

Abstract
Prp16p is a DEAH-box ATPase that transiently associates with the spliceosome to promote the structural transition required for the second chemical step. Yeast strains carrying the cold-sensitive allele prp16-302 stall the release of Prp16p at low temperatures, yet splice precursors with aberrant branchpoints at increased frequency. To identify new factors involved in the regulation of splicing fidelity, we sought suppressors of the prp16-302 growth phenotype. Deletion of the nonessential ISY1 gene (1) improves growth of prp16-302 strains, (2) alleviates stalling, and (3) restores fidelity of branchpoint usage to wild-type levels. Isy1p is a subunit of the NineTeen Complex containing Prp19p, an essential E3 ubiquitin ligase homolog required for splicing. Notably, Δisy1 PRP16 strains display reduced fidelity of 3′-splice site selection. Consistent with a recent two-state model of the spliceosome, our genetic and biochemical results suggest that Isy1p acts together with U6 snRNA to promote a spliceosomal conformation favorable for first-step chemistry. We propose that deletion of ISY1 favors the premature release of Prp16p, thus promoting second-step chemistry of precursors with inappropriate 3′-splice sites.
Keywords: Isy1p, Prp16p, pre-mRNA splicing, fidelity
Nuclear pre-mRNA splicing, the process that removes introns from primary transcripts in eukaryotic cells, is an essential step in gene expression. Splicing is catalyzed by the spliceosome, an extraordinarily complex ribonucleoprotein (RNP) machine composed of five small nuclear RNAs (snRNAs)—U1, U2, U4, U5, and U6—and about a hundred proteins (Burge et al. 1998; Jurica and Moore 2003). The pre-mRNA splicing reaction consists of two sequential transesterifications involving the intron-defining sequences at the 5′-splice site, 3′-splice site, and branchpoint. In the first chemical step, the conserved adenosine at the branch site attacks the 5′-splice site, producing a branched lariat intermediate and a free 5′-exon. In the second step, the 5′-exon attacks the 3′-splice site, resulting in ligation of 5′- and 3′-exons and release of the intron lariat. Because single-nucleotide precision is required, cells must have mechanisms to control the fidelity of pre-mRNA splicing.
Enzymes belonging to the DExH/D-box superfamily of RNA-dependent ATPases remodel contacts within RNPs by promoting the exchange of RNA–RNA or RNA–protein pairing partners (Staley and Guthrie 1998; Tanner and Linder 2001). Prp16p is an essential DEAH-box ATPase that associates transiently with the spliceosome to activate the second catalytic step of splicing (Schwer and Guthrie 1991). ATP hydrolysis by Prp16p is required for release of the enzyme from the spliceosome and subsequent second-step catalysis (Schwer and Guthrie 1991), and promotes a conformational rearrangement of the spliceosome (Schwer and Guthrie 1992).
Prp16p was originally identified in a screen for factors that allow splicing of pre-mRNAs containing branch-point mutations (Couto et al. 1987; Burgess et al. 1990). Interestingly, this relaxation in the fidelity of branch-point recognition invariably correlates with mutations decreasing Prp16p ATPase activity in vitro (Burgess and Guthrie 1993b). This led to a kinetic proofreading model in which the rate of ATP hydrolysis by Prp16p acts as a timer to regulate the outcome of two competing events involving the lariat intermediates produced in the first step of splicing (Burgess and Guthrie 1993b). Within the interval that Prp16p is associated with the spliceosome, lariat intermediates can either undergo a transition necessary to proceed to the second catalytic step or be otherwise discarded and degraded. Thus, aberrant lariat intermediates are actively discarded in wild-type cells, but are spared from the degradative pathway in prp16 mutants, which allow more time for the necessary transition to occur prior to ATP hydrolysis.
Two additional spliceosomal factors with a demonstrated role in splicing fidelity are the U6 snRNA and Prp8p. U6 snRNA is a key component of the catalytic core (Madhani and Guthrie 1992). In particular, various alleles have been shown to affect the accuracy of splice site choice (Lesser and Guthrie 1993b; McPheeters 1996; Chang and McPheeters 2000; Query and Konarska 2004). Notably, substitutions of the highly conserved nucleotide at position 57 in U6 display opposing effects on the two steps of splicing, and affect fidelity of both branch-point and 3′-splice site recognition (McPheeters 1996; Chang and McPheeters 2000; Query and Konarska 2004).
Extensive genetic analyses of PRP8 have long supported a major role for this U5 snRNP protein in spliceosomal fidelity. Numerous prp8 mutant alleles initially shown to suppress splicing defects due to mutations at the 3′-splice site were subsequently found to also suppress mutations at the 5′-splice site and the branchpoint (Umen and Guthrie 1995; Collins and Guthrie 1999; Siatecka et al. 1999; Query and Konarska 2004). In all cases, the efficiency of the second catalytic step was improved. Interestingly, the pattern of suppression was highly specific with respect to the position, not sequence, of the mutation being suppressed. Based on these and other findings (Collins and Guthrie 2001), it was proposed that Prp8p functions to stabilize an RNA tertiary structure juxtaposing sequences within U6 with the 5′- and 3′-splice sites (Collins and Guthrie 1999, 2000; Siatecka et al. 1999).
More recently, based on an elegant study of genetic interactions among U6-U57, prp8, and prp16 alleles, it has been proposed that complex patterns of allele-specific suppression of mutant splice signals can be most simply explained by positing an equilibrium between two conformational states of the spliceosome, one competent for first-step and the other for second-step chemistry (Query and Konarska 2004). In analogy to current models of translational decoding, the accuracy of splicing is seen to be perturbed by mutations that affect the relative stability of the alternative states.
To identify additional factors involved with Prp16p in the regulation of splicing fidelity, we sought suppressors of the cold-sensitive growth defect of prp16-302. Deletion of ISY1 suppresses the defects of the prp16-302 allele. Firstly, it improves growth at low temperatures. Secondly, it restores the fidelity of branchpoint recognition to wild-type levels. Lastly, while mutant Prp16p is stalled on lariat intermediate-containing spliceosomes, removal of Isy1p alleviates this stall. Notably, deletion of ISY1 in wild-type PRP16 cells reduces the efficiency of the first step of splicing, and decreases the fidelity of 3′-splice site recognition. We propose that loss of Isy1p favors the release of Prp16p, eliminating the stall that allows inappropriate branchpoint usage, and promoting second-step chemistry of 3′-splice site mutants. These results lend strong support to the two-state model of the catalytically active spliceosome and suggest that Isy1p affects the conformational changes catalyzed by Prp16p in the transition between the two steps of splicing. Interestingly, Isy1p is a component of the multi-subunit NineTeen Complex (NTC), which contains a homolog of U-box E3 ubiquitin ligases, Prp19p. We discuss the implications of these findings for ATP-dependent spliceosomal rearrangements and the regulation of fidelity.
Results
Mutations in the ATPase domain of Prp16p block the second catalytic step and lead to increased association with the spliceosome
To facilitate the identification of factors involved in the Prp16p-dependent step, we focused our analysis on the most cold-sensitive of PRP16 alleles, prp16-302 (Madhani and Guthrie 1994; Noble and Guthrie 1996). Sequencing of this previously unmapped allele identified mutations in two highly conserved residues within the ATPase domain, R456K and G691R (Fig. 1A). While the former substitution alone does not confer an apparent growth defect, substitution of the last glycine in motif VI confers a cold-sensitive growth defect (data not shown). Growth impairment at 16°C is most severe when the two mutations are combined (Fig. 1A). The original prp16-302 allele containing both mutations was used throughout the course of this study.
Figure 1.

Mutations in the ATPase domain of Prp16p block the second catalytic step and lead to increased association with the spliceosome. (A, top) Schematic diagram of the domain organization of Prp16p. Motifs I–VI in the ATPase domain are represented by vertical bars. The location of the two mutations in the prp16-302 allele is indicated. (Bottom) Serial dilutions of the wild-type PRP16 and mutant prp16-302 strains were spotted on YPD plates and grown for 8 d at the restrictive temperature of 16°C or for 3 d at the permissive temperature of 25°C. (B) In vitro splicing of radiolabeled actin pre-mRNA in extracts from the indicated strains was performed for 30 min at 18°C and subsequently continued for an additional 20 min at 18°C or at 30°C. From top to bottom, the position of the lariat intermediate, lariat intron, precursor, mRNA, and 5′-exon are indicated schematically on the left. (C) Coimmunoprecipitation of splicing intermediates with Prp16p from in vitro splicing reactions performed for 40 min at 18°C. The position of the RNA species is indicated schematically on the left. Unlabeled bands represent degradation products. Signals from at least three independent experiments were quantitated normalizing to levels of the input reaction and expressed relative to the wild-type arbitrarily set to 100.
Consistent with a loss of Prp16p function at the restrictive temperature, extract from prp16-302 cells displayed a conditional block prior to the second step in an in vitro splicing assay performed at low temperature (18°C) (Fig. 1B). This defect could be reversed by raising the incubation temperature to 30°C in the presence of ATP, leading to release of the reaction products (Fig. 1B). Thus, the cold-sensitive growth defect observed in prp16-302 cells correlated with conditional loss of the biochemical function in vitro. The in vitro splicing efficiency of prp16-302 extracts was also lower than wild-type at 30°C, although the lower activity is not rate-limiting for growth at this temperature.
Wild-type Prp16p associates only transiently with the spliceosome, and ATP hydrolysis is required for release of the protein from the spliceosome and for second-step catalysis (Schwer and Guthrie 1991). We tested how the prp16-302 mutation affects association of the mutant Prp16p with the spliceosome in vitro. Splicing reactions performed at the restrictive temperature were immunoprecipitated with antibodies against Prp16p, and the coimmunoprecipitated RNA species were analyzed on a denaturing gel and quantified (Fig. 1C). To take into account differences due to the relative levels of splicing intermediates, data were normalized relative to the levels of the input reaction (see Materials and Methods). We reproducibly observed a roughly 2.5-fold increase in the association of Prp16-302p with first-step products compared to the wild-type control. A similar twofold increased association of Prp16-302p was found at 30°C (data not shown). We conclude that the prp16-302 mutation affects the release of the protein from the spliceosome.
Deletion of the nonessential ISY1 gene suppresses prp16-302 defects
Previous genetic screens have proven successful at identifying targets of DExD/H-box proteins (J.Y. Chen et al. 2001; Kistler and Guthrie 2001). We therefore performed an open-ended screen for extragenic suppressors of the cold-sensitive phenotype of the prp16-302 allele. Yeast cells carrying the prp16-302 allele were EMS-mutagenized and screened for recovery of growth at the restrictive temperature of 16°C (see Materials and Methods). We isolated one recessive extragenic suppressor and found it to be allelic to the nonessential ISY1 gene. The suppressing mutation was caused by a transversion creating a premature termination codon resulting in a truncated protein of 74 out of 235 amino acids. We subsequently verified in a reconstruction experiment that a complete deletion of ISY1 also suppressed the cold-sensitive growth defect of prp16-302 (Fig. 2A), while expression of ISY1 from a plasmid restored the conditional lethal phenotype (data not shown).
Figure 2.

Deletion of the nonessential ISY1 gene suppresses prp16-302 defects. (A) Serial dilutions of the indicated strains were spotted on YPD plates and grown for 8 d at 16°C or for 3 d at 25°C. (B) In vitro splicing of radiolabeled actin pre-mRNA in extracts from the indicated strains was performed for 40 min at 18°C (18°C panel) or for 20 min at 25°C (25°C panel). The position of the RNA species is indicated schematically on the left. (C) Coimmunoprecipitation of splicing intermediates with Prp16p from in vitro splicing reactions performed for 40 min at 18°C. The position of the RNA species is indicated schematically on the left. Unlabeled bands represent degradation products. Signals were quantitated as in Figure 1C.
Isy1p is a component of the Prp19p-associated complex (NTC), a complex required for catalysis that binds the spliceosome at an early assembly step and stabilizes the association of the U5 and U6 snRNAs (Tarn et al. 1994; Chan et al. 2003). Because of this unprecedented connection between Prp16p and the NTC, we tested strains with deletion of other nonessential components of this complex for the ability to suppress prp16-302 cold sensitivity. Deletions of SNT309, NTC20, and SYF2 were all unable to rescue prp16-302 growth at the restrictive temperature (data not shown), strongly supporting the specificity of the genetic interaction between ISY1 and PRP16.
We reasoned that if Isy1p is involved in the Prp16p-dependent remodeling step, then its deletion should suppress the diverse phenotypes resulting from impairment of this step in the prp16-302 mutant strain. Indeed, when tested in in vitro splicing assays in the cold, an extract prepared from the prp16-302, Δisy1 suppressor strain reproducibly displayed a roughly twofold increase in splicing efficiency and mRNA yields (Fig. 2B, 18°C panel), consistent with at least partial suppression of the second-step defect due to prp16-302 and the relief of a defect limiting for growth. Additionally, we observed that deletion of ISY1 alone decreased overall splicing efficiency, suggesting an early block to splicing (Fig. 2B, 25°C panel; data not shown).
We then asked if deletion of ISY1 would favor the dissociation of Prp16-302p from the spliceosome. Splicing reactions performed at the restrictive temperature were immunoprecipitated with antibodies against Prp16p, and the coimmunoprecipitated RNA species were analyzed on a denaturing gel and quantified (Fig. 2C). Indeed, we found that deletion of ISY1 reduced the association of Prp16-302p with first-step intermediates to nearly wild-type levels. Additionally, wild-type Prp16p association with spliceosomal intermediates was also reduced in the absence of Isy1p. Similar results were observed at 30°C (data not shown).
The above data prompted us to test whether stalled complexes could also be detected in vivo, especially when mutant cells are shifted to the cold, that is, under conditions in which dissociation of Prp16-302p from spliceosomes is inhibited the greatest. We performed tandem affinity purification of TAP-tagged Prp16p and Prp16-302p from yeast cells grown at 28°C or shifted for 7 h to the restrictive temperature of 16°C, a time sufficient to elicit a detectable splicing defect in vivo (data not shown). Purified complexes were analyzed by SDS-PAGE and Western blotting. Both at 28°C and at 16°C, Isy1p copurified with TAP-tagged Prp16-302p, showing strong enrichment in the mutant strain with respect to its wild-type counterpart (Fig. 3). Similarly, we found that Ntc20p, which is in a subcomplex with Isy1p in the NTC (Chen et al. 2002), was also enriched with Prp16-302p, especially at 16°C.
Figure 3.
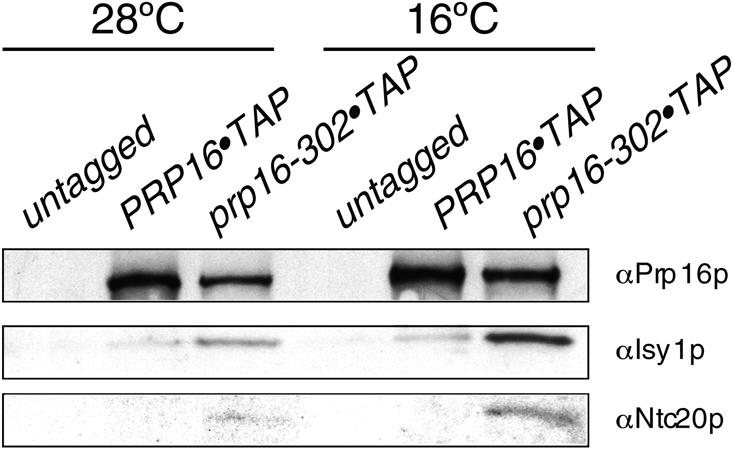
Isy1p copurifies with Prp16-302p. Western blotting of proteins purified by tandem affinity purification from the indicated strains grown at the indicated temperatures. Antibodies used for detection are indicated on the right.
Δisy1 suppresses the relaxed fidelity of branchsite recognition of prp16-302 and decreases accuracy of 3′-splice site usage
Mutations in PRP16 impairing ATP hydrolysis decrease splicing fidelity by allowing use of an inappropriate branchpoint (Burgess and Guthrie 1993b). Because the Δisy1 mutation suppressed prp16-302 splicing defects, we asked if it also prevented the use of mutant introns. We therefore determined the fidelity phenotypes conferred by the prp16-302 and Δisy1 mutations, alone and in combination. To test fidelity, we used the ACT1-CUP1 reporter, which provides a quantitative readout of splicing efficiency in vivo. Removal of the intron from the reporter pre-mRNA is required for metallothionein protein to be produced, thereby conferring copper resistance to yeast (Fig. 4; Lesser and Guthrie 1993a). Strains were transformed with reporters containing mutations at the 5′-splice site (G1A and G5A), branchpoint (C256A and A259C), or 3′-splice site (A302G and A302U), as well as with a wild-type reporter, and the concentration of copper tolerated by each strain was measured. The assay was performed at 30°C, that is, under conditions in which stalling of Prp16-302p on lariat intermediates is not rate-limiting for growth, yet the protein is not fully functional. Primer extension analyses of the reporter RNA levels were used to validate the results of the copper assay (data not shown).
Figure 4.

Δisy1 suppresses the relaxed fidelity of branch site recognition of prp16-302 and decreases accuracy of 3′-splice-site usage. Strains indicated in the legend were transformed with the ACT1-CUP1 reporter containing intron mutations. Serial dilutions of the strains were then grown on plates containing different mM concentrations of CuSO4 for 5 d at 30°C. Bars indicate the highest millimolar concentration of copper tolerated by strains transformed with the indicated reporter. Tested concentrations are shown on the Y-axis. The reporter is depicted schematically at the bottom with the location of the different mutations indicated according to numbering of the actin intron.
As expected, we found that prp16-302 exhibited a relaxed fidelity phenotype by allowing splicing of introns with branchpoint mutations, as judged by increased tolerance to copper compared to a wild-type strain. In contrast, the splicing efficiency of the reporter carrying the A259C substitution of the branch nucleotide, which strongly inhibits the first step of splicing (Vijayraghavan et al. 1986), showed a twofold decrease in a Δisy1 strain, indicating a negative effect of the ISY1 deletion on the first step. In the double-mutant strain, deletion of ISY1 strongly antagonized the prp16-302 reduced fidelity phenotype and restored wild-type accuracy of branchpoint recognition. Thus, we conclude that deletion of ISY1 not only suppresses the splicing deficiencies of prp16-302 but also its fidelity defects.
The prp16-302 mutation did not affect splicing of 5′-splice-site and 3′-splice-site mutant reporters. Very similar or identical results were observed with the previously isolated fidelity alleles prp16-1 and prp16-101 (Burgess and Guthrie 1993b; data not shown). In contrast, we reproducibly found a twofold increase in copper tolerance of Δisy1 strains transformed with the 3′-splice-site mutant reporters. Thus, deletion of ISY1 affects splicing fidelity by decreasing accuracy of 3′-splice-site selection.
Interestingly, we also observed that the double-mutant prp16-302, Δisy1 strain displayed a twofold increase in copper tolerance over each single mutant strain for the 5′-splice-site and 3′-splice-site mutant reporters G1A and A302G, suggesting a combined effect of prp16-302 and Δisy1 on suppression of these mutations. Altogether, these data uncover a role for Isy1p in the regulation of splicing fidelity during the Prp16p-dependent transition toward the second step of splicing.
Genetic and functional interactions between Δisy1 and U6 alleles connected to prp16-302 activity
Because of the known genetic interactions of several U6 alleles with prp16 fidelity mutants (Madhani and Guthrie 1994; McPheeters 1996; Query and Konarska 2004), we tested the interactions of U6 alleles with the Δisy1 mutant. The U6-U57C allele appears to have an opposite effect to Δisy1, in that it inhibits second-step catalysis while favoring the first step (McPheeters 1996; Query and Konarska 2004; data not shown). Indeed, U6-U57C is synthetically lethal with a prp16-302 mutation, consistent with the exacerbation of a second-step defect (McPheeters 1996; data not shown). Because the combination of mutants with opposing effects on splicing should result in reciprocal suppression, we tested whether deletion of ISY1 suppressed the U6-U57C slow-growth phenotype at 37°C. This phenotype was efficiently suppressed by combination of the U6-U57C and Δisy1 mutations (Fig. 5A). On the other hand, the U6-U57A allele phenocopies a Δisy1 strain. U6-U57A relaxes the fidelity of 3′-splice site recognition, decreases splicing of substrates containing a C-branch site, and suppresses the cold-sensitivity of prp16-302 (McPheeters 1996; Chang and McPheeters 2000; data not shown). We therefore expected the combination of the Δisy1 and U6-U57A mutations to result in a synergistic effect. We, indeed, found that the double-mutant strain displayed synthetic lethality at 37°C (Fig. 5A). U6 mutants carrying single-nucleotide deletions of residues 40–46 (U6-Δ40 to U6-Δ46) have also been identified as suppressors of the prp16-302 cold-sensitive phenotype (Madhani and Guthrie 1994). We predicted that, similar to U6-U57A, these alleles should be synthetic in combination with Δisy1. As previously reported (Madhani and Guthrie 1994), U6-ΔC43 and U6-ΔA44 are the strongest suppressors of prp16-302 among the single-deletion alleles of U6, although we found them to be weaker suppressors than a deletion of ISY1 (data not shown). While neither U6-ΔC43 nor U6-ΔA44 alone showed a growth defect at any temperature, combination with Δisy1 resulted in lethality and slow growth at 37°C, respectively (Fig. 5A). These findings reinforced the notion of a strong genetic and functional connection between Isy1p and the U6 snRNA related to the activity of Prp16p.
Figure 5.

Genetic and functional interactions between Δisy1 and U6 alleles connected to prp16-302 activity. (A) Serial dilutions of the indicated strains were spotted on YPD plates and grown for 3 d at 25°C or at 37°C. (B) Primer extension analysis of RP51A RNA from indicated strains following growth at 25°C or a shift for 2.5 h to 37°C. RNA species are indicated schematically on the left. U14 snoRNA was used as a loading standard. The bottom portions of the gel are shown at a lighter exposure than the top part. Strains are denoted above lanes as follows: (Δ) Δisy1; (+) ISY1; (wt) U6; (43) U6-ΔC43; (44) U6-ΔA44; (A) U6-U57A; (C) U6-U57C. (C) Primer extension analysis of U3 snoRNA from indicated strains following growth at 25°C. PGK1 was used as a loading standard. Strains are denoted above the lanes as in B.
Because U6-U57A, U6-ΔC43, and U6-ΔA44 all suppress prp16-302 and are synthetic lethal with Δisy1, we expected the U6-ΔC43 and U6-ΔA44 mutations to also inhibit splicing of precursors with branchpoint mutations. Using the copper assay described above, we, indeed, found that both mutants reduced by fivefold the splicing efficiency of reporters carrying branchpoint mutations, consistent with an inhibitory effect on the first step of splicing (data not shown). In contrast, mutations at the 5′- and 3′-splice sites were not affected by U6-ΔC43 and U6-ΔA44 (data not shown).
To more directly test the impact of Isy1p and the U6 snRNA on the first step of splicing, we analyzed by primer extension the steady-state level of the intron-containing RP51A RNA (Fig. 5B), as well as that of the U3 snoRNA (Fig. 5C), from strains carrying each of the U6 alleles described above, alone or in combination with the deletion of ISY1. In agreement with our genetic results, we found that the Δisy1 mutation in combination with U6-ΔC43, U6-ΔA44, and U6-U57A, but importantly not with U6-U57C, led to the accumulation of unspliced precursors both at 25°C (Fig. 5B [cf. lanes 2–4 and 5], C) or following a shift to 37°C for 2.5 h (Fig. 5B, cf. lanes 12–14 and 15), consistent with the exacerbation of a defect in first-step catalysis. We conclude that interactions between Isy1p and U6 promote the first step of splicing.
Discussion
To avoid splicing errors, surveillance mechanisms must exist to ensure proper selection of the branchpoint and 5′-splice site in the first step, and of the 3′-splice site in the second step. The Prp16p ATPase transiently associates with the spliceosome to promote the structural transition required for second-step chemistry. In prp16 mutants with reduced ATPase activity, the release of Prp16p required for the second step is inhibited, yet splicing of precursors with altered branchpoint sequences occurs with increased frequency. This is also the case in strains carrying the cold-sensitive allele prp16-302, where the second step of splicing is conditionally blocked at low temperatures and the mutant Prp16p remains associated with the spliceosome (Fig. 1). To identify additional factors involved with Prp16p in the regulation of splicing fidelity, we searched for suppressors of the cold-sensitive growth phenotype of prp16-302. If slowing the rate of the Prp16p-dependent conformational rearrangement required for the second step is necessary for the reduced fidelity phenotype, we reasoned that mutations that promote this rearrangement might restrict the use of mutant introns. In principle, such mutations might arise as second-site changes within PRP16 that improve ATP binding/hydrolysis. Alternatively, suppressors of the cold-sensitive growth phenotype might identify factors that promote the release of mutant Prp16p, thereby allowing activation of second-step chemistry with improved efficiency and accuracy even in the absence of fully functional Prp16p. We show that deletion of the ISY1 gene suppresses the cold-sensitive growth defect of prp16-302, improves the second step of splicing, and reduces the association of Prp16-302p with the spliceosome (Fig. 2). As predicted, fidelity of branchpoint recognition is restored to wild-type levels in the double-mutant strain (Fig. 4). Interestingly, in the Δisy1 single-mutant strain, fidelity of 3′-splice-site selection is reduced.
Isy1p is a conserved subunit of the Prp19p-associated complex, or NTC, which binds the spliceosome after U1 and U4 dissociation, allowing the stable association of the U5 and U6 snRNAs with the spliceosome (Chan et al. 2003). While Isy1p is nonessential, Prp19p is a U-box E3 ubiquitin ligase required for growth and splicing (Cheng et al. 1993; Hatakeyama et al. 2001). Within the NTC, Isy1p/Ntc30p is part of a subcomplex together with Ntc20p, Syf1p/Ntc90p, and Syf2p/Ntc31p (Chen et al. 2002). Based on genetic analyses, a functional overlap was proposed between Isy1p and Ntc20p, and possibly Syf1p (Chen et al. 2002). However, to date, none of these factors has been extensively characterized. Previous analyses of Δisy1 strains suggested an auxiliary role in splicing, possibly through modulation of the NTC function (Dix et al. 1999; C.H. Chen et al. 2001). We show that extracts from a Δisy1 strain display an overall decrease in splicing efficiency, consistent with an early block to splicing (Fig. 2). We provide evidence that Δisy1 mutants depress the first step of splicing in vivo, both alone and especially in combination with specific alleles of the U6 snRNA (Fig. 5). Finally, we show that Δisy1 mutants exacerbate the defect of precursors carrying a mutation of the branch nucleotide that inhibits the first step (Fig. 4). These results indicate that the wild-type function of Isy1p is required for the first step of splicing.
Molecular frameworks for splicing fidelity
Kinetic proofreading was first proposed on theoretical grounds as a mechanism for fidelity enhancement by the use of multiple discrimination events (Hopfield 1974; Ninio 1974). A hallmark of such strategies is the coupling of sequential inspections by an irreversible step, providing the opportunity for a discard branch. In the model of Burgess and Guthrie (1993b), the irreversible step is the hydrolysis of ATP by Prp16p. The lariat intermediate (LI) is thus partitioned between two fates, one productive and the other not, by the timing of ATP hydrolysis. In the productive pathway, a conformational rearrangement (LI to LI*) results in release of Prp16p from the spliceosome concomitant with ATP hydrolysis, allowing the second chemical step to proceed. Conversely, if ATP hydrolysis occurs prior to the rearrangement, the lariat intermediate is rejected via the discard branch (Fig. 6A). The model assumes that an unfavorable interaction between the mutant branchpoint and some spliceosomal component(s) hinders the structural rearrangement. Slowing the rate of ATP hydrolysis by mutation in the DEAH domain provides more time to undergo the LI-to-LI* rearrangement, thus sparing mutant substrates from the degradative discard pathway. According to this view, deletion of ISY1 could improve the accuracy of prp16-302 spliceosomes by promoting the LI-to-LI* transition.
Figure 6.

Molecular frameworks for splicing fidelity. (A) The previously proposed kinetic proofreading model for Prp16p function (Burgess and Guthrie 1993b) is integrated with the two-state model of the spliceosome recently proposed by Query and Konarska (2004). The transition between the first-step (LI)- and second-step (LI*)-specific conformations of the spliceosome is influenced by the Δisy1 and prp16-302 mutants as discussed in the text. (B) Elements of the catalytic core at the time of second-step activation. Nucleotides having genetic interaction with PRP16 are indicated in bold and italicized letters. Black dots represent tertiary interactions. The upstream ACA trinucleotide in U6 is boxed.
Based on the recent results of Query and Konarska (2004), we can now discuss this Prp16p-dependent transition as the rearrangement between two conformational states that promote the first-step or second-step chemical reactions, respectively. According to this view, slowed Prp16p action results in improved first-step catalysis for mutant substrates while Δisy1 improves the fidelity of prp16-302 by favoring the “second-step” (or LI*) conformation (Fig. 6A). In principle, the wild-type function of Isy1p could be stabilization of LI or destabilization of LI*. Alternatively, Isy1p could be a negative regulator of the Prp16p ATPase. While further experiments are required to distinguish among these possibilities, we favor the first model for several reasons. First, Δisy1 mutants by themselves decrease the efficiency of the first step of splicing, consistent with a role for Isy1p in maintaining the conformation required for the first chemical reaction. Second, this phenotype is strongly exacerbated by alleles of U6 that are themselves important for the first step of splicing (see below). Finally, Δisy1 reduces fidelity of 3′-splice site recognition, as would be predicted if progression to the second-step spliceosomal conformation is promoted. However, a more direct role of Isy1p, and possibly other NTC components, in surveillance of 3′-splice site selection at the exon ligation step also remains possible. This possibility is suggested by the genetic and physical interactions of NTC components with second-step factors (Ben-Yehuda et al. 2000; Makarov et al. 2002; Ohi et al. 2002), and by the observation that the related human CDC5/Prp19 complex is required for the second step based on a depletion/complementation study (Ajuh et al. 2000).
Our data showing that removal of Isy1p favors release of Prp16p from the spliceosomal complex also offers a mechanistic interpretation of how Prp16p may govern the discard pathway for aberrant lariat intermediates. In agreement with a previous hypothesis (Madhani and Guthrie 1994), we find that mutations in the ATPase domain of the protein affect its dissociation from first-step products. We propose that when Prp16p is stalled on the spliceosome, it stabilizes the mutant lariat intermediate, sparing it from a degradative pathway. Indeed, a Dbr1p-dependent degradation pathway for lariat intermediates has recently been identified (Hilleren and Parker 2003). This study showed that the event triggering decay is debranching of the intermediate, which, in turn, allows its export to the cytoplasm and digestion by exonucleases. Consistent with the view that retention of faulty substrates within the spliceosomes prevents or significantly slows down access of degradative enzymes, Dbr1p cannot generally act on introns if the spliceosome has not yet disassembled (Martin et al. 2002). Thus, deletion of ISY1 counteracts the stabilization of lariat intermediates both by triggering release of Prp16p, which allows access to Dbr1p, and by disfavoring first-step catalysis.
Interactions of Isy1p with U6 are required for the first step of splicing
The strong synthetic lethality between the Δisy1 mutation and several U6 mutations (U6-U57A, U6-ΔC43, or U6-ΔA44), and their combined negative effect on the first step of splicing (Fig. 5) indicate that Isy1p functionally interacts with the U6 snRNA during the first step. We propose that the first-step role of Isy1p is to favor the observed NTC-dependent stable association of U6 with the spliceosome (Chan et al. 2003). A functional connection between residues 42–44 in U6, Isy1p, and possibly other components of the NTC, finds support in a series of elegant studies by Cheng and coworkers showing that NTC binding to the spliceosome promotes the proper interactions between U6 and the 5′-splice site necessary for first-step catalysis (Chan et al. 2003; S.-C. Cheng, pers. comm.). Specifically, NTC association with the spliceosome favors switching of an early interaction between the UGU at positions +4 to +6 in the intron from the upstream ACA trinucleotide element at positions 42–44 in U6 to a later interaction with the downstream ACA at positions 47–49 of U6 within the ACAGAG motif (Fig. 6B; Chan et al. 2003). Because this switch leads to catalytic activation of the spliceosome, the negative effect on first-step catalysis by the Δisy1, U6-ΔC43, and U6-ΔA44 mutations could be readily explained by the synergistic destabilization of U6 and NTC interactions with the spliceosome.
In a similar fashion, the first-step inhibition by the Δisy1 and U6-U57A mutations may result from the weakening of additional contacts involving Isy1p that the NTC may engage with U2/U6 helix Ia (Fig. 6B). However, nucleotide U57 in U6 is clearly playing a more complex, yet poorly understood function. This is suggested by the opposing effects of the U6-U57A and U6-U57C alleles on the two steps of splicing and their accordingly opposite patterns of genetic interactions with Δisy1 (Fig. 5), and with prp16-302 (McPheeters 1996; Chang and McPheeters 2000; Query and Konarska 2004). One possibility is that physical contacts between U6-U57 and the NTC play a role in modulating the transition between the two conformational states of the spliceosome.
In any case, our data suggest that Prp16p remodels an interaction involving Isy1p in the transition between the two steps of splicing. Because neither the deletion of ISY1 or U6 mutants alone, nor their combination, were able to bypass the requirement for Prp16p (T. Villa and C. Guthrie, unpubl.), Isy1p is unlikely to be the only target of Prp16p activity. Rather, more complex interactions involving U6 and the NTC are likely remodeled by Prp16p between the two steps of splicing. We speculate that this rearrangement is necessary for the establishment of tertiary interactions among the 5′- and 3′-splice sites, the branchpoint, and the ACAGAG motif of U6 that promote second-step catalysis (Fig. 6B; Collins and Guthrie 1999, 2001; Query and Konarska 2004). Suppression of the prp16-302 second-step block in the Δisy1, U6-U57A, U6-ΔC43, and U6-ΔA44 mutants likely results in all cases from the destabilization of antagonistic U6/NTC interactions that are required for the first step of splicing.
Implications
As proposed by Burgess and Guthrie (1993a), each ATP-dependent step in splicing can be envisioned as the rearrangement between two alternative conformations where coupling with the irreversible step of ATP hydrolysis provides the opportunity for proofreading. Two other ATPases governing early steps of spliceosome remodeling, Sub2p and Prp5p, have been shown to involve nonessential spliceosomal factors (Mud2p and Cus2p, respectively) (Perriman and Ares 2000; Kistler and Guthrie 2001). It is tempting to speculate that the involvement of dispensable factors in remodeling steps reflects a general evolutionary strategy that allows the cell to modulate the rates of these events, conferring plasticity to the system. More generally, since the rate of the transition between two alternative states is influenced by the relative stability of each state, we expect that spliceosomal rearrangements will depend on the activity and/or concentration of general factors acting at each end of the remodeling transition. This idea is consistent with the recent finding that variations in the concentration of several core components of the spliceosome can modulate alternative splicing events (Park et al. 2004).
In light of our findings, it will be particularly interesting to investigate the largely uncharacterized function of the conserved Prp19p-associated complex, which has been implicated in different rearrangements during the splicing cycle (Makarov et al. 2002; Chan et al. 2003). We anticipate that other NTC components may also modulate splicing accuracy. Notably, the founding member of the NTC, Prp19p, belongs to the U-box family of E3 ubiquitin ligases (Hatakeyama et al. 2001). A point mutation in the Prp19p U-box domain impairs both ubiquitin ligase activity and splicing (Cheng et al. 1993; Ohi et al. 2003). Additional findings provide support for a cycle of ubiquitination and de-ubiqitination during splicing. Proteomic analyses have identified three ubiquitinated splicing factors, including the tri-snRNP assembly factor Sad1p and the U5 snRNP protein Snu114p (Peng et al. 2003). Notably, Sad1p contains a ubiquitin hydrolase domain. Recent genetic evidence suggests a strong functional link between SNU114, SAD1, PRP19, and, additionally, PRP8 (Brenner and Guthrie 2005). Interestingly, Prp8p contains a Jab/MPN domain associated with ubiquitin removal (Grainger and Beggs 2005). Taken together, these observations raise the intriguing possibility that post-translational modification of the spliceosomal active site may influence fidelity.
Materials and methods
Yeast strains and plasmids
All Saccharomyces cerevisiae strains are in the S288C background and are listed in Supplementary Table 1. Yeast manipulations were carried out using standard laboratory procedures. Disruption of ISY1 was generated by one-step PCR using pFA6a-KanMX6 as a template (Petracek and Longtine 2002) and confirmed by PCR analysis. Similarly, pFA6a-TAP-HIS3MX6 (Ghaemmaghami et al. 2003) was used for introduction of the TAP tag at the C terminus of PRP16 and prp16-302 on plasmids pSB58 (Burgess and Guthrie 1993b) and pHM115 (Madhani and Guthrie 1994) to generate plasmids pTV102 and pTV107, respectively. Correct integration was confirmed by PCR analysis and by sequencing, and the expression of TAP-tagged proteins was tested by Western blotting. Plasmids pSB2, pSX6U, pSX6T, and derivatives containing the U6-ΔC43 and U6-ΔA44 mutations were described elsewhere (Madhani et al. 1990; Burgess and Guthrie 1993b; Madhani and Guthrie 1994). Plasmids containing the U6-U57A and U6-U57C mutations (McPheeters 1996) were a kind gift of David McPheeters (Case Western University, Cleveland, OH). ACT1-CUP1 reporter plasmids were described previously (Burgess and Guthrie 1993b; Lesser and Guthrie 1993a; Umen and Guthrie 1995; Collins and Guthrie 1999).
Screen for prp16-302 suppressors
Strain yTV198 containing prp16-302 on plasmid pHM115 was mutagenized with ethyl methane sulfonate (EMS) to 18.8% survival. Cells were plated on 100 YPD plates at 5000 cells/plate for 10 d at 16°C. 31 colonies were isolated that retested for growth at the restrictive temperature (cs+) and had also acquired a thermal-sensitive (ts–) phenotype at 37°C. After transformation of these cells with fresh pHM115 and backcrossing to a PRP16 shuffle strain, five recessive extragenic suppressors and three intragenic suppressors were identified. After curing the backcrossed diploid strain from the PRP16 plasmid on 5-FOA, and following analysis of tetrads, only three extragenic suppressors showed cosegregation of the cs+ and ts– phenotypes. These extragenic suppressors were transformed with a YCp50-based genomic library (Rose et al. 1987), and plasmids were isolated from colonies that reverted to a cold-sensitive growth at 16°C. For all three suppressors, sequencing of the library plasmids revealed overlapping fragments from chromosome X, which were narrowed down to the ISY1 gene by subcloning. Crossing of the suppressor strains among each other confirmed that the suppressors belonged to the same complementation group. Genomic DNA from the suppressor strains was prepared and the ISY1 gene was cloned and sequenced, in all cases revealing a single-nucleotide change (C to T) at position +220 in the open reading frame that creates a premature termination codon resulting in a truncated protein of 74 out of 235 amino acids.
Copper assays
Triplicate cultures were grown to mid-log phase in –Leu medium to maintain ACT1-CUP1 reporter plasmids. Cultures were then serially diluted in fivefold decrements starting from 0.3 OD600, and equal volumes were dropped onto plates containing different concentration of CuSO4, ranging from 0.007 to 2.0 mM (Lesser and Guthrie 1993a). Plates were scored and photographed after 5 d at 30°C.
Splicing
Extracts were prepared and radiolabeled actin pre-mRNA was spliced as described (Umen and Guthrie 1995). A 100-fold excess of unlabeled pre-mRNA was added to splicing reactions incubated in the cold before shifting to high temperature to prevent de novo spliceosome assembly on the labeled precursor.
Immunoprecipitations and TAP purifications
Immunoprecipitations using the polyclonal anti-Prp16p antibodies were performed as previously described (Schwer and Guthrie 1991) except that all IP buffers contained 0.05% NP-40. Western blotting was used to confirm that equal amounts of wild-type and mutant Prp16p were immunoprecipitated. Signals from coimmunoprecipitated RNA species were quantitated by phosphorimage analysis and expressed relative to the wild-type control arbitrarily set to 100, according to [(LI + E1)/P]IP/[(LI + E1)/P]rxn, where P is the precursor, LI is the lariat intermediate, and E1 is the 5′ exon.
Tandem affinity purifications were performed as described (Rigaut et al. 1999), using splicing extracts from cells grown at 28°C or shifted for 7 h to 16°C. Complexes were analyzed by Western blotting with the following rabbit polyclonal antibodies: anti-Prp16p (1:2000), anti-Ntc30p (1:1000), and anti-Ntc20p (1:1000). The anti-Ntc30p and anti-Ntc20p (C.H. Chen et al. 2001) were a kind gift of Soo-Chen Cheng (Institute of Molecular Biology, Academia Sinica, Nankang, Taiwan, Republic of China).
RNA analysis
RNA was extracted from exponentially growing yeast cultures by the hot-phenol method (Schmitt et al. 1990). Primer extension analyses were performed on 10 μg of total RNA as described (Boorstein and Craig 1989). Data were quantitated by phosphorimage analysis and internally controlled using U14 snoRNA levels as a standard.
Acknowledgments
This work is dedicated to the memory of Luigi Villa (1934–2001). We thank John Abelson, Tomas Aragon, Richard Bennett, Soo-Chen Cheng, Magda Konarska, Hiten Madhani, Charles Query, Stephen Rader, Tobias Walther, and the Guthrie lab for helpful discussions and critical reading of the manuscript; Soo-Chen Cheng and David McPheeters for sharing of reagents; and Soo-Chen Cheng, Magda Konarska, and Charles Query for communication of results prior to publication. We are grateful to Michael Dinglasan for excellent technical support, and to Judy Piccini for administrative assistance. T.V. was supported by fellowships from the Human Frontier Science Program Organization and from the American Heart Association. C.G. is an American Cancer Society Research Professor of Molecular Genetics. This work was supported by NIH grant GM21119 to C.G.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1336305.
References
- Ajuh P., Kuster, B., Panov, K., Zomerdijk, J.C., Mann, M., and Lamond, A.I. 2000. Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J. 19: 6569–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yehuda S., Dix, I., Russell, C.S., McGarvey, M., Beggs, J.D., and Kupiec, M. 2000. Genetic and physical interactions between factors involved in both cell cycle progression and pre-mRNA splicing in Saccharomyces cerevisiae. Genetics 156: 1503–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boorstein W.R. and Craig, E.A. 1989. Primer extension analysis of RNA. Methods Enzymol. 180: 347–369. [DOI] [PubMed] [Google Scholar]
- Brenner T.J. and Guthrie, C. 2005. Genetic analysis reveals a role for the C-terminus of the S. cerevisiae GTPase Snu114 during spliceosome activation. Genetics (in press). [DOI] [PMC free article] [PubMed]
- Burge C.B., Tuschl, T.H., and Sharp, P.A. 1998. Splicing of precursors to mRNAs by the spliceosomes. In RNA world II (eds. R.F. Gesteland et al.), pp. 525–560. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Burgess S.M. and Guthrie, C. 1993a. Beat the clock: Paradigms for NTPases in the maintenance of biological fidelity. Trends Biochem. Sci. 18: 381–384. [DOI] [PubMed] [Google Scholar]
- ___. 1993b. A mechanism to enhance mRNA splicing fidelity: The RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell 73: 1377–1391. [DOI] [PubMed] [Google Scholar]
- Burgess S., Couto, J.R., and Guthrie, C. 1990. A putative ATP binding protein influences the fidelity of branchpoint recognition in yeast splicing. Cell 60: 705–717. [DOI] [PubMed] [Google Scholar]
- Chan S.P., Kao, D.I., Tsai, W.Y., and Cheng, S.C. 2003. The Prp19p-associated complex in spliceosome activation. Science 302: 279–282. [DOI] [PubMed] [Google Scholar]
- Chang J.S. and McPheeters, D.S. 2000. Identification of a U2/U6 helix la mutant that influences 3′ splice site selection during nuclear pre-mRNA splicing. RNA 6: 1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.H., Tsai, W.Y., Chen, H.R., Wang, C.H., and Cheng, S.C. 2001. Identification and characterization of two novel components of the Prp19p-associated complex, Ntc30p and Ntc20p. J. Biol. Chem. 276: 488–494. [DOI] [PubMed] [Google Scholar]
- Chen J.Y., Stands, L., Staley, J.P., Jackups Jr., R.R., Latus, L.J., and Chang, T.H. 2001. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol. Cell 7: 227–232. [DOI] [PubMed] [Google Scholar]
- Chen C.H., Yu, W.C., Tsao, T.Y., Wang, L.Y., Chen, H.R., Lin, J.Y., Tsai, W.Y., and Cheng, S.C. 2002. Functional and physical interactions between components of the Prp19p-associated complex. Nucleic Acids Res. 30: 1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.C., Tarn, W.Y., Tsao, T.Y., and Abelson, J. 1993. PRP19: A novel spliceosomal component. Mol. Cell. Biol. 13: 1876–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C.A. and Guthrie, C. 1999. Allele-specific genetic interactions between Prp8 and RNA active site residues suggest a function for Prp8 at the catalytic core of the spliceosome. Genes & Dev. 13: 1970–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 2000. The question remains: Is the spliceosome a ribozyme? Nat. Struct. Biol. 7: 850–854. [DOI] [PubMed] [Google Scholar]
- ____. 2001. Genetic interactions between the 5′ and 3′ splice site consensus sequences and U6 snRNA during the second catalytic step of pre-mRNA splicing. RNA 7: 1845–1854. [PMC free article] [PubMed] [Google Scholar]
- Couto J.R., Tamm, J., Parker, R., and Guthrie, C. 1987. A trans-acting suppressor restores splicing of a yeast intron with a branch point mutation. Genes & Dev. 1: 445–455. [DOI] [PubMed] [Google Scholar]
- Dix I., Russell, C., Yehuda, S.B., Kupiec, M., and Beggs, J.D. 1999. The identification and characterization of a novel splicing protein, Isy1p, of Saccharomyces cerevisiae. RNA 5: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S., Huh, W.K., Bower, K., Howson, R.W., Belle, A., Dephoure, N., O'Shea, E.K., and Weissman, J.S. 2003. Global analysis of protein expression in yeast. Nature 425: 737–741. [DOI] [PubMed] [Google Scholar]
- Grainger R.J. and Beggs, J.D. 2005. Prp8 protein: At the heart of the spliceosome. RNA 11: 533–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama S., Yada, M., Matsumoto, M., Ishida, N., and Nakayama, K.I. 2001. U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276: 33111–33120. [DOI] [PubMed] [Google Scholar]
- Hilleren P.J. and Parker, R. 2003. Cytoplasmic degradation of splice-defective pre-mRNAs and intermediates. Mol. Cell 12: 1453–1465. [DOI] [PubMed] [Google Scholar]
- Hopfield J.J. 1974. Kinetic proofreading: A new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. 71: 4135–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurica M.S. and Moore, M.J. 2003. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell 12: 5–14. [DOI] [PubMed] [Google Scholar]
- Kistler A.L. and Guthrie, C. 2001. Deletion of MUD2, the yeast homolog of U2AF65, can bypass the requirement for sub2, an essential spliceosomal ATPase. Genes & Dev. 15: 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser C.F. and Guthrie, C. 1993a. Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics 133: 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 1993b. Mutations in U6 snRNA that alter splice site specificity: Implications for the active site. Science 262: 1982–1988. [DOI] [PubMed] [Google Scholar]
- Madhani H.D. and Guthrie, C. 1992. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 71: 803–817. [DOI] [PubMed] [Google Scholar]
- ____. 1994. Genetic interactions between the yeast RNA helicase homolog Prp16 and spliceosomal snRNAs identify candidate ligands for the Prp16 RNA-dependent ATPase. Genetics 137: 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani H.D., Bordonne, R., and Guthrie, C. 1990. Multiple roles for U6 snRNA in the splicing pathway. Genes & Dev. 4: 2264–2277. [DOI] [PubMed] [Google Scholar]
- Makarov E.M., Makarova, O.V., Urlaub, H., Gentzel, M., Will, C.L., Wilm, M., and Luhrmann, R. 2002. Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome. Science 298: 2205–2208. [DOI] [PubMed] [Google Scholar]
- Martin A., Schneider, S., and Schwer, B. 2002. Prp43 is an essential RNA-dependent ATPase required for release of lariatintron from the spliceosome. J. Biol. Chem. 277: 17743–17750. [DOI] [PubMed] [Google Scholar]
- McPheeters D.S. 1996. Interactions of the yeast U6 RNA with the pre-mRNA branch site. RNA 2: 1110–1123. [PMC free article] [PubMed] [Google Scholar]
- Ninio J. 1974. A semi-quantitative treatment of missense and nonsense suppression in the strA and ram ribosomal mutants of Escherichia coli. Evaluation of some molecular parameters of translation in vivo. J. Mol. Biol. 84: 297–313. [DOI] [PubMed] [Google Scholar]
- Noble S.M. and Guthrie, C. 1996. Identification of novel genes required for yeast pre-mRNA splicing by means of cold-sensitive mutations. Genetics 143: 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi M.D., Link, A.J., Ren, L., Jennings, J.L., McDonald, W.H., and Gould, K.L. 2002. Proteomics analysis reveals stable multiprotein complexes in both fission and budding yeasts containing Myb-related Cdc5p/Cef1p, novel pre-mRNA splicing factors, and snRNAs. Mol. Cell. Biol. 22: 2011–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi M.D., Vander Kooi, C.W., Rosenberg, J.A., Chazin, W.J., and Gould, K.L. 2003. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Biol. 10: 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.W., Parisky, K., Celotto, A.M., Reenan, R.A., and Graveley, B.R. 2004. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc. Natl. Acad. Sci. 101: 15974–15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J., Schwartz, D., Elias, J.E., Thoreen, C.C., Cheng, D., Marsischky, G., Roelofs, J., Finley, D., and Gygi, S.P. 2003. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21: 921–926. [DOI] [PubMed] [Google Scholar]
- Perriman R. and Ares Jr., M. 2000. ATP can be dispensable for prespliceosome formation in yeast. Genes & Dev. 14: 97–107. [PMC free article] [PubMed] [Google Scholar]
- Petracek M.E. and Longtine, M.S. 2002. PCR-based engineering of yeast genome. Methods Enzymol. 350: 445–469. [DOI] [PubMed] [Google Scholar]
- Query C.C. and Konarska, M.M. 2004. Suppression of multiple substrate mutations by spliceosomal prp8 alleles suggests functional correlations with ribosomal ambiguity mutants. Mol. Cell 14: 343–354. [DOI] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko, A., Rutz, B., Wilm, M., Mann, M., and Seraphin, B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17: 1030–1032. [DOI] [PubMed] [Google Scholar]
- Rose M.D., Novick, P., Thomas, J.H., Botstein, D., and Fink, G.R. 1987. A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene 60: 237–243. [DOI] [PubMed] [Google Scholar]
- Schmitt M.E., Brown, T.A., and Trumpower, B.L. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18: 3091–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B. and Guthrie, C. 1991. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature 349: 494–499. [DOI] [PubMed] [Google Scholar]
- ____. 1992. A conformational rearrangement in the spliceosome is dependent on PRP16 and ATP hydrolysis. EMBO J. 11: 5033–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siatecka M., Reyes, J.L., and Konarska, M.M. 1999. Functional interactions of Prp8 with both splice sites at the spliceosomal catalytic center. Genes & Dev. 13: 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley J.P. and Guthrie, C. 1998. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell 92: 315–326. [DOI] [PubMed] [Google Scholar]
- Tanner N.K. and Linder, P. 2001. DExD/H box RNA helicases: From generic motors to specific dissociation functions. Mol. Cell 8: 251–262. [DOI] [PubMed] [Google Scholar]
- Tarn W.Y., Hsu, C.H., Huang, K.T., Chen, H.R., Kao, H.Y., Lee, K.R., and Cheng, S.C. 1994. Functional association of essential splicing factor(s) with PRP19 in a protein complex. EMBO J. 13: 2421–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen J.G. and Guthrie, C. 1995. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes & Dev. 9: 855–868. [DOI] [PubMed] [Google Scholar]
- Vijayraghavan U., Parker, R., Tamm, J., Iimura, Y., Rossi, J., Abelson, J., and Guthrie, C. 1986. Mutations in conserved intron sequences affect multiple steps in the yeast splicing pathway, particularly assembly of the spliceosome. EMBO J. 5: 1683–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]