical association reactions at the microscopic level requires efficient sampling of the configurations of the reactants and solvent as a function of the reaction pathways. In most cases, the associating ligand and receptor have complementary interlocking shapes. Upon association, loosely connected or disconnected solvent cavities at and around the binding site are formed. Disconnected solvent regions lead to severe statistical sampling problems when simulations are performed with explicit solvent. It was recently proposed that, when such limitations are encountered, they might be overcome by the use of the grand canonical ensemble. Here we investigate one such case and report the association free energy profile (potential of mean force) between trypsin and benzamidine along a chosen reaction coordinate as calculated using the grand canonical Monte Carlo method. The free energy profile is also calculated for a continuum solvent model using the Poisson equation, and the results are compared to the explicit water simulations. The comparison shows that the continuum solvent approach is surprisingly successful in reproducing the explicit solvent simulation results. The Monte Carlo results are analyzed in detail with respect to solvation structure. In the binding site channel there are waters bridging the carbonyl oxygen groups of Asp189 with the NH2 groups of benzamidine, which are displaced upon inhibitor binding. A similar solvent-bridging configuration has been seen in the crystal structure of trypsin complexed with bovine pancreatic trypsin inhibitor. The predicted locations of other internal waters are in very good agreement with the positions found in the crystal structures, which supports the accuracy of the simulations.
Full text
PDF

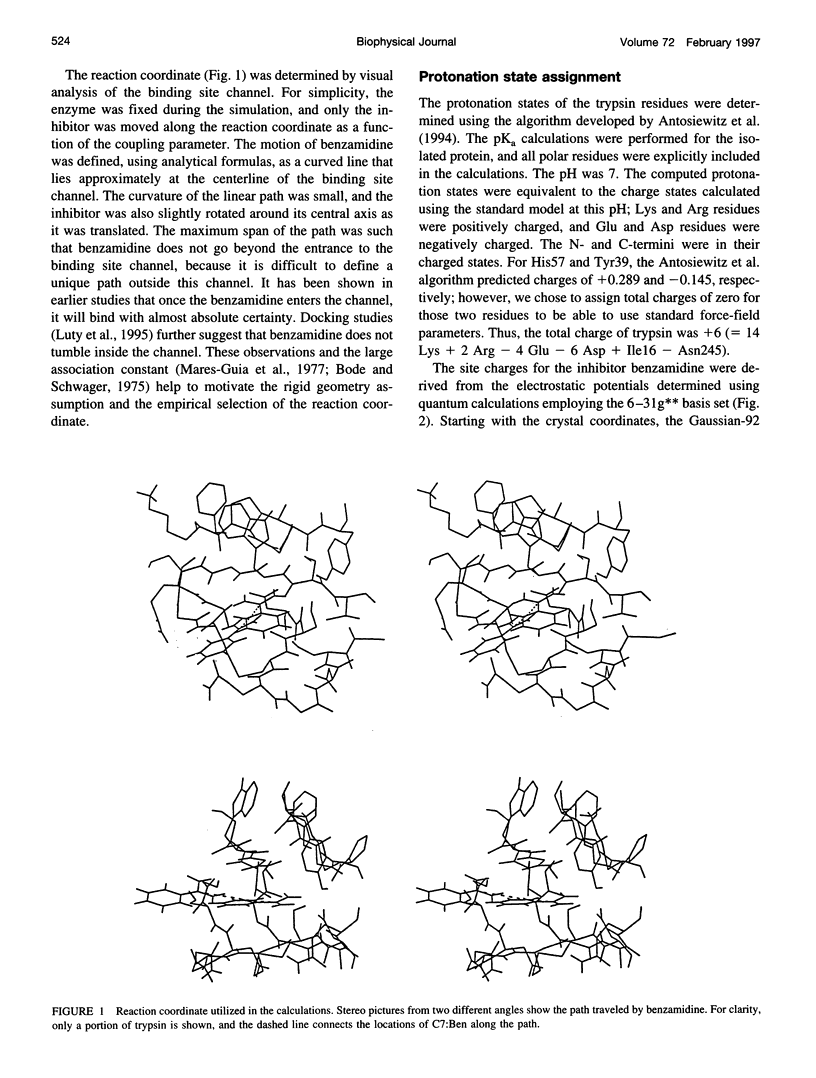

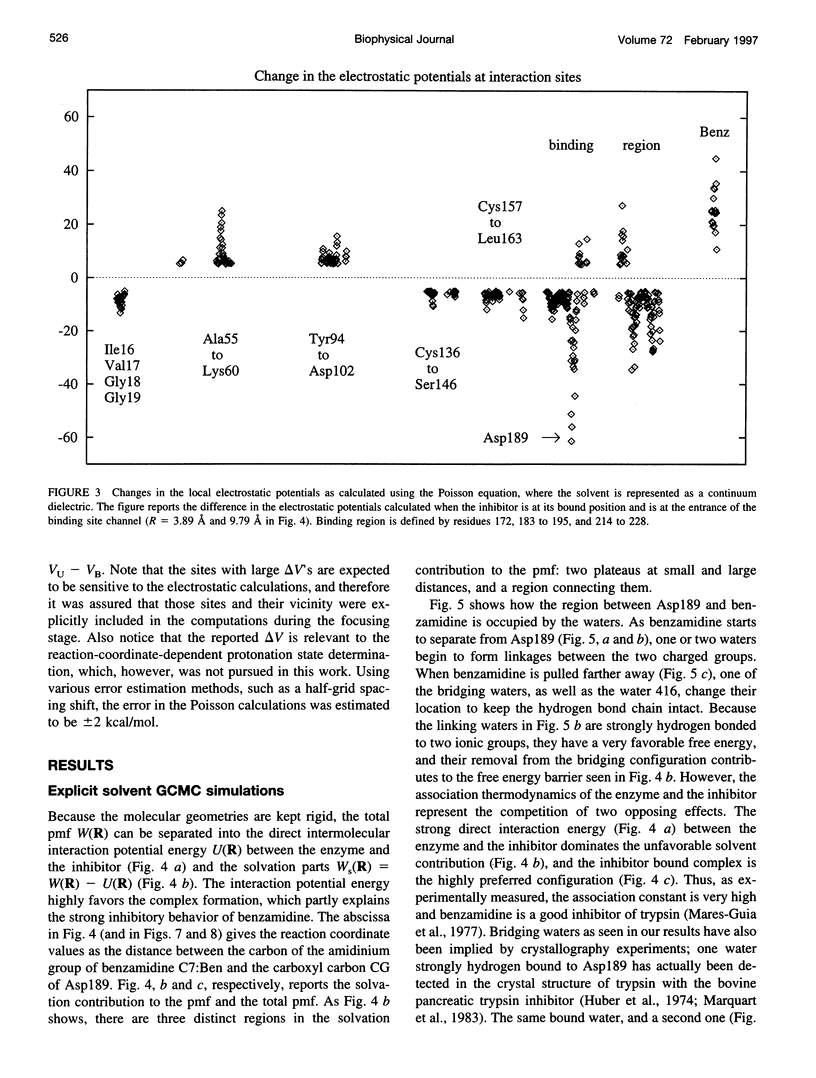
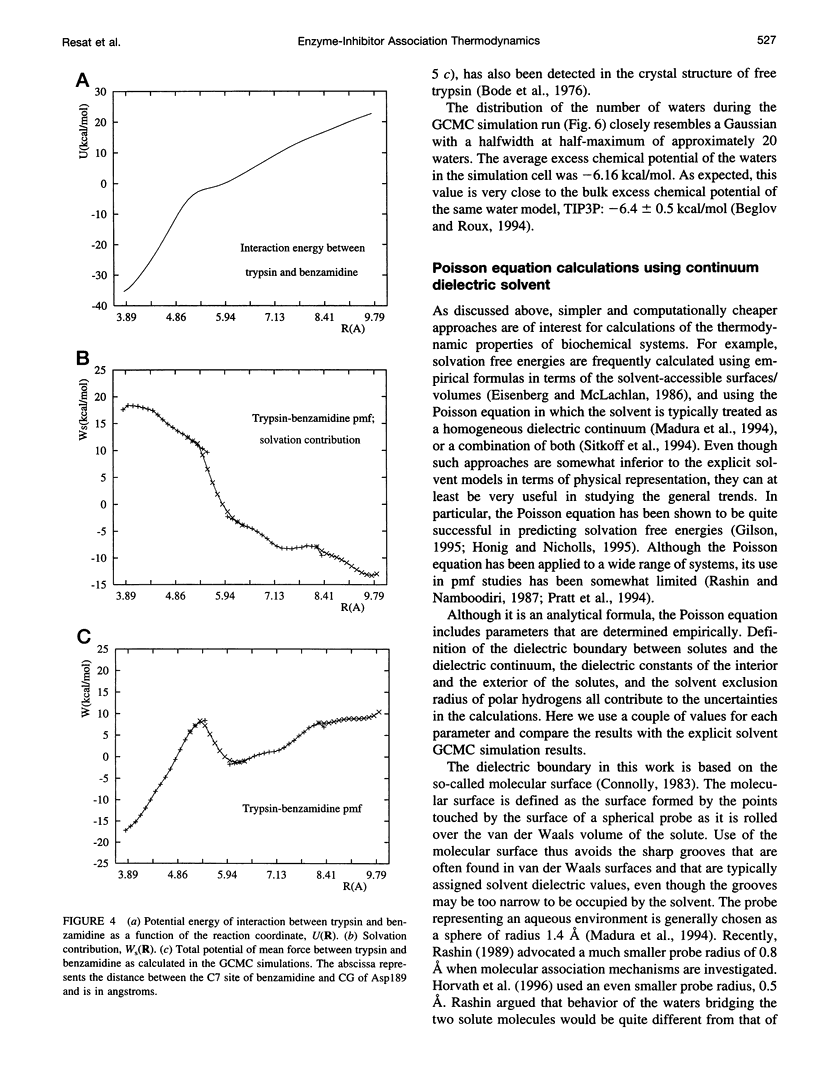

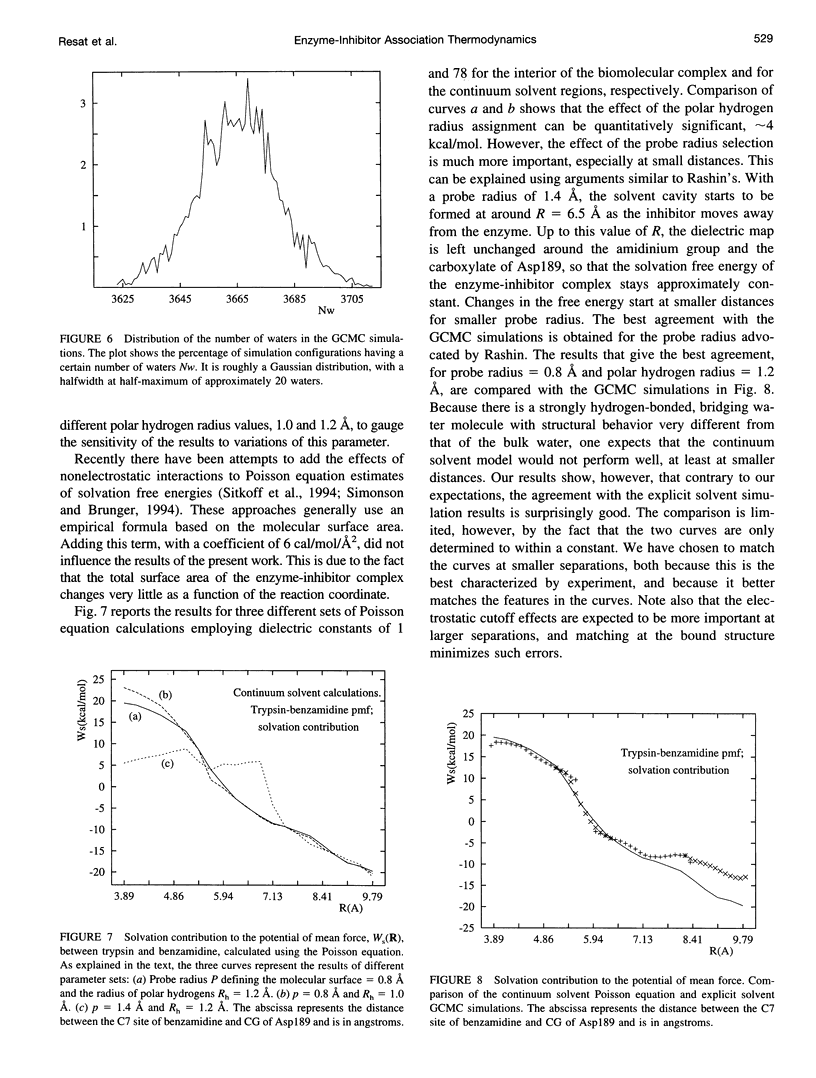



Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Antosiewicz J., McCammon J. A., Gilson M. K. Prediction of pH-dependent properties of proteins. J Mol Biol. 1994 May 6;238(3):415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- Bode W., Schwager P., Huber R. The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. The refined crystal structures of the bovine trypsinogen-pancreatic trypsin inhibitor complex and of its ternary complex with Ile-Val at 1.9 A resolution. J Mol Biol. 1978 Jan 5;118(1):99–112. doi: 10.1016/0022-2836(78)90246-2. [DOI] [PubMed] [Google Scholar]
- Bode W., Schwager P. The refined crystal structure of bovine beta-trypsin at 1.8 A resolution. II. Crystallographic refinement, calcium binding site, benzamidine binding site and active site at pH 7.0. J Mol Biol. 1975 Nov 15;98(4):693–717. doi: 10.1016/s0022-2836(75)80005-2. [DOI] [PubMed] [Google Scholar]
- Eisenberg D., McLachlan A. D. Solvation energy in protein folding and binding. Nature. 1986 Jan 16;319(6050):199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- Finer-Moore J. S., Kossiakoff A. A., Hurley J. H., Earnest T., Stroud R. M. Solvent structure in crystals of trypsin determined by X-ray and neutron diffraction. Proteins. 1992 Mar;12(3):203–222. doi: 10.1002/prot.340120302. [DOI] [PubMed] [Google Scholar]
- Gilson M. K. Theory of electrostatic interactions in macromolecules. Curr Opin Struct Biol. 1995 Apr;5(2):216–223. doi: 10.1016/0959-440x(95)80079-4. [DOI] [PubMed] [Google Scholar]
- Honig B., Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995 May 26;268(5214):1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Huber R., Kukla D., Bode W., Schwager P., Bartels K., Deisenhofer J., Steigemann W. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. II. Crystallographic refinement at 1.9 A resolution. J Mol Biol. 1974 Oct 15;89(1):73–101. doi: 10.1016/0022-2836(74)90163-6. [DOI] [PubMed] [Google Scholar]
- Lounnas V., Pettitt B. M. A connected-cluster of hydration around myoglobin: correlation between molecular dynamics simulations and experiment. Proteins. 1994 Feb;18(2):133–147. doi: 10.1002/prot.340180206. [DOI] [PubMed] [Google Scholar]
- Lybrand T. P. Ligand-protein docking and rational drug design. Curr Opin Struct Biol. 1995 Apr;5(2):224–228. doi: 10.1016/0959-440x(95)80080-8. [DOI] [PubMed] [Google Scholar]
- Mares-Guia M., Nelson D. L., Rogana E. Electronic effects in the interaction of para-substituted benzamidines with trypsin: the involvement of the pi-electronic density at the central atom of the substituent in binding. J Am Chem Soc. 1977 Mar 30;99(7):2331–2336. doi: 10.1021/ja00449a051. [DOI] [PubMed] [Google Scholar]
- Mezei M., Beveridge D. L. Free energy simulations. Ann N Y Acad Sci. 1986;482:1–23. doi: 10.1111/j.1749-6632.1986.tb20933.x. [DOI] [PubMed] [Google Scholar]
- Resat H., Mezei M. Grand canonical ensemble Monte Carlo simulation of the dCpG/proflavine crystal hydrate. Biophys J. 1996 Sep;71(3):1179–1190. doi: 10.1016/S0006-3495(96)79322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soman K., Yang A. S., Honig B., Fletterick R. Electrical potentials in trypsin isozymes. Biochemistry. 1989 Dec 26;28(26):9918–9926. doi: 10.1021/bi00452a007. [DOI] [PubMed] [Google Scholar]