

Abstract
Diabetes has been reported to increase CYP2E1 (cytochrome P450) and CYP2B1 expression at both the mRNA and protein levels in rat livers. This increase has been attributed to mRNA stabilization and can be reversed by daily insulin treatment. In a previous study, we showed that this hormone directly down-regulates CYP2E1 and 2B1 expression through a post-transcriptional mechanism in rat hepatoma cell lines. We then aimed to identify the molecular mechanisms involved in this regulation. We first identified a 16-mer sequence that we later showed to be the actual functional target of insulin on the rat CYP2E1 mRNA. Similar work was performed with CYP2B1. We first investigated the presence of mRNA–protein interactions. Using cytoplasmic proteins of Fao cells treated or not with insulin (0.1 μM) and the full-length CYP2B1 mRNA as a probe, a major CYP2B1 RNA–protein complex was observed with RNase T1 protection experiments. With the use of different CYP2B1 mRNA probes and by means of competition experiments with antisense oligonucleotides, a protein fixation site was located on a 16-nt sequence in the 5′ part of the coding region. This sequence has a hairpin loop structure, shows 80% sequence identity with a structure previously identified on CYP2E1 and is also responsible for the post-transcriptional effects of insulin on this mRNA. Protein(s) bound to both CYP2B1 and CYP2E1 sequences are cytosolic and have an apparent molecular mass of 60 kDa. The protein(s) that bind(s) to both these sequences and the insulin transduction signal involved in this regulation remain(s) to identified.
Keywords: cis-acting element, cytochrome P450 (CYP), helix–loop, insulin, mRNA, post-transcriptional regulation
Abbreviations: AREs, adenylate/uridylate-rich elements; CAR, constitutive androstane receptor; CYP, cytochrome P450; DRB, 5, 6-dichloro-1-β-D-ribofuranosylbenzimidazole; hnRNP, heterogeneous nuclear ribonucleoprotein; P, penetratin; PXR, pregnane X receptor; RT, reverse transcriptase; UTR, untranslated region
INTRODUCTION
Insulin plays a key role in glucose homoeostasis and in the control of fat and protein metabolism. The hormone exerts its effects on tissues involved in the regulation of fuel metabolism such as muscle, adipose tissue and liver. In addition to post-translational effects, insulin modulates the expression of a variety of genes, which leads to the regulation of metabolic pathways [1] by several mechanisms, including post-transcriptional regulation. Insulin also controls the expression of several genes encoding CYPs (cytochromes P450) [2] that are involved in the metabolism of xenobiotics and some endogenous compounds [3]. Although important, the mechanisms and the biological implications of this regulation are still poorly understood.
CYPs are the primary enzymes involved in hepatic phase I metabolic reactions. The CYP2 family is the largest one and comprises 30 subfamilies (CYP2A–CYP2AD) [David Nelson's Cytochrome P450 Homepage (http://drnelson.utmem.edu/cytochromeP450.html)]. The first five subfamilies (CYP2A–CYP2E) are the best characterized and most studied ones in mammalian species such as rat, rabbit and human. Among them, rat CYP2E1 and CYP2B1 are clearly regulated by insulin [2]. CYP2E1 metabolizes clinically important drugs such as acetaminophen, isoniazid, tamoxifen, enflurane and halothane [5]. In addition, because of its ability to catalyse the metabolism of acetone, it is postulated to be involved in the intermediate steps of the gluconeogenic pathway [5]. CYP2E1 is inducible by many compounds, including ethanol. The regulation of CYP2E1 expression is complex and involves transcriptional activation, mRNA stabilization, increased mRNA translatability and decreased protein degradation [6]. In chemically induced and spontaneously diabetic rats, an increase in hepatic CYP2E1 expression has been reported [7]. This elevation of mRNA levels in the diabetic state in vivo has been attributed to mRNA stabilization [8] and can be reversed by daily insulin treatment [9].
CYP2B1 is mainly expressed in the liver, where it catalyses the metabolism of several important pharmaceutical agents (cyclophosphamide, tricyclic antidepressants etc.), various endogenous compounds of physiological importance, such as testosterone, and certain procarcinogens, into, ultimately, carcinogens. CYP2B gene regulation has been studied mainly at the transcriptional level. It is inducible in the liver by several drugs (barbiturates, pesticides), and the CAR (constitutive androstane receptor) appears to be one of the main factors involved in transcriptional induction of the CYP2B genes, although PXR (the pregnane X receptor), the glucocorticoid receptor and other nuclear receptors may also be required for optimal regulation [10]. Nevertheless, as in the case of CYP2E1, CYP2B1 mRNA stability was shown to be increased by diabetes in the rat liver [7].
Previous results showed that insulin directly down-regulates the expression of CYP2B1 and CYP2E1 through a post-transcriptional mechanism in rat hepatoma cell lines [2] as well as in primary cultured rat hepatocytes [11]. The mechanisms of insulin action have been largely described for some enzymes involved in glucose and lipid metabolism and were found to be mostly transcriptional [1]. In contrast, little is known about the mechanism of post-transcriptional regulation involved in the control of gene expression. This regulation implies specific RNA–protein interactions which mediate RNA processing (7mGpppN capping, polyadenylation, splicing etc.), RNA nucleocytoplasmic transport and RNA stabilization [12]. Indeed, some RNA sequences may serve as binding sites for proteins that could play a regulatory role in gene expression. Such interactions often occur in the 5′-UTR (5′-untranslated region) or 3′-UTR of the mRNA and rarely within the coding region [12]. In the case of insulin, studies on regulation of mRNA turnover have focused on CYP2E1. We have recently demonstrated that the binding of cytoplasmic proteins from rat hepatoma cell lines to a 16-mer hairpin loop, within the 5′ part of gene coding region, was involved in CYP2E1 mRNA destabilization by insulin [13]. Little is known about the mechanism through which this sequence controls mRNA turn-over. In addition, whether similar sequences are present and functional in other CYP mRNAs and whether they could be involved in insulin regulation is not known.
In order to answer this question, the molecular mechanisms involved in CYP2B1 mRNA destabilization by insulin were studied. We show here that a 16-mer sequence within the CYP2B1 mRNA is required for insulin regulation of this mRNA stability. This sequence, predicted to form a hairpin structure, displays 80% identity with the regulatory sequence of CYP2E1 and can bind a hepatic cytosolic protein with an apparent molecular mass of 60 kDa.
EXPERIMENTAL
Cell culture and treatments
Fao cells were grown in monolayer culture as described previously [14]. Cells were exposed to insulin (0.1 μM), the usual supplement to a primary hepatocyte culture medium, for 6 h during their exponential phase of growth [15]. Untreated cultures were used as controls.
Preparation of cytoplasmic extracts
After trypsinization, a pellet of cells from five culture dishes (30×106 cells), was resuspended in 25 mM Tris (pH 7.9)/0.5 mM EDTA. After four cycles of freezing in ethanol/solid CO2 and thawing at 37 °C for 5 min, the samples were then centrifuged at 4 °C at 15000 g for 15 min; the supernatant was divided into portions and stored at −80 °C until use. The protein concentration was determined, using the bicinchoninic acid (‘BCA’) (Pierce) procedure, with BSA as standard, according to the manufacturer's instructions.
Preparation of rat liver extracts
The cellular fractions were prepared by differential centrifugation. All manipulations were performed at 4 °C. The Zucker-rat liver kindly provided by Dr Chantal Benelli (INSERM U530, Hôpital des Enfants Malades, Paris, France) was thawed in iso-osmotic NaCl, diced with scissors, washed in iso-osmotic NaCl two or three times, ground (1 g/5 ml) in buffer (250 mM sucrose/100 mM Tris/HCl/1 mM EDTA, pH 7.4) with an Ultra-Turrax® grinder (IKA-Labortechnik, Staufen, Germany) for 10–20 s and homogenized with a Potter–Elvehjem-type device (Fisher Bioblock Scientific, Illkirch, France). The homogenate was successively centrifuged at 800 g for 10 min to isolate the nuclear pellet, and at 9000 g for 20 min to isolate the mitochondrial pellet and the S9 supernatant. The S9 supernatant was then centrifuged at 105000 g for 60 min to obtain the microsomal pellet and the cytosolic supernatant. The microsomes were suspended in 100 mM sodium phosphate/20% (v/v) glycerol. The fractions were stored at −80 °C. The protein concentration was determined as described above.
Plasmids
Dr. Frank J. Gonzalez (National Cancer Institute, National Institutes of Health, Bethesda, MD, U.S.A.) generously provided us with a partial rat CYP2B1 cDNA lacking the 5′-UTR. To obtain full-length CYP2B1 cDNA, this partial cDNA was reamplified by PCR, using a primer containing the additional 5′-UTR. EcoRI sites were added in both primers for cloning into pcDNAI/Amp (Invitrogen) (Figure 1). This plasmid was named pcDNAI/CYP2B1. The plasmid pcDNAI/CYP2E1 was previously described [13].
Figure 1. Map of full-length rat CYP2B1 cDNA and sizes of the RNA probes.

Full-length rat CYP2B1 cloned into pcDNAI at EcoRI sites and mRNAs with different sizes (PstI, AflIII, and NotI) are located. ATG, start of coding region; TGA, stop codon.
The Renilla reniformis (sea pansy) luciferase expression plasmid p-αglob-RL, which was named pRL, contains the proximal promoter of the α-globin gene [13]. To maintain the open reading frame, an 18-mer oligonucleotide (CTTCCCCTCTTGGGGAAC), corresponding to the binding site of cytoplasmic proteins found in the CYP2B1 sequence, was inserted in pRL in the coding sequence of the Renilla gene at the AflIII site, 183 bases after the ATG codon. The new plasmid was named pRL-Seq2B16. The insertion was analysed by sequencing.
Northern-blot analysis
Total RNA from Fao cells was extracted using the RNeasy Mini kit (Qiagen, Courtaboeuf, France). Subsequently, 10–20 μg of total RNA was subjected to electrophoresis in denaturing formaldehyde/1.2% agarose gels and transferred to nylon membranes. These membranes were prehybridized and then hybridized with several 32P-labelled cDNA probes [rat CYP2E1 and CYP2B1, and 18 S rRNA (an endogenous housekeeping gene)]. After hybridization, the filters were washed at 65 °C for 30 min successively with 2×SSC (standard saline citrate; 1×SSC is 150 mM NaCl/15 mM sodium citrate, pH 7.0), 0.1% SDS, then 0.5×SSC, 0.1% SDS, and, sometimes, 0.1×SSC and 0.1% SDS, and then were processed for autoradiography. The relative intensities of the hybridization signals were determined by scanning with a Storm™ 840 phosphoimager (Amersham Biosciences).
In vitro transcription
After linearization of the pcDNAI/CYP2B1 with either the PstI or AflIII or NotI restriction enzymes (Figure 1), the products were transcribed with T7 RNA polymerase in the presence or in the absence of [32P]UTP (800 Ci/mmol; Amersham Biosciences) using standard protocols (Stratagene, Amersham Biosciences). Transcripts were separated from unincorporated nucleotides on a Sephadex G-50 column, and specific radioactivity was determined by counting an aliquot in a Packard liquid-scintillation counter.
RNA–protein binding reactions (RNase T1 protection)
The reaction was carried out as previously described [13]. In competition experiments, the unlabelled RNA (50–100-fold excess) was preincubated at room temperature for 10 min with cytoplasmic proteins before the addition of the radiolabelled probe. In competition experiments with antisense oligonucleotides, the antisense oligonucleotide (3 μg) was preincubated with the 32P-labelled RNA probe to allow base-pairing for 5 min at 65 °C, followed by 10 min at room temperature before the addition of cytoplasmic proteins.
Two 16-mer RNAs [CYP2E1 RNA (UUCCCAUCCUUGGGAA) and CYP2B1 RNA (UUCCCCUCUUGGGGAA)] were synthesized by Proligo (Paris, France) and 5′-labelled with [γ-32P]ATP by the T4 polynucleotide kinase. Incubations and competition experiments with antisense oligonucleotides were performed as described above for RNA–protein binding reactions without addition of RNase T1, but with UV-cross-linking reaction on ice (one run of 8 min, followed by three runs at 120000 μJ using a UV Stratalinker® 1800 instrument from Stratagene) performed after cytoplasmic protein addition.
UV cross-linking analysis
The reaction was carried out in a final volume of 50 μl. Cytoplasmic proteins (40 μg) and 106 c.p.m. of 32P-labelled RNA probes were incubated at room temperature for 10 min in a binding buffer [13 mM Hepes/40 mM KCl/53 mM MgCl2/1 mM dithiothreitol/5% (v/v) glycerol], and 2 μg/μl yeast tRNA to prevent non-specific binding. The reaction mixture was exposed to UV light on ice (one run of 8 min, followed by three at 120000 μJ; UV Stratalinker® 1800 instrument), then treated with RNase A (30 μg for 20 min at 37 °C). A Laemmli buffer was then added [0.006% Bromophenol Blue/125 mM Tris/4% (w/v) SDS/5% (v/v) glycerol] and the reaction mixture was incubated at 95 °C for 10 min and subjected to SDS/10%-(w/v)-PAGE with 15 g/l Tris/72 g/l glycine/5 g/l SDS as running buffer. Protein–32P mRNA complexes were detected by autoradiography of the dried gel and quantified with the phosphoimager.
Antisense internalization
A CYP2B 26-mer antisense oligonucleotide containing the 16-mer antisense sequence coupled with penetratin (AS2B16-P: GGAGGUUCCCCAAGAGGGGAAGGGGA), a 26-mer control sequence, without hairpin loop, coupled with penetratin (AS control-P: GGAGGUUAGUCAAGAUCAGAAGGGGA) and free penetratin (P) were purchased from Q-Biogene (Illkirch, France). The oligonucleotide–penetratin conjugation efficiency on a 0.1% SDS/15%-polyacrylamide gel was guaranteed by the manufacturer.
At 1 day before transfection, Fao cells (106 cells/well) were seeded into the culture medium in a six-well dish. Concentrated solutions of AS2B26-P, AS2Bcontrol-P and P (800 nM) were prepared in culture medium and added to a final concentration of 400 nM. After 30 min, the cells were treated or not treated with insulin (0.1 μM). After 6 h, two identically treated wells were mixed and total mRNAs were extracted.
Transfection experiments
At 1 day prior to the transfection, cells were seeded (4×105 cells/well) into the culture medium in a six-well dish. Each well was transfected with 100 μl of a medium containing 2 μg of pRL or 2 μg of pRL-Seq2B16, 3 μl of FuGENE 6™ Transfection Reagent (Roche) and serum free-medium.
mRNA half-life determinations
mRNA turnover was assessed at different times after treatment of cells with the transcription inhibitor DRB (5,6-dichloro-1-β-D-ribofuranosylbenzimidazole) [16]. At this dose, DRB treatment for 15 h did not affect the number or viability of Fao cells. Fao cells were treated with insulin (0.1 μM) or left untreated for 3 h and were then treated with 100 μM DRB. Total RNA was isolated 0, 3, 6, 9, 12 and 15 h after DRB treatment.
Reverse transcription was performed on 2 μg of RNA samples using the cDNA High-Capacity Archive Kit (Applied Biosystems, Courtaboeuf, France) in a final volume of 50 μl, according to the manufacturer's instructions. R. reniformis luciferase specific oligonucleotide primers were designed using oligoexplorer software (Oligosoftware) (primer sense: AGGAAACGGATGATAACTGG; primer antisense: GCTACTGGCTCAATATGTGG). 18 S rRNA was used as reference gene (primer sense: TCCCCCAACTTCTTAGAGG; primer antisense: CTTATGACCCGCACTTACTG). For each primer pair, we performed a no-template control and a no-RT (reverse transcriptase) control (RT-negative assays), which produced negligible signals, suggesting no primers–dimer formation. Amplicons were 182 and 234 nts long for R. reniformis luciferase and 18 S rRNA respectively. Gel electrophoresis was performed to verify the size of the specific amplications and the absence of additional PCR fragments. Quantitative changes in mRNA expression were assessed with a real-time quantitative PCR method performed in an ABI Prism 7900 Sequence Detector System (Applied Biosystems, Foster City, CA, U.S.A.). Reaction mixtures for real-time PCR using SYBR-Green detection consisted of SYBR® Green PCR Master Mix (ABGene, Courtaboeuf, France), 300 nM of each primer and 4 ng of cDNA in a final volume of 10 μl. PCR cycles proceeded as follows: denaturation 15 min at 95 °C, followed by 40 cycles at 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. Rat 18 S rRNA was used as an internal control. Relative quantification of the mRNA expression levels of target genes was calculated using the 2−ΔΔCT method, where:
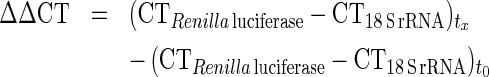 |
Each sample determination was performed in triplicate. The amount of mRNA at zero time (t0, the time of DRB addition) in each group (pRL and pRL-Seq2B16 control, pRL and pRL-Seq2B16 treated with insulin) was assigned as reference compared with the amount of mRNA at time x (tx, where x=3, 6, 9, 12 or 15 h).
Statistical analysis
Student's two-tailed t tests were performed using Statview software (Abacus Concepts, Berkeley, CA, U.S.A.).
RESULTS
It was previously shown that a 6 h insulin treatment decreased the amount of CYP2B1 mRNA by 60% in the highly differentiated Fao hepatoma cells by a post-transcriptional mechanism [2]. In order to determine the mechanisms of CYP2B1 mRNA stability and its regulation, RNA–protein interactions were first studied and the protein-binding sites were mapped on the CYP2B1 mRNA. Such binding sites were detected by the ability of bound proteins to protect a labelled RNA probe from complete degradation by RNase. Using Fao cells, cytoplasmic proteins and the full-length CYP2B1 mRNA as a probe, a major CYP2B1 RNA–protein complex was observed (Figure 2A, lanes 3 and 5). Treatment of the binding reaction mixture with proteinase K completely abolished the formation of the complex (results not shown), indicating that the complex was due to the interaction between RNA and proteins. Moreover, the formation of this complex was prevented by a 100-fold excess of unlabelled full-length CYP2B1 mRNA as competitor (Figure 2A, lanes 4 and 6). The pattern of the labelled complex formed was similar with cytoplasmic proteins from either control or insulin-treated cells (Figure 2A, lanes 3 and 5), indicating that protein(s) was (were) present in both situations. In order to map the protein-binding site on the CYP2B1 mRNA, various truncated mRNA probes were prepared and used for binding reactions [1–1700 nt (Figure 2B) and 1–165 nt (Figure 2C)]. The RNA–protein complex was observed with both probes (Figures 2B and 2C, lanes 3 and 5) and was competed out in each case by an excess of homologous unlabelled mRNA (Figures 2B and 2C, lanes 4 and 6). Treatment with insulin did not modify the binding pattern. In order to demonstrate that the complexes formed with the different probes were related, cross-competition experiments were performed. Figure 2(D) shows that excess unlabelled 1–165 CYP2B1 RNA prevents the binding of cytoplasmic protein to the full-length mRNA probe (Figure 2D, lanes 4 and 6). All these results suggest that the RNA–protein complex is located within this 1–165-nt fragment of the CYP2B1 mRNA.
Figure 2. Complexes of cytoplasmic proteins from Fao cells, treated or not treated with insulin, on full-length 32P-labelled CYP2B1 mRNA (A, D) and on partial 32P-CYP2B1 mRNA probes obtained by transcription in vitro from pcDNAI/CYP2B linearized by AflIII (B, 1–1700-nt fragment) and PstI (C, 1–165-nt fragment).

The reaction was carried out in a final volume of 20 μl containing 105 c.p.m. of 32P-labelled CYP2B mRNA (A and D, full-length fragment; B, fragment 1–1700 nt; C, 1–165-nt fragment) (lanes 1–6) and 40 μg of cytoplasmic proteins from Fao cells untreated (lanes 3 and 4) or treated with 0.1 μM insulin (lanes 5 and 6). For competition experiments (lanes 4 and 6) a 50–100-fold excess of unlabelled CYP2B1 mRNA (A, full-length fragment; B, 1–1700-nt fragment; C and D, 1–165-nt fragment) were incubated with cytoplasmic proteins before addition of the radiolabelled probe. A portion (100 units) of RNase T1 was added to the reaction mixture (lanes 2–6) to degrade the mRNA unprotected by protein binding. Complexes were visualized after PAGE and autoradiography.
In order to map the RNA–protein complex more precisely, several consecutive and non-overlapping antisense oligonucleotides covering the 1–165-nt fragment were synthesized (Figure 3A). Preincubation of the fragment [32P]1–165-nt CYP2B1 mRNA with antisense oligonucleotides AS1–AS4 did not prevent complex formation (Figure 3B, lanes 2–5). In contrast, oligonucleotides AS5 (Figure 3B, lane 6) prevented protein from binding to the 1–165-nt fragment. Similar results were obtained using cytoplasmic proteins from cells treated (or not) with insulin (results not shown), indicating that the RNA–protein interaction is independent of insulin treatment. RNAdraw©, a predictive secondary-structure software [16a], was used to predict the putative secondary structure of the 1–165-nt CYP2B1 mRNA and showed the presence of a hairpin loop (Figure 4), which exhibits 80% similarity with the one described for CYP2E1 mRNA [13]. It was thus hypothesized that this putative hairpin loop could be the binding site for cytoplasmic protein(s). Therefore, a 16-mer antisense oligonucleotide, complementary to this hairpin loop (AS2B16), was synthesized. As shown in Figure 5 (lane 5), AS2B16 was indeed able to block the formation of the full-length CYP2B1 mRNA–cytoplasmic protein complex. These data suggest that the CYP2B1 16-mer is necessary for the formation of the RNA–protein complex. In order to test whether this hairpin alone is sufficient to bind cytoplasmic proteins, a conventional gel-mobility-shift assay was performed using a 32P-labelled 16-mer RNA covering the loop region and Fao cytoplasmic proteins. The formation of the 16-mer RNA–protein complex was shown to be dose-dependent and was competed by a 100-fold excess of unlabelled 16-mer CYP2B1 probe (results not shown). The formation of the 16-mer RNA–protein complex (Figure 6A, lane 1) was displaced by a homologous antisense oligomer (AS2B16) (Figure 6A, lane 3) and was unaffected by a non-specific antisense oligomer (ASns) (Figure 6A, lane 2).
Figure 3. (A) Localization and sequence of antisense oligonucleotides complementary to the CYP2B cDNA from EcoRI to PstI sites (fragment 1–165 nt CYP2B1 mRNA) and (B) inhibition of complex formation using antisense competitors.

AS1–AS5 correspond to the EcoRI site. The reaction was carried out in a final volume of 20 μl containing 105 c.p.m. of 1-165-nt fragment 32P-CYP2B mRNA probe (lanes 1–6) and 40 μg of cytoplasmic proteins from Fao cells (lanes 1–6). For competition experiments, 3 μg of antisense (AS) oligonucleotide was incubated with 1-165-nt fragment 32P-labelled CYP2B1 mRNA probe (lanes 1–6) before the addition of the cytoplasmic proteins. A portion (100 units) of RNase T1 was added to the reaction mixture (lanes 1–6) to degrade mRNA unprotected by protein binding. Complexes were visualized, after PAGE, on the phosphoimager.
Figure 4. (A) Predictive secondary structure of the CYP2B1 mRNA 1–165-nt fragment, and localization of antisense oligonucleotides complementary to this sequence, and (B) CYP2B1 and CYP2E1 16-mer helix loops.

Bases non-homologous in the two helix loops are boxed. The numbering starts from ATG.
Figure 5. Complexes of cytoplasmic proteins from untreated Fao cells on full-length 32P-CYP2B1 mRNA.

The reaction was carried out in a final volume of 20 μl containing 105 c.p.m. of full-length 32P-CYP2B mRNA (lanes 1–6) and 40 μg of cytoplasmic proteins from Fao cells (lanes 3–6). For competition experiments, 3 μg of antisense oligonucleotides (ASns, lane 4; AS5-2B, lane 5; AS2B16, lane 6) were incubated with the full-length 32P-labelled CYP2B1 mRNA probe before the addition of the cytoplasmic proteins. A portion (100 units) of RNase T1 was added to the reaction mixture (lanes 2–6) to degrade the mRNA unprotected by protein binding. Complexes were visualized after PAGE and autoradiography.
Figure 6. Reconstitution (A) and UV-cross-linking analysis (B) of the 16-mer RNA–protein binding complex by incubation of the CYP2B and CYP2E 16-mer RNA with cytoplasmic proteins from untreated Fao cells.

(A) The reaction was carried out in a final volume of 20 μl containing 105 c.p.m. of 32P-labelled CYP2B 16-mer RNA probe (lanes 1–4) or 32P-labelled 16-mer CYP2E RNA probe (lanes 5–8) and 40 μg of cytoplasmic proteins from untreated Fao cells (lanes 1–8). For competition experiments, 3 μg of antisense oligonucleotides (ASns, lanes 2 and 6; AS2B16, lanes 3 and 8; AS1,2-2E, lanes 4 and 7) were incubated with the 32P-labelled 16-mer RNA probe before addition of the cytoplasmic proteins. Complexes were visualized, after PAGE, on the phosphoimager. (B) The reaction was carried out in a final volume of 50 μl containing 106 c.p.m. of fragment 32P-labelled 16-mer CYP2B and CYP2E mRNA (lanes 1–8) and 40 μg of cytoplasmic proteins from Fao cells (lanes 2–4 and 6–8). For competition experiments (lanes 3, 4, 7 and 8), 3 μg of antisense oligonucleotides (ASns, lanes 3 and 7; AS2B16, lane 4; AS1,2-2E, lane 8) were incubated with 32P-labelled 16-mer CYP2B1 (lanes 1–4) and CYP2E1 (lanes 5–8) mRNA probes before the addition of the cytoplasmic proteins. Reaction mixtures were placed on ice and exposed to UV light for 8 min, followed by three runs at 120000 μJ. Complexes were visualized, after PAGE, by autoradiography.
As the CYP2B1 and CYP2E1 hairpin loops share an 80% homology, binding experiments were conducted using each mRNA in the presence of specific antisense oligonucleotides directed against either the CYP2E1 or the CYP2B1 hairpin loop. Figure 6(A) shows that the formation of the 16-mer CYP2B1 RNA–protein complex was displaced by the homologous antisense oligomer (AS2B16, lane 3), but also by a CYP2E1-specific antisense oligomer (AS1,2-2E, lane 4). Similar results were obtained with the CYP2E1: the formation of the 16-mer CYP2E1 RNA–protein complex was displaced by a CYP2E1-specific antisense oligomer (AS1,2-2E, lane 7) and also by a CYP2B1-specific antisense oligomer (AS2B16, lane 8). The protein(s) bound to both CYPs may be similar, or they might even be the same protein. We hypothesized that the same protein was able to bind to both CYP mRNA sequences. This was suggested by the similar migration in polyacrylamide gels (Figure 6A) and was in agreement with the cellular localization of the protein and its molecular mass. Binding assays using fractions from successive differential centrifugations of a rat liver extract were performed using both of the 16-mer 32P-labelled RNA probes. No complexes were found with the nucleus and the mitochondrial proteins (Figure 7). A complex was only observed with the cytosolic fraction and with the S9 supernatant, which contains the cytosol. It was thus concluded that the proteins are cytosolic. A UV-cross-linking experiment confirmed that complexes with either the CYP2E1 and/or CYP2B1 sequences migrate similarly on SDS/PAGE with a similar apparent molecular mass of 60 kDa (Figure 6B). This complex is likely to comprise a single protein, although the possibility cannot be excluded that another minor protein with a molecular mass of 43 kDa is also present. These experiments suggest that similar, or even identical, proteins are bound to the CYP2E1 and CYP2B1 hairpin loops.
Figure 7. Reconstitution of 16-mer RNA–protein binding complex by incubation of the CYP2B and CYP2E 16-mer RNA with different cellular fractions from rat liver.

The reaction was carried out in a final volume of 20 μl containing 105 c.p.m. of 32P-labelled 16-mer RNA probe (lanes 1–5) and 40 μg of rat liver differential-centrifugation fractions (lanes 1, nuclear pellet; lanes 2, S9 fraction; lanes 3, mitochondrial pellet; lanes 4, cytosolic fraction; lanes 5, microsomal pellet). Complexes were visualized, after PAGE, on the phosphoimager.
The functional consequences of the presence of a binding site for cytoplasmic proteins in the CYP2B1 mRNA were then studied. Indeed, RNA-binding proteins could play a role in the destabilization of this mRNA by insulin, although the results described above suggest that the binding of the cytoplasmic proteins to the CYP2B1 mRNA is not significantly altered by insulin. However, other functions of these proteins could be the target of the hormone. Consequently, additional experiments were undertaken to demonstrate the actual functionality of the 16-mer sequence in the destabilization of CYP2B1 mRNA by insulin. To investigate this question, the antisense oligonucleotide (AS2B16) was introduced in the Fao cells and the CYP2B1 mRNA was quantified in the presence or absence of insulin. A control using 18 S mRNA, which is not regulated by insulin, was used to correct loading variability. A CYP 26-mer antisense oligonucleotide containing the AS2B16 sequence was coupled to P in order to ensure an efficient delivery of the oligonucleotides in the cultured cells. Figure 8 shows that insulin significantly decreased (P<0.01) the amount of CYP2B1 mRNA, in untreated Fao cells, in cells treated with free penetratin and in cells transfected with the P-linked antisense control-P described in the Experimental section. In contrast, in cells transfected with the P-linked AS2B16, the CYP2B1 mRNA expression was no longer affected by insulin, suggesting that the 16-mer sequence is necessary for insulin effect on the amount of endogenous mRNA.
Figure 8. Effects of a 6 h 0.1 μM insulin treatment on the amount of CYP2B1 mRNA.

Fao cells were treated with 400 nM free P or Ascontrol-P or AS2B16-P and, 30 min later, cells were treated or not treated with 0.1 μM insulin for 6 h. CYP2B and 18 S mRNAs were quantified by Northern blotting. Bars 1, Fao cells (n=5); bars 2, Fao cells treated with free P (n=5); bars 3, Fao cells treated with P coupled with antisense control (AS control-P) (n=5); bars 4, Fao cells treated with P coupled with AS2B16 (AS2B16-P) (n=5). The results are expressed as percentages of the relative CYP2B1 mRNA expression, and 100% corresponds to the value in control cells. Data are means±S.E.M. For each treatment, statistical differences with the control values are indicated by **P<0.01.
The ability of this sequence to function in a heterologous environment was also tested by a half-life determination. We introduced the 16-mer CYP2B1 sequence in the coding sequence of the Renilla luciferase because this gene is not regulated by insulin and because the half-life of its mRNA is the same order of magnitude as that of CYP2B1. Following transfection of the different constructs into Fao cells, the effects of insulin (0.1 μM) on the Renilla luciferase mRNA expression were evaluated by quantitative RT-PCR (Figure 9) in each group (pRL and pRL-Seq2B16). Insulin did not modify the half-life of wild-type Renilla luciferase [10.3 h (r2=0.81) without insulin and 9.4 h (r2=0.87) with insulin]. Interestingly, the half-life of the chimaeric Renilla luciferase containing the 18-mer insertion was comparable with that of the wild-type (8.9 h, r2=0.94) and was significantly shortened to 5.3 h (r2=0.98) (P<0.05) in the presence of insulin. There was no significant difference between the decay rate constants of the pRL with or without insulin, and the pRL-Seq2B16 without insulin, curves.
Figure 9. Effects of insulin on the turnover rate of pRL and pRL-Seq2B16 mRNA.

Fao cells were treated with 0.1 μM insulin (○, pRL; △, pRL-Seq2B16) or left untreated (●, pRL; ▲, pRL-Seq2B16) for 3 h, then treated with 100 μM DRB (zero time). Total RNA was isolated at the indicated times. Renilla luciferase mRNA abundance was determined by quantitative RT-PCR (measurements at each point were performed in triplicate) and results were corrected with the corresponding 18 S rRNA level and normalized with respect to zero-time values. The first-order decay rate constants were derived and used to calculate the half-life values. The results were combined from two independent experiments.
DISCUSSION
The mechanisms through which insulin regulates gene transcription are now well understood [1]. However, insulin also regulates, at the post-transcriptional level, the expression of a number of genes, not only those involved in the glucose metabolic pathway, such as the those encoding phosphoenolpyruvate carboxykinase, glucose transporter type 4 (‘GLUT4’) and glycogen synthase, but also those involved in xenobiotic metabolism, such as those encoding CYP2B1 and CYP2E1 [1]. Most of the pathways by which the stability of these mRNAs is controlled by insulin remain unknown. However, Peng and Coon [17] showed that the 3′-UTR is involved in the regulation of the CYP2E1 mRNA by insulin. Moreover, our group showed that a 16-mer sequence is implicated in CYP2E1 mRNA destabilization by insulin [13], and Kawashima et al. [18] suggested that the down-regulation of resistin mRNA by insulin occurs through the synthesis of novel protein(s). Indeed, cycloheximide, a protein-synthesis inhibitor, completely blocks insulin-induced reduction of resistin mRNA. In the present study we examined the mechanisms of rat CYP2B1 regulation at the post-transcriptional level.
The importance of the post-transcriptional mechanisms in the regulation of gene expression has become increasingly clear, and recent studies have highlighted the role of specific RNA–protein interactions. This RNA-binding regulation requires both mRNA sequences (cis-acting elements) and RNA binding proteins (trans-acting factors). These proteins may influence RNA nuclear export, subcellular localization, stabilization and translation [19]. Several examples of CYP post-transcriptional regulation have been described previously: rat and rabbit CYP2E1 and rat CYP2B1 by insulin [2,17], mouse and rat CYP1A2 by 3-methylcholanthrene [20,21], human CYP1A1 by dehydroepiandrosterone [22], rat CYP1A1 and CYP1A2 by interferon-α/β [23] and CYP 2A5 by pyrazole [24,25]. The molecular mechanisms involved in this regulation have been only partly identified. For rabbit CYP2E1, mouse CYP2A5 and mouse CYP1A2, the cis-acting elements were shown to be localized in the 3′-UTR. AREs (adenylate/uridylate-rich elements), the most common determinant of RNA stability in mammalian cells, have been identified in these regions [26]. The AREs have been extensively investigated as cis-acting determinants of rapid mRNA turnover. Such ARE elements are not present either in the 5′- or the 3′-UTR of rat CYP2B1 and CYP2E1.
In the present study, we showed that cytoplasmic protein(s) was (were) able to bind to a 16-nucleotide sequence within the CYP2B1 mRNA and that this sequence is implicated in CYP2B1 mRNA destabilization by insulin.
This sequence is located in the 5′ part of the coding region; it is 80% similar to the one mediating the post-transcriptional effect of insulin in rat CYP2E1 mRNA, which is also located in the 5′ part of its coding region. Whereas most of the sequences involved in the regulation of mRNA stability were identified in the 3′-UTR, a few cases of regulatory sequences in the open reading frame have been reported in the mRNA of human c-fos, c-myc and interferon-β [27–29], mouse β-tubulin [30] and Saccharomyces cerevisiae (baker's yeast) PPR1 transcripts [31]. Moreover, two destabilizing elements in the protein-coding region of fushi tarazu mRNA are essential for Drosophila embryogenesis [32].
The 16-mer sequences involved in both CYP2B1 and CYP2E1 regulation by insulin have homologous sequences, a hairpin loop stucture (RNAdraw©) and display similar binding properties. Indeed, antisense oligonucleotides directed against either the CYP2B1 or CYP2E1 16-mer sequence were able to prevent the binding of cytoplasmic proteins to the CYP2B1 mRNA as well as to the CYP2E1 mRNA (Figure 6A). The sequence and structure of regulatory RNA motifs are critical for their function [33] as described for the iron-responsive element (‘IRE’) and for the CYP2a5 mRNA [25]. There are numerous examples of the importance of such secondary structures in the literature. The presence and integrity of a stem loop in the 3′ end of histone RNA is essential for the post-transcriptional regulation of the mRNA [34]. Yeast ribosomal protein L30 binds to an asymmetric, purine-rich internal loop in its transcript to repress its own splicing and translation [35].
Recently, a number of trans-acting RNA-binding proteins interacting with AREs have been described [36]. Some of these proteins appear to promote destabilization of the RNA upon binding to the ARE, whereas other proteins binding to the ARE contribute to RNA stabilization. Two nuclear hepatic proteins of 37 and 46 kDa exhibit a high affinity for a poly(U) motif in the 3′-UTR of the mouse CYP1a2 mRNA, and their binding efficiencies are altered by 3-methylcholanthrene [20]. Few investigations have been conducted on mouse CYP2a5 mRNA. A set of proteins from mouse liver is specifically bound to the 3′-UTR of this transcript, and a 44 kDa protein is suggested to be involved in the stabilization of CYP2A5 mRNA by controlling the length of the poly(A) tail [37]. Pyrazole treatment increases the binding of the 44 kDa protein and is associated with mRNA stabilization [24]. More recently, this protein has been identified as hnRNPA1 (heterogeneous nuclear ribonucleoprotein A1) [38]. In our work, UV-cross-linking experiments showed the binding of a major protein (with an apparent molecular mass of about 60 kDa) to either CYP2E1 or CYP2B1 mRNA. Similar electrophoretic properties of both complexes on SDS/PAGE and competitive experiments suggest that the same protein(s) [or very closely related protein(s)] was (were) able to bind the two 16-mer sequences. Furthermore, reconstitution experiments performed with different rat liver subcellular fractions showed that the protein(s) bound to the two 16-mer sequences is (are) both essentially expressed in the cytosol.
Insulin has no effect on the binding of this (these) protein(s) to either the CYP2B1 or the CYP2E1 16-mer sequence. The RNA binding ability and/or function of certain RNA-binding proteins can be modulated by means of conformational changes induced by amino acid modifications, such as post-translational phosphorylation. Raffalli-Mathieu et al. [20] showed that phosphatase treatment of the nuclear extracts reduced the binding of the 46 kDa protein to CYP1A2 mRNA, suggesting that the binding activity of the protein may depend on its phosphorylation status or on some intermediary factors necessary for binding. Likewise, the phosphorylation of the C-terminus of p68 RNA helicase abolishes its RNA binding, whereas the dephosphorylation restores it. Consequently, regulation of RNA-binding by the phosphorylation/dephosphorylation cycle could control the functions and the enzymatic activities of p68 RNA helicase in cells [39]. No differences in the localization or the pattern of the RNA–protein complexes were observed in experiments performed with cytoplasmic extracts from Fao cells treated, or not treated, with insulin. Thus, in the case of CYP2B1 mRNA, as previously shown for CYP2E1, the critical regulatory steps are probably not the binding of proteins to the target RNA sequence. It is possible that other functions, such as protein–protein interactions mediating the destabilization of the mRNA, are regulated. Alternatively, this RNA-binding protein may constitute a scaffold upon which the actual target of insulin can bind and exert an RNA destabilization function. It is known that hnRNP K recruits different molecular partners (e.g. mRNA, transcriptional repressors, inducible kinases etc.) and plays an important role in different pathways, such as RNA processing [40]. Furthermore, Ostrowski et al. [41] demonstrated that insulin-induced phosphorylation of hnRNP K protein alters K protein–protein and K protein–RNA interactions.
However, we were able to show the functional role of this sequence in the destabilization of CYP2B1 mRNA by insulin. Indeed, an antisense oligonucleotide against this sequence impaired the action of insulin on CYP2B1 mRNA concentration in Fao cells (Figure 8). Moreover, the insertion of the 16-mer CYP2B1 sequence in a heterologous gene unresponsive to insulin, namely that encoding the Renilla luciferase, led to the destabilization of its mRNA in the presence of insulin (Figure 9). These results are perfectly superimposable on those obtained with CYP2E1 [13]. These results showed that the modulation of the protein binding to the 16-mer sequences is not the mechanism by which insulin destabilizes CYP2B1 and CYP 2E1 mRNA.
In conclusion, we have shown that the binding of cytosolic proteins to a 16-mer hairpin loop was responsible for CYP2B1 and CYP2E1 mRNA turnover by insulin. These are the first identifications of functional RNA sequences that are the target of insulin action. In man, the equivalents of rat CYP2B1 and CYP2E1 are, respectively, human CYP2B6 and CYP2E1. We have recently obtained RNA–protein complexes by using full human RNA CYP2B6 and CYP2E1 probes and human cytosolic proteins from an HEPG2 cell culture. Our future investigations will seek to identify the proteins that bind to this sequence. Identification and characterization of these proteins could provide a considerable insight into the mechanisms involved in regulated mRNA turnover by insulin, not only for CYP expression, but also for other genes that are post-transcriptionally regulated by this hormone. The identification of the insulin pathway used in this regulation will complete our understanding of the role of these proteins in the whole cellular process initiated by insulin.
Acknowledgments
This work was supported by the l'Institut National de la Santé et de la Recherche Médicale (INSERM), the University ParisV René Descartes, the Région Ile de France (Soutien aux Equipes Scientifiques pour l'Acquisition de Moyens Expérimentaux), the Ligue contre le Cancer, the Association de Recherches contre le Cancer and the Fondation pour la Recherche Médicale.
References
- 1.Hall R. K., Granner D. K. Insulin regulates expression of metabolic genes through divergent signaling pathways. J. Basic Clin. Physiol. Pharmacol. 1999;10:119–133. doi: 10.1515/jbcpp.1999.10.2.119. [DOI] [PubMed] [Google Scholar]
- 2.De Waziers I., Garlatti M., Bouguet J., Beaune P. H., Barouki R. Insulin down-regulates cytochrome P450 2B and 2E expression at the post-transcriptional level in the rat hepatoma cell line. Mol. Pharmacol. 1995;47:474–479. [PubMed] [Google Scholar]
- 3.Nelson D. R., Koymans L., Kamataki T., Stegeman J. J., Feyereisen R., Waxman D. J., Waterman M. R., Gotoh O., Coon M. J., Estabrook R. W., et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 4. Reference deleted.
- 5.Lieber C. S. Cytochrome P-4502E1: its physiological and pathological role. Physiol. Rev. 1997;77:517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- 6.Koop D. R., Tierney D. J. Multiple mechanisms in the regulation of ethanol-inducible cytochrome P450IIE1. Bioessays. 1990;12:429–435. doi: 10.1002/bies.950120906. [DOI] [PubMed] [Google Scholar]
- 7.Dong Z. G., Hong J. Y., Ma Q. A., Li D. C., Bullock J., Gonzalez F. J., Park S. S., Gelboin H. V., Yang C. S. Mechanism of induction of cytochrome P-450ac (P-450j) in chemically induced and spontaneously diabetic rats. Arch. Biochem. Biophys. 1988;263:29–35. doi: 10.1016/0003-9861(88)90610-8. [DOI] [PubMed] [Google Scholar]
- 8.Song B. J., Matsunaga T., Hardwick J. P., Park S. S., Veech R. L., Yang C. S., Gelboin H. V., Gonzalez F. J. Stabilization of cytochrome P450j messenger ribonucleic acid in the diabetic rat. Mol. Endocrinol. 1987;1:542–547. doi: 10.1210/mend-1-8-542. [DOI] [PubMed] [Google Scholar]
- 9.Yamazoe Y., Murayama N., Shimada M., Yamauchi K., Kato R. Cytochrome P450 in livers of diabetic rats: regulation by growth hormone and insulin. Arch. Biochem. Biophys. 1989;268:567–575. doi: 10.1016/0003-9861(89)90324-x. [DOI] [PubMed] [Google Scholar]
- 10.Wang H., Negishi M. Transcriptional regulation of cytochrome p450 2B genes by nuclear receptors. Curr. Drug. Metab. 2003;4:515–525. doi: 10.2174/1389200033489262. [DOI] [PubMed] [Google Scholar]
- 11.Woodcroft K. J., Hafner M. S., Novak R. F. Insulin signaling in the transcriptional and posttranscriptional regulation of CYP2E1 expression. Hepatology. 2002;35:263–273. doi: 10.1053/jhep.2002.30691. [DOI] [PubMed] [Google Scholar]
- 12.Day D. A., Tuite M. F. Post-transcriptional gene regulatory mechanisms in eukaryotes: an overview. J. Endocrinol. 1998;157:361–371. doi: 10.1677/joe.0.1570361. [DOI] [PubMed] [Google Scholar]
- 13.Moncion A., Truong N. T., Garrone A., Beaune P., Barouki R., De Waziers I. Identification of a 16-nucleotide sequence that mediates post-transcriptional regulation of rat CYP2E1 by insulin. J. Biol. Chem. 2002;277:45904–45910. doi: 10.1074/jbc.M207841200. [DOI] [PubMed] [Google Scholar]
- 14.Pave-Preux M., Aggerbeck M., Veyssier C., Bousquet-Lemercier B., Hanoune J., Barouki R. Hormonal discrimination among transcription start sites of aspartate aminotransferase. J. Biol. Chem. 1990;265:4444–4448. [PubMed] [Google Scholar]
- 15.Guillouzo A., Morel F., Ratanasavanh D., Chesné C., Guguen-Guillouzo C. Long-term culture of fonctional hepatocytes. Toxicol. in vitro. 1990;4:415–427. doi: 10.1016/0887-2333(90)90092-8. [DOI] [PubMed] [Google Scholar]
- 16.Zandomeni R., Mittleman B., Bunick D., Ackerman S., Weinmann R. Mechanism of action of dichloro-β-D-ribofuranosylbenzimidazole: effect on in vitro transcription. Proc. Natl. Acad. Sci. U.S.A. 1982;79:3167–3170. doi: 10.1073/pnas.79.10.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a.Matzura O., Wennborg A. RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. CABIOS Comput. Appl. Biosci. 1996;12:247–249. doi: 10.1093/bioinformatics/12.3.247. [DOI] [PubMed] [Google Scholar]
- 17.Peng H. M., Coon M. J. Regulation of rabbit cytochrome P450 2E1 expression in HepG2 cells by insulin and thyroid hormone. Mol. Pharmacol. 1998;54:740–747. [PubMed] [Google Scholar]
- 18.Kawashima J., Tsuruzoe K., Motoshima H., Shirakami A., Sakai K., Hirashima Y., Toyonaga T., Araki E. Insulin down-regulates resistin mRNA through the synthesis of protein(s) that could accelerate the degradation of resistin mRNA in 3T3-L1 adipocytes. Diabetologia. 2003;46:231–240. doi: 10.1007/s00125-002-1022-3. [DOI] [PubMed] [Google Scholar]
- 19.Dreyfuss G., Kim V. N., Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell. Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 20.Raffalli-Mathieu F., Geneste O., Lang M. A. Characterization of two nuclear proteins that interact with cytochrome P-450 1A2 mRNA. Regulation of RNA binding and possible role in the expression of the Cyp1a2 gene. Eur. J. Biochem. 1997;245:17–24. doi: 10.1111/j.1432-1033.1997.00017.x. [DOI] [PubMed] [Google Scholar]
- 21.Silver G., Krauter K. S. Expression of cytochromes P-450c and P-450d mRNAs in cultured rat hepatocytes. 3-Methylcholanthrene induction is regulated primarily at the post-transcriptional level. J. Biol. Chem. 1988;263:11802–11807. [PubMed] [Google Scholar]
- 22.Ciolino H. P., Yeh G. C. The steroid hormone dehydroepiandrosterone inhibits CYP1A1 expression in vitro by a post-transcriptional mechanism. J. Biol. Chem. 1999;274:35186–35190. doi: 10.1074/jbc.274.49.35186. [DOI] [PubMed] [Google Scholar]
- 23.Delaporte E., Renton K. W. Cytochrome P4501A1 and cytochrome P4501A2 are downregulated at both transcriptional and post-transcriptional levels by conditions resulting in interferon-α/β induction. Life Sci. 1997;60:787–796. doi: 10.1016/s0024-3205(97)00006-4. [DOI] [PubMed] [Google Scholar]
- 24.Thulke-Gross M., Hergenhahn M., Tilloy-Ellul A., Lang M., Bartsch H. Pyrazole-inducible proteins in DBA/2 mouse liver bind with high affinity to the 3′-untranslated regions of the mRNAs of coumarin hydroxylase (CYP2A5) and c-jun. Biochem. J. 1998;331:473–481. doi: 10.1042/bj3310473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tilloy-Ellul A., Raffalli-Mathieu F., Lang M. A. Analysis of RNA–protein interactions of mouse liver cytochrome P4502A5 mRNA. Biochem. J. 1999;339:695–703. [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C. Y., Shyu A. B. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein P. L., Herrick D. J., Prokipcak R. D., Ross J. Control of c-myc mRNA half-life in vitro by a protein capable of binding to a coding region stability determinant. Genes Dev. 1992;6:642–654. doi: 10.1101/gad.6.4.642. [DOI] [PubMed] [Google Scholar]
- 28.Schiavi S. C., Wellington C. L., Shyu A. B., Chen C. Y., Greenberg M. E., Belasco J. G. Multiple elements in the c-fos protein-coding region facilitate mRNA deadenylation and decay by a mechanism coupled to translation. J. Biol. Chem. 1994;269:3441–3448. [PubMed] [Google Scholar]
- 29.Paste M., Huez G., Kruys V. Deadenylation of interferon-β mRNA is mediated by both the AU-rich element in the 3′-untranslated region and an instability sequence in the coding region. Eur. J. Biochem. 2003;270:1590–1597. doi: 10.1046/j.1432-1033.2003.03530.x. [DOI] [PubMed] [Google Scholar]
- 30.Yen T. J., Gay D. A., Pachter J. S., Cleveland D. W. Autoregulated changes in stability of polyribosome-bound β-tubulin mRNAs are specified by the first 13 translated nucleotides. Mol. Cell. Biol. 1988;8:1224–1235. doi: 10.1128/mcb.8.3.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kebaara B., Nazarenus T., Taylor R., Forch A., Atkin A. L. The Upf-dependent decay of wild-type PPR1 mRNA depends on its 5′-UTR and first 92 ORF nucleotides. Nucleic Acids Res. 2003;31:3157–3165. doi: 10.1093/nar/gkg430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito J., Jacobs-Lorena M. Functional mapping of destabilizing elements in the protein-coding region of the Drosophila fushi tarazu mRNA. J. Biol. Chem. 2001;276:23525–23530. doi: 10.1074/jbc.M102965200. [DOI] [PubMed] [Google Scholar]
- 33.Dandekar T., Hentze M. W. Finding the hairpin in the haystack: searching for RNA motifs. Trends Genet. 1995;11:45–50. doi: 10.1016/s0168-9525(00)88996-9. [DOI] [PubMed] [Google Scholar]
- 34.Williams A. S., Marzluff W. F. The sequence of the stem and flanking sequences at the 3′ end of histone mRNA are critical determinants for the binding of the stem-loop binding protein. Nucleic Acids Res. 1995;23:654–662. doi: 10.1093/nar/23.4.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White S. A., Hoeger M., Schweppe J. J., Shillingford A., Shipilov V., Zarutskie J. Internal loop mutations in the ribosomal protein L30 binding site of the yeast L30 RNA transcript. RNA. 2004;10:369–377. doi: 10.1261/rna.2159504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Staton J. M., Thomson A. M., Leedman P. J. Hormonal regulation of mRNA stability and RNA–protein interactions in the pituitary. J. Mol. Endocrinol. 2000;25:17–34. doi: 10.1677/jme.0.0250017. [DOI] [PubMed] [Google Scholar]
- 37.Geneste O., Raffalli F., Lang M. A. Identification and characterization of a 44 kDa protein that binds specifically to the 3′-untranslated region of CYP2a5 mRNA: inducibility, subcellular distribution and possible role in mRNA stabilization. Biochem. J. 1996;313:1029–1037. doi: 10.1042/bj3131029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glisovic T., Ben-David Y., Lang M. A., Raffalli-Mathieu F. Interplay between hnRNP A1 and a cis-acting element in the 3′ UTR of CYP2A5 mRNA is central for high expression of the gene. FEBS Lett. 2003;535:147–152. doi: 10.1016/s0014-5793(02)03893-0. [DOI] [PubMed] [Google Scholar]
- 39.Yang L., Yang J., Huang Y., Liu Z. R. Phosphorylation of p68 RNA helicase regulates RNA binding by the C-terminal domain of the protein. Biochem. Biophys. Res. Commun. 2004;314:622–630. doi: 10.1016/j.bbrc.2003.12.129. [DOI] [PubMed] [Google Scholar]
- 40.Bomsztyk K., Van Seuningen I., Suzuki H., Denisenko O., Ostrowski J. Diverse molecular interactions of the hnRNP K protein. FEBS Lett. 1997;403:113–115. doi: 10.1016/s0014-5793(97)00041-0. [DOI] [PubMed] [Google Scholar]
- 41.Ostrowski J., Kawata Y., Schullery D. S., Denisenko O. N., Higaki Y., Abrass C. K., Bomsztyk K. Insulin alters heterogeneous nuclear ribonucleoprotein K protein binding to DNA and RNA. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9044–9049. doi: 10.1073/pnas.161284098. [DOI] [PMC free article] [PubMed] [Google Scholar]