

Abstract
Aberrant alternative splicing and abnormal alternative splicing events (ASEs) in glioblastoma multiforme (GBM) remain largely elusive. The prognostic-associated ASEs in GBM were identified and summarized into 123 genes using GBM and LGG datasets from ASCancer Atlas and TCGA. The eleven genes (C2, COL3A1, CTSL, EIF3L, FKBP9, FN1, HPCAL1, HSPB1, IGFBP4, MANBA, PRKAR1B) were screened to develop an alternative splicing prognostic risk score (ASRS) model through machine learning algorithms. The model was trained on the TCGA-GBM cohort and validated with four external datasets from CGGA and GEO, achieving AUC values of 0.808, 0.814, 0.763, 0.859, and 0.836 for 3-year survival rates, respectively. ASRS could be an independent prognostic factor for GBM patients (HR > 1.8 across three datasets) through multivariate Cox regression analysis. The high-risk group demonstrated poorer prognosis, elevated immune scores, increased levels of immune cell infiltration, and greater differences in drug sensitivity. We found that FN1, used for model construction, contained 4 abnormal ASEs resulting in high expression of non-canonical transcripts and the presence of premature termination codon. These abnormal ASEs may be regulated by tumour-related splicing factors according to the PPI network. Furthermore, both mRNA and protein levels of FN1 were highly expressed in GBM compared to LGG, correlating with poor prognosis in GBM. In conclusion, our findings highlight the role of ASEs in affecting the progression of GBM, and the model showed a potential application for prognostic risk of patients. FN1 may serve as a promising splicing biomarker for GBM, and mechanisms of processes of aberrant splicing need to be revealed in the future.
Keywords: Glioblastoma, Alternative splicing, ASRS prognostic model, FN1
Subject terms: Cancer, Computational biology and bioinformatics
Introduction
Glioblastoma multiforme (GBM) is the most prevalent malignant primary brain tumour in adults1, with a 1-year survival rate of 40.9% and a 5-year survival rate of 6.6%2,3. Clinical research results in China are consistent with observations in other countries4,5. The poor prognosis of GBM patients, which is in part due to the high heterogeneity of tumour cells and the primary or acquired resistance of tumour cells to chemotherapy, immunotherapy, and radiotherapy6,7. Distinct mRNA types produced by alternative splicing are an important mechanism for resistance to these therapies8.
Alternative splicing is an important post-transcriptional regulation process by which different combinations of exons are joined together and result in the production of multiple forms of mRNA from a single pre-mRNA9, which in turn may affect gene expression levels and mRNA translation into proteins, conferring different functional properties10–13. It has been shown that splicing regulatory abnormality is highly correlated with tumourigenesis and progression14–16, and that dysregulation of alternative splicing exhibit distinct patterns in different tumours and cells17–20.
In glioma, increased splicing burden leads to an increase in abnormal alternative splicing events (ASE), such as upregulation of REST that mediates the splicing of NF1 into a less active isoform and leads to activation of the RAS/MAPK pathway, which reduces the survival of glioblastoma patients21. Processes of the splicing regulation may provide targets for cancer therapy, such as targeted splicing factors, antisense oligonucleotide targeted Splicing Factor-RNA interactions, and activation of the Nonsense-Mediated mRNA Decay (NMD) mechanism22–26. Therefore, investigating genes subjected to aberrant alternative splicing as GBM prognostic markers is a promising research direction.
In this study, we integrated alternative splicing data and gene expression data to identify genes that are regulated by abnormal splicing in GBM progression with prognostic value, and constructed a prognostic risk model. The model was used to predict the prognosis of GBM patients, the tumour microenvironment differences and potentially effective anti-cancer drugs. FN1, used for the risk model construction, which contained 4 abnormal ASEs in the GBM and LGG comparison, and one ASE is a known cancer-associated splicing event, which was reported in other tumours27–30. Finally, we focused on FN1, and the comprehensive analysis showed this gene may be a potential splicing biomarker for GBM. The flow chart of this study is shown in Fig. 1.
Fig. 1.

Workflow of this study.
Methods
Data collection and pre-processing
RNA-seq data collection
We used the "TCGAbiolinks" package to obtain RNA-seq data and corresponding clinical information for TCGA-LGG and TCGA-GBM samples from The Cancer Genome Atlas database (TCGA, https://portal.gdc.cancer.gov/)31. A total of 546 LGG samples and 168 GBM samples were obtained. The original CGGA-693 cohort with 249 GBM samples and CGGA-325 cohort with 139 GBM samples downloaded from the Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org), were used to validate model performance32.
Microarray data collection
To further validate the performance and generalizability of the ASRS model, microarray datasets of GSE4412 and GSE43378 were downloaded from the GEO database, comprising a total of 84 and 49 GBM samples, respectively.
Protein array data collection
The GBM and LGG reverse phase protein array (RPPA) dataset were downloaded from TCGA. RPPA data was available on 232 GBM samples and 429 LGG samples.
Alternative splicing events data acquisition
The ASEs data were obtained from ASCancer Atlas (https://ngdc.cncb.ac.cn/ascancer/home). The GBM dataset comprised 69,324 ASEs from 164 samples, while the LGG dataset included 60,213 ASEs from 515 samples33. The dataset contained information on various aspects of each ASE, such as the type of ASE, exon loci, gene symbols, and the Percent Spliced In (PSI, Ψ) value. These two datasets contained five major types of AS, including exon skipping (ES), alternative 5' splice site (A5SS), alternative 3' splice site (A3SS), intron retention (IR), and mutually exclusive exons (MEX or MXE, ME).
Splicing factor data collection
We integrated the splicing factors from the literature, the SpliceAid2 database (www.introni.it/spliceaid.html), and the results of retrieving Gene Ontology (http://www.geneontology.org) with the keyword of "mRNA splicing", then we identified 529 genes with mRNA splicing function or associated with splicing regulation as the splicing factor gene list dataset in this study (Table S1)34–38.
Data pre-processing
All cases with TCGA and CGGA data that meet the following criteria included: 1. An available histological diagnosis of GBM; 2. Patients with available ASCancer Atlas data; 3. Patients with follow-up information and survival status more than 30 days after the initial diagnosis. A total of 156 CGGA-693 samples and 127 CGGA-325 samples were subsequently used to validate model performance. The TCGA-GBM gene expression data, clinical information and ASEs data from ASCancer Atlas contained 150 common samples across these datasets, and 515 samples for TCGA-LGG. To ensure data quality, ASEs of GBM with over 80% of sample "nulls" and PSI standard deviation less than 0.01 were removed. A total of 52,370 ASEs of GBM as clean data for prognostically critical genes analysis. The original GBM and LGG ASEs data were intersected to obtained 36,923 common ASEs for differentially alternative splicing events analysis. RPPA data and mRNA expression data from GBM were extracted according to sample ID, and a total of 72 samples were used for analyzing the correlation between mRNA and protein expression. All primary tumour samples from GBM and LGG RPPA data were employed for a comparative analysis of fibronectin expression.
Analysis of differentially expressed splicing factors
The "TCGAbiolinks" package was used to perform differential expression analysis on GBM and LGG datasets. Differentially expressed genes (DEGs) were filtered by |log2FC|> 1 (FC, Fold Change) and adjusted P-value < 0.05. The DEGs were intersected with the splicing factor list to obtain the set of abnormally expressed splicing factors (DEG-SF).
Identification of prognostically critical genes regulated by abnormal splicing in GBM
ASEs associated with overall survival (OS) were screened using univariate Cox proportional hazards regression analysis and the gene involved in the above ASEs was defined as an ASE prognostic-associated gene (ASE-PAG). Pearson's correlation analysis (|cor|> 0.5 and P-value < 0.01) was performed on the DEG-SF and ASE-PAG to screen out ASE-PAG, which were considered to be regulated by DEG-SFs associated with glioblastoma progression.
Construction and validation of prognostic model
The gene expression data of prognostic key genes regulated by abnormal splicing was extracted from TCGA-GBM RNA-seq dataset as the training set for constructing the prognostic model. Firstly, The "randomForestSRC" (Random Survival Forest, RSF) and "xgboost" (Xtreme Gradient Boosting, XGBoost) packages were used to screen the features in the training set, and the common features were selected for subsequent analysis. Then, the LASSO regression model was further screened for the above common features, and the result was obtained by the "glmnet" package39. Finally, the screened features were utilized to construct an Alternative Splicing Risk Score (ASRS) prognostic model according to the following formula:
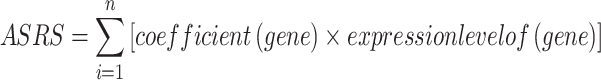 |
According to the median ASRS, all samples were divided into a high-risk and a low-risk group, Kaplan‒Meier (K‒M) survival analysis was performed using the "survival" package to compare the difference in Overall Survival (OS) between the two groups. The receiver operating characteristic (ROC) curves were drawn using the "timeROC" package to evaluate the performance and prognostic value of the ASRS. In addition, four independent datasets, CGGA-693, CGGA-325, GSE4412 and GSE43378 cohorts, were used to test the model performance.
Analysis of clinical characteristics and enrichment pathways based on ASRS
To investigate ASRS as an independent prognostic factor for GBM patients, univariate and multivariate Cox proportional hazard regression analyses were performed in the CGGA-693 dataset along with seven characteristics, including gender, age, IDH mutation, radiation therapy, chemotherapy, 1p/19q co_deletion, and MGMTp methylation status. The "rms" and "regplot" packages were used to construct a nomogram, and calibration curves and ROC curves were utilized to evaluate the performance. Finally, we used the "GSVA" package to carry out gene set enrichment analysis (GSEA) between the two groups40.
Analysis of tumour environment
The "ESTIMATE" package was used to assess the immune scores. Six algorithms in "TIMER 2.0" were used to calculate immune cell infiltration scores41. Subsequently, the correlation coefficients (Spearman's correlation P < 0.05) were calculated across the cellular immune infiltration scores and the ASRS. In addition, survival analyses were conducted to evaluate the prognostic impact of cellular immune infiltration scores on GBM prognosis using Kaplan‒Meier curves. We identified differentially expressed immune checkpoint-related genes (ICGs) sourced from a previous study between the risk groups42, and calculated correlation coefficients between these differential ICGs and 11 features. Additionally, drug sensitivity analysis was performed using the "oncoPredict" package to identify drugs that showed significant differences in sensitivity between the risk groups (P < 0.05)43.
Analysis of DASEs
The differential ASEs (DASEs) between GBM and LGG were identified using the "pischomcis" package44,45. The filtering criteria for significantly DASEs had adjusted P-value < 0.05 (Wilcoxon's signed-ranks test and Benjamini–Hochberg method to adjust the P-value for multiple testing) and delta Median PSI ≥ 0.05. GEPIA2 (http://gepia2.cancer-pku.cn/) was used to analyse the expression of FN1 and CTSL transcripts46. Gene information about exon coordinates, transcript ID, and exon ID was retrieved from the Ensembl database (http://www.ensembl.org/) (GRCh37.p13)47,48.
Analysis of PPI network for DEG-SFs
A protein–protein interaction (PPI) network was constructed in Cytoscape (v3.9.1) for selected DEG-SFs according to correlation coefficients (|cor|> 0.5 and P-value < 0.01) between DEG-SFs and genes with DASEs49. Protein interactions were downloaded from the BioGRID50, IntAct51, MINT52, and Uniprot53 databases via Cytoscape. Subsequently, gene ID conversion was performed using the "biomaRt" package. Pathway modules were analysed using the "ClueGO" App in Cytoscape, with biological pathways data sourced from KEGG and REACTOME databases54–57. Finally, 198 critical genes associated with glioblastoma were mapped into the PPI network. These critical genes were curated from the literature by BioGRID curators.
All analyses in this study were carried out using the R platform and packages, and the statistical significance is P < 0.05 (T-test or Wilcoxon's signed-ranks test).
Results
Identification of prognostically critical genes regulated by abnormal splicing in GBM
Identification of genes associated with prognostic ASEs
We identified 4,337 ASEs significantly associated with GBM prognosis (P < 0.05) from 52,370 ASEs of the TCGA-GBM dataset. These prognostically associated ASEs were categorized into five major types of alternative splicing, with the highest percentage of ES events at 31.75%, followed by IR events at 30.30%, and the other at 22.34%, 13.95%, and 1.66% for A3SS, A5SS, and MEX events, respectively (Table 1). In total, 2,267 genes contained the 4,337 ASEs, which we defined as ASE prognostic-associated gene (ASE-PAG).
Table 1.
Statistical table of prognostic alternative splicing events.
| Type of ASE | Number of ASE | Number of Gene | ASE Ratio (%) |
|---|---|---|---|
| A3SS | 969 | 782 | 22.34 |
| A5SS | 605 | 535 | 13.95 |
| ES | 1,377 | 866 | 31.75 |
| IR | 1,314 | 983 | 30.30 |
| MEX | 72 | 35 | 1.66 |
Identification of differentially expressed splicing factors
A total of 2,683 DEGs were identified between TCGA-LGG and TCGA-GBM, including 1,185 up-regulated genes and 1,498 down-regulated genes in GBM (Fig. 2A). In total, 27 differentially expressed splicing factors (DEG-SFs) were also extracted from DEGs (Fig. 2B, Table S2).
Fig. 2.

Identification of DEG-SF in GBM and enrichment analysis of 123 prognostic related genes potentially regulated by DEG-SF. A The differentially expressed genes showed by volcano plot between TCGA-GBM and TCGA-LGG. The top 10 up- and down-regulated genes were labeled gene symbols. B Hierarchical clustering analysis of 27 DEG-SFs in the TCGA-GBM and TCGA-LGG combined gene expression data. C KEGG enrichment analysis of 123 genes with prognostic value and related to DEG-SF regulation. D GO enrichment analysis of 123 genes with prognostic value and related to DEG-SF regulation. DEG-SFs means differentially expressed splicing factors.
Identification and functional analysis of the critical genes regulated by abnormal splicing
Correlation analysis of 27 DEG-SFs and 2,267 ASE-PAGs was performed and 1,006 genes were regulated by abnormal splicing in GBM. Further univariate Cox regression analysis obtained 123 prognostic critical genes regulated by DEG-SFs (Table S3). These critical genes were enriched in 12 KEGG pathways and 142 GO Terms (Fig. 2C-D, Tables S4 and S5). Enriched KEGG pathways such as ECM-receptor interaction, Focal adhesion, and Proteoglycans in cancer, along with enriched GO terms such as Cytoplasmic translation, Extracellular matrix (ECM), are related to tumour development, progression, metastasis, and treatment58–64.
Construction and assessment of an AS-based prognostic risk model for GBM
In the results from the RSF, we identified 76 genes with importance scores greater than 0 (Fig. 3A-B), while the top 20 genes ranked by importance scores were selected by XGBoost (Fig. 3C). Both algorithms yielded a total of 14 common feature genes (Fig. 3D). Next, There were 11 features, C2, COL3A1, CTSL, EIF3L, FKBP9, FN1, HPCAL1, HSPB1, IGFBP4, MANBA, and PRKAR1B, determined by LASSO when the regularization parameter λ reached its minimum value (Fig. 3E-F). Finally, a multivariate Cox regression risk model was constructed as follows:
 |
Fig. 3.

Screening of splicing-associated prognostic genes. A-B Random survival forest (RSF) error rate versus the number of classification trees and the relative importance of 76 genes with importance score more than 0 (Top 40 genes with absolute importance score > 0 are shown). C Top 20 important genes in XGBoost screening. D Venn diagram showed 14 genes screened by both RSF and XGBoost. E the cross-validation result of LASSO, and the dotted line on the left indicated the value of the harmonic parameter log(λ) when the error of the model is minimized. Eleven genes were selected. F The LASSO coefficient profiles of the 14 genes.
Based on the median ASRS, Kaplan‒Meier survival curves showed that TCGA-GBM patients in the high-risk group had a poorer prognosis (Fig. 4A,C). ROC curve showed that the prediction rates of 1-, 2- and 3-year survival in the training set were all around 0.8, indicating a robust model prediction (Fig. 4D). The expression levels of the 11 features were significantly different in the high- and low-score groups, for example, FN1 and CTSL were upregulation in the high-score group (Fig. 4B). It has been reported that overexpression of FN1 predicts poorer prognosis in GBM65. The four external test sets produced similar results, and GSE43378 dataset exhibited the best performance with the prediction accuracy of more than rates 0.85 for 1-, 2- and 3-year OS (Fig. 4). The favorable performance of the ASRA model in external retrospective datasets indicated that it not only avoided overfitting but also demonstrated potential for clinical applications.
Fig. 4.

Construction and validation of the ASRS model. A Risk score distribution in the high-risk and low-risk groups of the TCGA-GBM training set, and four external test sets (CGGA-693, CGGA-325, GSE43378 and GSE4412 cohorts). B Heatmap of the expression of 11 model genes in the high-risk and low-risk groups of all cohorts. C Kaplan‒Meier curves of overall survival for all cohorts. Blue color means low-risk score, red color high-risk score. D Time-dependent ROC curves of all cohorts.
Analysis of clinical characteristics and enrichment pathways based on ASRS
The univariate and multivariate Cox regression analysis results suggested that GBM patients with high ASRS indicated a high risk of death in CGGA-693 dataset (HR=1.957 and 2.081, Fig. 5A,B). The nomogram spots could accurately quantify the survival rate (Fig. 5C), and the ROC curves showed reliable performance with AUC of 0.683, 0.788, and 0.814 for 1-, 2- and 3-year survival rates, respectively (Fig. 5D). The results of calibration curves showed that the nomogram predicted OS was close to the actual OS probability (Fig. 5E). Meanwhile, we obtained similar results in TCGA-GBM and CGGA-325 datasets (Figs. S1, S2). KEGG enriched pathways resulted by GSEA revealed that there were more immune activation-related pathways in the high-risk group, for example, complement and coagulation cascades, cytokine-cytokine receptor interaction, NOD-like receptor signaling pathway, cell adhesion molecules, and chemokine signaling pathway (Fig. 5F,G, and Table S6).
Fig. 5.

ASRS-based nomogram construction and biological functions analysis in the CGGA-693 cohort. A The results of univariate Cox regression analysis of clinical characteristics and ASRS. B The results of multivariate Cox regression analysis of clinical characteristics and ASRS. C Nomogram based on ASRS and clinical characteristics. "*" means P-value < 0.05, "**" P-value < 0.01, and "***" P-value < 0.001. D ROC curve analysis results. E Calibration curve analysis results. F Enrichment KEGG pathways of the high-risk and low-risk groups by GSEA analysis. G Enrichment GO terms of the high-risk and low-risk groups by GSEA analysis.
Tumour microenvironment analysis based on ASRS
The ESTIMATE analysis results showed that the high-risk group had significantly higher scores than the low-risk group (Fig. 6A), indicating a notable difference in the level of immune infiltration in tumours between risk groups of GBM based on ASRS. CIBERSORT analysis revealed seven immune cell types with significant differences in immune infiltration between risk groups. Specifically, regulatory T cell (Tregs), resting NK cell, and M0 Macrophage were significantly higher in the high-risk group. In contrast, B cell, activated NK cell, M1 and M2 macrophage were significantly lower in the high-risk group (Fig. 6B).
Fig. 6.

ASRS-based analysis of the tumour immune microenvironment. A ESTIMATE analysis for calculating stromal score, immune score, and overall ESTIMATE score in the high-risk and low-risk groups in the TCGA-GBM cohort. B Significant immune cell infiltration by CIBERSORT analysis in the high-risk and the low-risk groups. "*" means P-value < 0.05, and "**" P-value < 0.01. C The correlation analysis between ASRS and immune cell fraction calculated by 6 algorithms wrapped in TIMER2. D Kaplan‒Meier curves of overall survival for Macrophage M2 cell, Macrophage cell, Class-switched memory B cell and Cancer associated fibroblast cell infiltration in the TCGA-GBM cohort.
The concordance results of six TIMER2.0 algorithms showed that infiltration scores of several immune cell types were significantly correlated with ASRS, such as Macrophage, M0, M1 and M2 macrophage (Fig. 6C). and high infiltration level of M2 macrophage, Macrophages, Class-switched memory B cells, Cancer associated fibroblast was significantly associated with poorer prognosis in GBM patients (Fig. 6D).
Additionally, the enrichment score of the ICGs was significantly higher in the high-risk group (Fig. S3A). At the gene expression level, there were 9 ICGs, CD40, HLA-DOA, HLA-E, ICOSLG, TDO2, TIGIT, TNFRSF9, TNFSF4, and TNFSF14, which was significantly higher in the high-risk group (Fig. S3B). The expression levels of FN1 and CTSL were significantly correlated with the above ICGs (Fig. S3C). The results of drug sensitivity showed significant differences between two risk groups, such as SB216763, and Tozasertib (Fig. S3D).
All these results demonstrated that the ASRS is significantly associated with the tumour microenvironment, immune checkpoint function, and drug sensitivity characteristics, while some of the genes used for the modeling may involved in these functions.
Analysis of abnormal splicing regulation of FN1
A total of 4,812 significantly DASEs were obtained from GBM and LGG comparison. There were 8 DASEs in the CTSL, EIF3L, and FN1 (Table S7). FN1 contained 9 ES events (Table S8), three of which were DASEs, and one other event was also considered (delta Median PSI = 0.049). The exon 25 skipping event in FN1 (chr2:216257654-216257926, ENSE00000965897, 273 bp) had a more than twofold difference in the median PSI between GBM and LGG, suggesting that exon 25 is more inclusion in the transcript of FN1 in GBM (Fig. 7A-B). This DASE may affect the expression of FN1 transcripts, resulting in more transcripts production, then leading to higher expression levels in GBM, such as FN1-002 (ENST00000323926) and FN1-012 (ENST00000432072) (Fig. 7C). It's a cancer-associated splicing event (CASE) defined by ASCancer Atlas, which was found in lung cancer, prostate cancer, breast cancer, and colon cancer (Table S7)27–30.
Fig. 7.

Exploration of significant alternative splicing events in FN1. A Transcript structure visualization for 4 abnormal ASEs of exon skipping in FN1. B PSI median values and the case of affected FN1 transcripts in the GBM and LGG of the four ASEs. C The expression analysis of four FN1 transcripts potentially altered through ASEs in the TCGA-GBM and TCGA-LGG datasets by GEPIA2. D Expression level of FN1 in GTEx, TCGA-LGG, and TCGA-GBM cohorts. "****" means P-value < 0.0001. E Correlation analysis between FN1 and DEG-SF, and the expression level of RBMS1 is positive correlated with FN1 and has the most highest Pearson's correlation coefficient value 0.648. ASE means alternative splicing event. DEG-SFs means differentially expressed splicing factors.
Interestingly, the ES event (chr2_216257654:216257924) also occurred in exon 25, but this event retained the 2 bases (AG) at the start position of the exon, resulting in a code-shift mutation in the transcript, which allowed the presence of a premature termination codon (PTC) (Fig. 7A). This DASE with a significantly higher median PSI value in GBM (GBM: 0.229, LGG: 0.105) may generate more aberrant mRNA transcripts, leading to produce truncated proteins (Fig. 7B). The exon 25 was reported as a cancer-specific exon with high expression in pediatric solid and brain tumours, which contains EDB domain. The EDB domain encodes tumour-associated antigens which can serve as chimeric antigen receptor targets, and the EDB-CAR T cell has anti-tumour activity against pediatric sarcoma66. Our findings suggested that although highly expressed of exon 25, the PTC may lead to the absence of EDB domain in truncated proteins and could be involved in GBM acquired immunotherapy resistance, subsequently.
Additionally, exon 40.2 (chr2:216236832-216237023, ENSE00001728548, 192 bp), and exon 40.1-40.2 (chr2:216236832-216237098, ENSE00003500512, 267 bp) skipping events were observed. Exon 40.2 had a significantly higher median PSI value in GBM (GBM:0.792, LGG:0.721) leading to the higher expression level of FN1-008 (ENST00000446046) in GBM (Fig. 7A-C). All these results suggested that the combined effect of the 4 abnormal ASEs resulted in higher expression of non-canonical transcripts in the GBM samples, such as FN1-002, FN1-008, and FN1-012 (Fig. 7C). In terms of gene expression, The expression levels of FN1 significantly increased stepwise in normal brain tissue, LGG, and GBM samples (Fig. 7D). Consistent results were also observed in the validation of protein expression levels between GBM and LGG comparison (P-value < 0.05) (Fig. S4A). There was a strong correlation between the mRNA and protein expression levels of FN1 in GBM (R = 0.549, P-value = 9.29e-07) (Fig. S4B). Furthermore, elevated levels of both mRNA and protein expression were associated with poor prognosis in GBM (Fig. S4C,D).
There were four A5SS DASEs in CTSL, and the median PSI values were all significantly lower in the GBM samples than those in the LGG samples (Table S7). These DASE could affect expression levels of CTSL transcripts such as CTSL-002 in GBM and LGG (Fig. S5A-C).
Discussion
Currently, Alternative splicing regulation is still not elucidated for glioblastoma, especially whether splicing differences between LGG and GBM influence tumor progression, prognosis, and shaping of the tumour microenvironment still not fully clarified. Research in the high-grade diffuse glioma has shown increased alternative splicing burden compared with the normal brain21, and this result suggested that splicing burden is highly correlated with tumour progression, which is also reflected by the number of ASEs we used for LGG (6,0213) and GBM (6,9324). Our previous study has revealed that abnormal ASEs between LGG and GBM could characterize certain heterogeneity of glioblastoma with prognostic indications67. In this study, we aimed to identify ASEs associated with glioblastoma progression to aid in prognostic model construction, so the DEG-SFs were identified between GBM and LGG. The survival-related ASEs were summarised to gene expression level to construct the ASRS model, followed by a comprehensive analysis of both the model's performance and the associated genes.
We identified 27 DEG-SF and 123 prognostic critical genes were potentially regulated by DEG-SFs. These critical genes were used to construct a prognostic model after rigorous screening. In the training set, an ASRS prognostic signature was constructed using 11 genes, including C2, COL3A1, CTSL, EIF3L, FKBP9, FN1, HPCAL1, HSPB1, IGFBP4, MANBA, and PRKAR1B. The ROC curve results showed prediction accuracy around 80% for 1-, 2- and 3-year OS. Two CGGA cohorts and two microarray datasets from GEO were used to validate the model performance, yielding the best prediction accuracy of more than 85% for 1-, 2- and 3-year OS of GSE43378. Moreover, the ASRS was highly correlated with the OS in all datasets, which means that the high-score group (or the high-risk group) had a poorer prognosis. Cox regression analyses showed that the ASRS independently predicts GBM patient prognosis, demonstrating good performance regardless of other clinical factors. Prognostic models have been constructed using ASEs in previous studies34,68–71, and the PSI value of ASEs were used to construct models in GBM or GBM + LGG datasets. In comparison to previous studies, the ASRS model demonstrates the following advantages: 1. We summarised survival-related ASEs into gene expression level. Quantifying gene expression presents a more straightforward approach than quantifying splicing, thereby facilitating the clinical validation and application of the model in future. 2. Modeling with a suitable number of genes yielded good performance. 3. The model demonstrated consistent performance on both the training set and external test sets, effectively avoiding overfitting. We validated the model using four external test sets, especially on microarray datasets, confirming its potential generalization capabilities and clinical application.
Patients of high-risk group exhibited distinct characteristics, including increased levels of immune cell infiltration, elevated immune scores, and greater differences in drug sensitivity. Immune-related pathways activation, such as chemokine signaling pathway, and ECM receptor interaction, enhanced the ability of glioblastoma cell differentiation, migration, and invasion and associated with tumour immune infiltration. For example, the infiltration levels of Tregs and Macrophage were observed significantly increased in the high-risk group. Tregs can induce and maintain immunosuppression by up-regulating PD-L1, and it also plays a key role in the emergence of drug resistance to GBM immunotherapy72,73. M0 macrophage infiltration in GBM correlated with a malignant phenotype and a poor prognosis74. Whereas, M2 macrophage infiltration was associated with the immunosuppression and angiogenesis processes in GBM75,76. Our results showed that ASRS positively correlated with macrophage and M2 macrophage infiltration levels. Higher infiltration levels are significantly linked to poor prognosis in GBM consistent with previous studies77,78. In terms of immune checkpoint functions, the TNFRSF family genes were highly expressed in the high-risk group, which is related to immune suppressive status. Drug sensitivity analysis results showed that SB216763 and Tozasertib had significantly increased sensitivity. Tozasertib (VX-680) is a therapeutic agent targeted in the TNFRSF family genes that induced tumour cell apoptosis and autophagy79,80. Due to higher expression levels of TNFRSF family genes in the high-risk group, this drug may have a role in treating GBM.
Since the heterogeneity and progression of glioma were significantly different, we further analysed the DASEs between GBM and LGG. There were 8 DASEs in 3 genes (CTSL, EIF3L, and FN1). The FN1 gene is known to have a total of 9 exon skipping (ES) events in the merged dataset, of which three were DASEs. The median PSI values of these splicing events were higher in the GBM samples. To investigate the potential abnormal splicing regulation of these DASEs, we calculated correlation coefficients between 27 DEG-SFs and these 3 genes. Several key splicing factors were extracted, such as CELF3, ELAVL2, IGF2BP3, LGALS3, RBM47, RBMS1, WTAP, YBX3 (Figs. 7E, S5D-G), which showed a high degree of association with progression in GBM21,81–84. To further elucidate the splicing regulation and progression effects of these key splicing factors in GBM, the PPI network (Node: 924, Edge: 1184) was constructed from public PPI databases (Fig. S6A). ClueGO analysis results showed that the network genes mainly enriched in tumour-associated pathways, such as signaling by FGFR, spliceosome, mRNA splicing, autophagy, oncogene induced senescence, nonsense-mediated decay pathway (Fig. S6B, Table S9). FN1 also appeared in the PPI network. As mentioned above, the expression level of FN1 was not only correlated with RBMS1, YBX3, and RBM47 but also might be regulated by WTAP, LGALS3, and IGF2BP3 according to the PPI network. These distinctions make FN1 valuable for in-depth research into the molecular mechanisms associated with GBM progression.
FN1 encodes fibronectin, a major ECM macro-molecule, which is involved in cell adhesion, cell motility, Integrin signaling pathway, and PI3K-Akt signaling pathway85,86, and maintains cellular homeostasis50. Overexpression of FN1 has been reported in several cancers, such as thyroid carcinoma87, breast cancer88, bladder cancer89, esophageal squamous cell carcinoma90. It is an oncogene of GBM, and highly expressed levels of FN1 indicate a poor prognosis in GBM65. We found that FN1 was the most up-regulated in GBM samples compared with LGG samples and normal brain samples. Consistent results were also observed in the validation of protein expression levels. Furthermore, abnormal alternative splicing regulation resulted in several DASEs in FN1, followed by the observation of non-canonical transcripts in GBM. Another meaningful finding is that an abnormal ASE occurred in exon 25 with additional 2 bases (AG) retained at the start of the exon, which allowed the presence of PTC in several non-canonical transcripts, such as FN1-002, FN1-012. The PTC may lead to the absence of EDB domain of exon 25 in truncated proteins and could be involved in GBM acquired immunotherapy resistance. On the one hand, the defects in post-transcriptional mRNA processing lead to the formation of PTCs, and if the aberrant mRNA transcripts are not identified and degraded, truncated proteins may be produced that cause damage to the organism25. On the other hand, the pre-mRNA splicing is strongly related to NMD, and if it is possible to degrade the defective mRNAs produced due to PTC by the monitoring of the NMD pathway, then a mechanism of tumor suppression could be achieved.
In this comprehensive study, FN1 with distinct characteristics emerged as the analysis progresses: 1. Both PSI values of ASEs and gene expression level of FN1 significantly associated with GBM prognosis. 2. The expression levels of FN1 significantly increased stepwise in normal brain tissue, LGG, and GBM samples. 3. Consistent results were also observed in the validation of protein expression levels. 4. FN1 was used for ASRS model construction. 5. FN1 contained at least 4 abnormal splicing events which alter the expression of different transcripts. One of the most important ASE occurred in exon 25 resulting in the presence of PTC. 6. Several cancer-related splicing factors may play role in aberrant alternative splicing regulation of FN1. 7. FN1 was reported as a prognosis biomarker in other tumours and linked to tumour progression.
Based on the above findings, we conclude that FN1 may become a potential splicing biomarker of GBM, and three inferences can be drawn: First, the overexpression of FN1 in GBM may be due to unproductive splicing91. Second, the aberrant transcript products of FN1 due to PTC may play a key role in the progression of GBM and involve in GBM acquired immunotherapy resistance. Third, the overexpression of FN1 in GBM suggests that NMD may not be functioning.
It is important to note that the conclusions reached regarding FN1 in this study were based on intricate analyses and correlations. Future research should include systematic in vivo and in vitro experiments at single-cell resolution to fully clarify the role of aberrant splicing regulation and abnormal ASEs of FN1 in the pathogenesis and progression of glioblastoma. Other limitations inevitably influenced the reliability of the current results. While the ASRS model has shown effectiveness in predicting survival outcomes in GBM patients, our study's validation used a limited number of patients from four external cohorts. All samples in the study were retrospective and lacked clinical samples to verify the accuracy of the ASRS model. Clinical and molecular data from the real world should be integrated to validate and optimize the ASRS model, ensuring its accuracy and reliability across different patient populations. In addition, it is insufficient to predict survival outcomes for GBM merely based on ASEs. Other omics data should be aggregated to develop a robust signature.
In summary, we developed an effective prognostic signature of ASEs for GBM, and the ASRS model demonstrated potential for application. This signature highlights the differences in the tumour microenvironment, responses to immunotherapy, and prognostic outcomes between risk groups. FN1 may serve as a promising splicing biomarker for GBM, and further in-depth research on this gene is expected to yield targets for GBM treatment and offer new insights into GBM progression.
Supplementary Information
Acknowledgements
We all authors sincerely acknowledge the contributions from the TCGA project, CGGA project and ASCancer Atlas. The authors thank all study participants for support and cooperation during the study.
Author contributions
Yang and Ren conceived the original study concept and design. Liu and Song performed the bioinformatical analysis and finished figures. Zhou, He, and Wu prepared the supplementary files. All authors read and approved the final manuscript.
Funding
This study was supported by the Health Commission of Guangdong Province (No. A2020177), Guangzhou Municipal Science and Technology Bureau (No. 202201010764), Traditional Chinese Medicine Bureau of Guangdong Province (No. 20231219), and The Special Fund Project for Science and Technology Innovation Strategy of Guangdong Province ("Climbing Plan" No. pdjh2023b0279).
Data availability
The datasets generated and/or analysed during the current study are available in the TCGA, CGGA, ASCancer Atlas, and GEO. The accession number for the utilized GEO datasets are GSE43378 and GSE4412.
Declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent to publish
Not applicable.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xi Liu and Jinming Song contributed equally to this work.
Contributor Information
Jin Yang, Email: y.jin04@gdpu.edu.cn.
Zhonglu Ren, Email: renzhonglu@gdpu.edu.cn.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-025-91038-4.
References
- 1.Miller, K. D. et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin.71, 381–406 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Ostrom, Q. T. et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2016–2020. Neuro Oncol.25, iv1–iv99 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostrom, Q. T. et al. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro Oncol.24, v1–v95 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang, P. et al. Management and survival rates in patients with glioma in China (2004–2010): A retrospective study from a single-institution. J. Neurooncol.113, 259–266 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Jiang, N. et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat. Commun.13, 1511 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osuka, S. & Van Meir, E. G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Invest.127, 415–426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan, A. C. et al. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin.70, 299–312 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Sciarrillo, R. et al. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updat.53, 100728 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Ule, J. et al. An RNA map predicting Nova-dependent splicing regulation. Nature444, 580–586 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Zhang, Y., Qian, J., Gu, C. & Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target Ther.6, 78 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Climente-Gonzalez, H., Porta-Pardo, E., Godzik, A. & Eyras, E. The functional impact of alternative splicing in cancer. Cell Rep.20, 2215–2226 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Zhou, J., Zhao, S. & Dunker, A. K. Intrinsically disordered proteins link alternative splicing and post-translational modifications to complex cell signaling and regulation. J. Mol. Biol.430, 2342–2359 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Weatheritt, R. J., Sterne-Weiler, T. & Blencowe, B. J. The ribosome-engaged landscape of alternative splicing. Nat. Struct. Mol. Biol.23, 1117–1123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bradley, R. K. & Anczukow, O. RNA splicing dysregulation and the hallmarks of cancer. Nat. Rev. Cancer23, 135–155 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivanova, O. M. et al. Non-canonical functions of spliceosome components in cancer progression. Cell Death Dis.14, 77 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi, S., Cho, N., Kim, E. M. & Kim, K. K. The role of alternative pre-mRNA splicing in cancer progression. Cancer Cell Int.23, 249 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng, R. et al. A pan-cancer analysis of alternative splicing of splicing factors in 6904 patients. Oncogene40, 5441–5450 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Kahles, A. et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell34(211–224), e216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi, F. et al. Significance of alternative splicing in cancer cells. Chin. Med. J. (Engl)133, 221–228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu, Z. & Rabadan, R. Computing the role of alternative splicing in cancer. Trends Cancer7, 347–358 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siddaway, R. et al. Splicing is an alternate oncogenic pathway activation mechanism in glioma. Nat. Commun.13, 588 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, D., Yu, W. & Lai, M. Targeting serine- and arginine-rich splicing factors to rectify aberrant alternative splicing. Drug Discov. Today28, 103691 (2023). [DOI] [PubMed] [Google Scholar]
- 23.Havens, M. A. & Hastings, M. L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res.44, 6549–6563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanley, R. F. & Abdel-Wahab, O. Dysregulation and therapeutic targeting of RNA splicing in cancer. Nat. Cancer3, 536–546 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patro, I., Sahoo, A., Nayak, B. R., Das, R., Majumder, S., & Panigrahi, G. K. Nonsense-mediated mRNA decay: Mechanistic insights and physiological significance. Mol. Biotechnol. (2023). [DOI] [PubMed]
- 26.Araki, S., Ohori, M. & Yugami, M. Targeting pre-mRNA splicing in cancers: Roles, inhibitors, and therapeutic opportunities. Front Oncol.13, 1152087 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Miguel, F. J. et al. A large-scale analysis of alternative splicing reveals a key role of QKI in lung cancer. Mol. Oncol.10, 1437–1449 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munkley, J., Li, L., Krishnan, S. R. G., Hysenaj, G., Scott, E., Dalgliesh, C., Oo, H. Z., Maia, T. M., Cheung, K., Ehrmann, I., et al. Androgen-regulated transcription of ESRP2 drives alternative splicing patterns in prostate cancer. Elife8 (2019). [DOI] [PMC free article] [PubMed]
- 29.Dutertre, M. et al. Exon-based clustering of murine breast tumor transcriptomes reveals alternative exons whose expression is associated with metastasis. Cancer Res.70, 896–905 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Gardina, P. J. et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics7, 325 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colaprico, A. et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res.44, e71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao, Z. et al. Chinese glioma genome atlas (CGGA): A comprehensive resource with functional genomic data from Chinese glioma patients. Genom. Proteom. Bioinform.19, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu, S. et al. ASCancer Atlas: A comprehensive knowledgebase of alternative splicing in human cancers. Nucleic Acids Res.51, D1196–D1204 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li, Y. & Guo, D. Genome-wide profiling of alternative splicing in glioblastoma and their clinical value. BMC Cancer21, 958 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seiler, M. et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep.23, 282-296 e284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong, Y. et al. Profiles of alternative splicing in colorectal cancer and their clinical significance: A study based on large-scale sequencing data. EBioMedicine36, 183–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piva, F., Giulietti, M., Burini, A. B. & Principato, G. SpliceAid 2: A database of human splicing factors expression data and RNA target motifs. Hum. Mutat.33, 81–85 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Gene Ontology, C., Aleksander, S. A., Balhoff, J., Carbon, S., Cherry, J. M., Drabkin, H. J., Ebert, D., Feuermann, M., Gaudet, P., Harris, N. L., et al. The gene ontology knowledgebase in 2023. Genetics224 (2023). [DOI] [PMC free article] [PubMed]
- 39.Tay, J. K., Narasimhan, B., & Hastie, T. Elastic net regularization paths for all generalized linear models. J. Stat. Softw.106. (2023) [DOI] [PMC free article] [PubMed]
- 40.Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform.14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res.48, W509–W514 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu, F. F., Liu, C. J., Liu, L. L., Zhang, Q., & Guo, A. Y. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief Bioinform.22 (2021). [DOI] [PubMed]
- 43.Maeser, D., Gruener, R. F., & Huang, R. S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform.22 (2021). [DOI] [PMC free article] [PubMed]
- 44.Saraiva-Agostinho, N. & Barbosa-Morais, N. L. psichomics: Graphical application for alternative splicing quantification and analysis. Nucleic Acids Res.47, e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saraiva-Agostinho, N. & Barbosa-Morais, N. L. Interactive alternative splicing analysis of human stem cells using psichomics. Methods Mol. Biol.2117, 179–205 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res.47, W556–W560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Birney, E. et al. An overview of Ensembl. Genome Res.14, 925–928 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin, F. J. et al. Ensembl 2023. Nucleic Acids Res.51, D933–D941 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res.13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oughtred, R. et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci.30, 187–200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Del Toro, N. et al. The IntAct database: Efficient access to fine-grained molecular interaction data. Nucleic Acids Res.50, D648–D653 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Licata, L. et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res.40, D857-861 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res.49, D480–D489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res.53, D672–D677 (2025). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci.28, 1947–1951 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res.28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Milacic, M. et al. The reactome pathway knowledgebase 2024. Nucleic Acids Res.52, D672–D678 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bao, Y. et al. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell Mol. Biol. Lett.24, 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang, Z. et al. A novel basement membrane-related gene signature for prognosis of lung adenocarcinomas. Comput. Biol. Med.154, 106597 (2023). [DOI] [PubMed] [Google Scholar]
- 60.Lin, X. et al. lncRNA ITGB8-AS1 functions as a ceRNA to promote colorectal cancer growth and migration through integrin-mediated focal adhesion signaling. Mol. Ther.30, 688–702 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karamanos, N. K. et al. Extracellular matrix-based cancer targeting. Trends Mol. Med.27, 1000–1013 (2021). [DOI] [PubMed] [Google Scholar]
- 62.Wade, A. et al. Proteoglycans and their roles in brain cancer. FEBS J.280, 2399–2417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Espinoza-Sanchez, N. A. & Gotte, M. Role of cell surface proteoglycans in cancer immunotherapy. Semin. Cancer Biol.62, 48–67 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Robichaud, N., Sonenberg, N., Ruggero, D., & Schneider, R. J. Translational control in cancer. Cold Spring Harb. Perspect. Biol.11 (2019). [DOI] [PMC free article] [PubMed]
- 65.Wu, S. et al. High expression of fibronectin 1 predicts a poor prognosis in glioblastoma. Curr. Med. Sci.42, 1055–1065 (2022). [DOI] [PubMed] [Google Scholar]
- 66.Shaw, T. I. et al. Discovery of immunotherapy targets for pediatric solid and brain tumors by exon-level expression. Nat. Commun.15, 3732 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li, Y. et al. Classification of glioma based on prognostic alternative splicing. BMC Med. Genomics12, 165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao, L. et al. Comprehensive characterization of alternative mRNA splicing events in glioblastoma: Implications for prognosis, molecular subtypes, and immune microenvironment remodeling. Front. Oncol.10, 555632 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang, M. et al. Identification and validation of a prognostic immune-related alternative splicing events signature for glioma. Front. Oncol.11, 650153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiu, J. et al. Transcriptome analysis and prognostic model construction based on splicing profiling in glioblastoma. Oncol. Lett.21, 138 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang, B., Wu, Q., Cheng, S. & Li, W. Systematic profiling of mRNA splicing reveals the prognostic predictor and potential therapeutic target for glioblastoma multiforme. J. Oncol.2021, 4664955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DiDomenico, J. et al. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology7, e1448329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amoozgar, Z. et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat. Commun.12, 2582 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang, L. et al. EFEMP2 indicates assembly of M0 macrophage and more malignant phenotypes of glioma. Aging (Albany NY)12, 8397–8412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tian, Y., Ke, Y. & Ma, Y. High expression of stromal signatures correlated with macrophage infiltration, angiogenesis and poor prognosis in glioma microenvironment. PeerJ8, e9038 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu, X. et al. Impact of ferroptosis-related risk genes on macrophage M1/M2 polarization and prognosis in glioblastoma. Front. Cell Neurosci.17, 1294029 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li, J., Guo, Q. & Xing, R. Construction and validation of an immune infiltration-related risk model for predicting prognosis and immunotherapy response in low grade glioma. BMC Cancer23, 727 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shan, X. et al. Prognostic value of a nine-gene signature in glioma patients based on tumor-associated macrophages expression profiling. Clin. Immunol.216, 108430 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Kulbay, M., Paimboeuf, A., Ozdemir, D. & Bernier, J. Review of cancer cell resistance mechanisms to apoptosis and actual targeted therapies. J. Cell Biochem.123, 1736–1761 (2022). [DOI] [PubMed] [Google Scholar]
- 80.Paladini, J., Maier, A., Habazettl, J. M., Hertel, I., Sonti, R., & Grzesiek, S. The molecular basis of Abelson kinase regulation by its alphaI-helix. Elife12 (2024). [DOI] [PMC free article] [PubMed]
- 81.Sun, C. et al. Identification of IGF2BP3 as an adverse prognostic biomarker of gliomas. Front. Genet.12, 743738 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin, D. I. et al. Expression and roles of Wilms’ tumor 1-associating protein in glioblastoma. Cancer Sci.103, 2102–2109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xi, Z. et al. WTAP expression predicts poor prognosis in malignant glioma patients. J. Mol. Neurosci.60, 131–136 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Ji, Q., Guo, Y., Li, Z. & Zhang, X. WTAP regulates the production of reactive oxygen species, promotes malignant progression, and is closely related to the tumor microenvironment in glioblastoma. Aging (Albany NY)16, 5601–5617 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Owens, R. J. & Baralle, F. E. Mapping the collagen-binding site of human fibronectin by expression in Escherichia coli. EMBO J.5, 2825–2830 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Calaycay, J. et al. Primary structure of a DNA- and heparin-binding domain (Domain III) in human plasma fibronectin. J. Biol. Chem.260, 12136–12141 (1985). [PubMed] [Google Scholar]
- 87.Chen, C. & Shen, Z. FN1 promotes thyroid carcinoma cell proliferation and metastasis by activating the NF-Kappa pathway. Protein Pept. Lett.30, 54–64 (2023). [DOI] [PubMed] [Google Scholar]
- 88.Chen, C., Ye, L., Yi, J., Liu, T. & Li, Z. FN1 mediated activation of aspartate metabolism promotes the progression of triple-negative and luminal a breast cancer. Breast Cancer Res. Treat.201, 515–533 (2023). [DOI] [PubMed] [Google Scholar]
- 89.Zhang, L. et al. Fibronectin 1 as a key gene in the genesis and progression of cadmium-related bladder cancer. Biol. Trace Elem. Res.201, 4349–4359 (2023). [DOI] [PubMed] [Google Scholar]
- 90.Ma, J., Chen, S., Su, M. & Wang, W. High FN1 expression is associated with poor survival in esophageal squamous cell carcinoma. Medicine (Baltimore)102, e33388 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fair, B. et al. Global impact of unproductive splicing on human gene expression. Nat. Genet.56, 1851–1861 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available in the TCGA, CGGA, ASCancer Atlas, and GEO. The accession number for the utilized GEO datasets are GSE43378 and GSE4412.