

Abstract
Thimet oligopeptidase (TOP) is a soluble metalloendopeptidase belonging to a family of enzymes including neurolysin and neprilysin that utilize the HEXXH metal-binding motif. TOP is widely distributed among cell types and is able to cleave a number of structurally unrelated peptides. A recent focus of interest has been on structure–function relationships in substrate selectivity by TOP. The enzyme's structural fold comprises two domains that are linked at the bottom of a deep substrate-binding cleft via several flexible loop structures. In the present study, fluorescence spectroscopy has been used to probe structural changes in TOP induced by the chemical denaturant urea. Fluorescence emission, anisotropy and collisional quenching data support a two-step unfolding process for the enzyme in which complete loss of the tertiary structure occurs in the second step. Complete loss of activity and loss of catalytic Zn(II) from the active site, monitored by absorption changes of the metal chelator 4-(2-pyridylazo)-resorcinol, are also connected with the second step. In contrast, the first unfolding event, which is linked to changes in the non-catalytic domain, leads to a sharp increase in kcat towards a 9-residue substrate and a sharp decrease in kcat for a 5-residue substrate. Thus a conformational change in TOP has been directly correlated with a change in substrate selectivity. These results provide insight into how the enzyme can process the range of structurally unrelated peptides necessary for its many physiological roles.
Keywords: denaturation, metalloendopeptidase, protein folding, substrate specificity, thimet oligopeptidase, urea
Abbreviations: Bk, bradykinin; DNP, 2,4-dinitrophenyl; DSC, differential scanning calorimetry; MCA, 7-methoxycoumarin-4-acetyl-Pro-Leu-GlyPro-Lys-dinitrophenol; mca-Bk, 7-methoxycoumarin-4-acetyl-[Ala7, Lys(DNP)9]-bradykinin; PAR, 4-(2-pyridylazo)resorcinol; TCEP, tris-(2-carboxyethyl)-phosphine; TOP, thimet oligopeptidase
INTRODUCTION
Thimet oligopeptidase (TOP) (EC 3.4.24.15; EP24.15) is a 78-kDa Zn(II)-dependent endopeptidase [1]. It is related to several metallopeptidases, including angiotension-converting enzyme, neprilysin and neurolysin, all of which share a common HEXXH metal-binding motif [1]. TOP has been isolated in a variety of cells types, notably the brain, testis and pituitary, and has various sequence requirements for substrates [2]. For these reasons, TOP is implicated in the neuropeptide regulation of several physiological processes, such as blood pressure control, reproduction and the immune response. However, its specific roles are difficult to determine, because of overlapping substrate specificities with other neuropeptidases.
Recently, the structure of the apo (substrate-free) form of the human variant of TOP was solved by X-ray diffraction [3]. The enzyme's three-dimensional fold is, indeed, quite similar to that of its closest relative, neurolysin, as previously proposed based on modelling studies [4]. TOP is primarily α-helical (Figure 1), and consists of two domains separated by a deep cleft containing the substrate-binding site. The primary sequence crosses back and forth between domain I and II several times, connecting the two domains mainly via loop regions located at the bottom of the cleft. Domain II contains the active-site Zn(II) and all the residues critical for peptide cleavage except Tyr612. This tyrosine residue has been shown to be involved in H-bond donation, stabilizing the transition-state structure, and is located in a loop region connecting the two domains [5,6]. Domain I does not contain any of the key active-site residues, but may play a role in limiting accessibility of substrates to the active site [3].
Figure 1. Tertiary structure of TOP showing the location of the active site zinc (white sphere) and the tryptophan residues (space filled) present in domains I and II.

Two unique features of substrate selectivity by TOP are of interest, length restriction and sequence variability. For instance, the longest peptide shown to be susceptible to hydrolysis by the enzyme is 17 residues in length [7], yet TOP is known cleave at specific sites, in vitro or in vivo, many peptides of differing sequences, such as Bk (bradykinin), gonadotropin-releasing hormone, somatostatin, α- and β-neoendorphin, dynorphin A and neurotensin [1,2,8–13]. On the basis of the X-ray structure and previous modelling studies, it has been proposed that the deep cleft formed at the interface between domains I and II limits the size of unstructured peptides that may have access to the active site. The cleft is lined with extended loop regions that are proposed to rearrange and interact with the substrate peptide near the scissile bond [3,14]. It has also been speculated that the enzyme may undergo a hingeing motion as a consequence of the movement of the flexible loop regions connecting the domains. It has been noted that at least some structural reorientation during substrate binding is necessary in order to position Tyr612 properly during catalysis [3,5]. It is possible that the channel could accommodate larger substrates if association between the two domains of the enzyme were interrupted. Furthermore, changes in the extensible loops may allow TOP to recognize peptides of variable sequence [3,14].
Fluorescence spectroscopy is a valuable tool for studying protein structure, because the emission spectrum of the enzyme is sensitive to changes in the environment surrounding the intrinsic side-chain fluorophores of endogenous tyrosine and tryptophan. TOP contains seven tryptophan residues distributed unevenly between the two domains. Trp335 and Trp614 reside in domain I, and tryptophan at positions 26, 124, 389, 511 and 513 are found in domain II. We here report on use of the denaturing agent urea as a tool to study partial unfolding of TOP, as monitored by fluorescence emission, collisional quenching and anisotropy. Unfolding occurs as a two-step process, with loss of the catalytic zinc occurring only with the second unfolding event. Complete loss of activity towards a 5-residue quenched fluorescent substrate MCA (7-methoxycoumarin-4-acetyl-Pro-Leu-Gly-Pro-Lys-dinitrophenol) is apparent at even low urea concentrations, but activity towards a 9-residue Bk-derived substrate is enhanced at low urea levels. These consistent results show that TOP undergoes conformational changes that differentially affect substrate recognition, and suggest a mechanism by which TOP can accommodate substrate variation.
EXPERIMENTAL
Materials
Glutathione–Sepharose, Sephacryl S-200 and PD-10 columns were obtained from Amersham Pharmacia Biotech (Piscataway, NJ, U.S.A.). Tris/glycine polyacrylamide gels (12%) were from Invitrogen (Carlsbad, CA, U.S.A.), and stained with Gelcode Blue from Pierce (Rockford, IL, U.S.A.). TCEP [tris-(2-carboxyethyl)phosphine] was also purchased from Pierce. The quenched fluorescent substrates MCA and modified bradykinin, mca-Bk {7-methoxycoumarin-4-acetyl-[Ala7, Lys(DNP)9]-bradykinin, where DNP is 2,4-dinitrophenyl}, were obtained from Bachem (King of Prussia, PA, U.S.A.). All other reagents were purchased from Sigma Chemical Co (St. Louis, MO, U.S.A.).
Recombinant TOP preparation
TOP (accession number P24155) was expressed and purified as described previously [15]. The enzyme concentration was determined using the molar absorption coefficient ε280=73.11 mM−1·cm−1, calculated based on the amino acid content of the protein using the automated ProtParam Tool on the SWISS ExPASy server [5,16].
Kinetic assays
Kinetic assays were performed using a Cary Eclipse spectrofluorimeter or PerkinElmer luminescence spectrometer LS 50 B. The cleavage of the fluorogenic MCA [17] or mca-Bk substrate was monitored by the increase in emission at 400 nm over time using an λexcitation of 325 nm. Substrate concentration was calculated based on the molar absorption coefficient ε365 (17.3 mM−1·cm−1) of the DNP. Product formation was determined to be linear with time under all conditions monitored, and less than 10% of the substrate was consumed during the assay. Assays were performed in duplicate at 23 °C in 25 mM Tris/HCl at pH 7.8, adjusted to a conductivity of 12 mS/cm2 with KCl and containing 10% glycerol. The temperature was chosen so as to minimize breakdown of both enzyme and substrate and to allow for sensitivity at low substrate concentrations; the pH represents the optimal value for kcat/Km for MCA [5]. TCEP (1 mM) was also added to the buffer to prevent protein dimerization [29]. For assays in urea, the above buffer and an identical buffer containing approx. 10 M urea were mixed in the appropriate ratio. The final urea concentration was determined based on the refractive index of the solution [18]. The change in fluorescence intensity over time was converted into rate of product formation using a standard curve calculated for the peptide products. Individual standard curves were prepared at each urea concentration. Although the intercept of the standard curve changed, the slope was found to be independent of urea concentration. The kinetic parameters Vmax and Km were determined using a hyperbolic fit {rate=Vmax[S]/(Km+[S])} to the plot of substrate concentration (μM) versus rate of product formation (μmol/s per μmol of enzyme) under conditions in which [S] is above and below Km.
HPLC analysis
Products of the enzymatic reactions with mca-Bk were analysed by HPLC on a Hewlett Packard 1090 apparatus. The reaction mixture, 200 μl total volume in Tris buffer or Tris buffer with 2 M urea, contained mca-Bk (0.4 mg/ml) and 0.05 μM enzyme. A sample was taken at 0 min (before initiation of the reaction) and after reacting for 30 min at 23 °C. The reaction was terminated with an equal volume of 1% trifluoroacetic acid in methanol.
A 20-μl aliquot was subjected to reverse-phase HPLC using a C18 3μ column (150 mm×4.6 mm; Alltech). Solvent A was acetonitrile and solvent B 0.1% trifluoroacetic acid in water. A linear gradient of 10–82% solvent A was applied, and the products were detected by absorbance monitored at 330 nm.
DSC (differential scanning calorimetry)
All experiments were performed using a VP-DSC microcalorimeter (MicroCal Inc., Northampton, MA, U.S.A.) at an upscan rate of 60 °C per h over the range 20° to 110 °C. Degassed buffer, identical with that diluting the sample, was used as the reference. The injection volume was 0.51 ml. Raw heat data were transformed and plotted as heat capacity as a function of temperature using the Origin for DSC software supplied by the manufacturer.
Urea-dependent unfolding of TOP
Structural changes as a function of urea were monitored by intrinsic fluorescence, collisional quenching and anisotropy. Buffer conditions were identical with those for activity assays, except that 15 mM TCEP was used as the reductant. In all experiments the final enzyme concentration was 0.25 μM. At this concentration the absorbance at 295 nm is less than 0.06 and errors due to the inner filter effect are minimized [19]. Samples were maintained at 23 °C using a circulating water bath.
For intrinsic fluorescence of tryptophan residues, the excitation (5 nm slit width) was set to 295 nm and the emission (10 nm slit width) intensity was monitored from 320 to 480 nm. The background fluorescence was subtracted using a blank solution at each concentration of urea. The appropriate incubation time for samples at each urea concentration was determined by monitoring the emission until no change was apparent after 1–2 h. Changes in emission at low urea (0–4 M) were found to be immediate. However, the changes in emission associated with the second unfolding event, at higher urea concentrations, required an incubation time of 12–14 h to reach equilibrium. The fractional changes in emission (Em) at 326 nm and 377 nm as a function of urea fit to the following double sigmoidal equations:
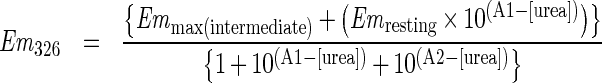 |
1 |
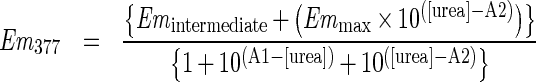 |
2 |
in which A1 and A2 represent the urea concentrations at the first and second transitions.
The degree of exposure of tryptophan in the unfolded states at 0 M, 3.5 M and 8 M urea was monitored by collisional quenching with I−. Samples at the desired concentration of I− were prepared by mixing two identical buffered urea solutions containing either 0.3 M KCl or 0.3 M KI. Stock TOP was added to a final concentration of 0.25 μM and incubated at 23 °C until reaching equilibrium. The integrated intensity from 320 to 420 nm was monitored as a function of the concentration of quencher. The data were then analysed according to the Stern–Volmer equation [20]:
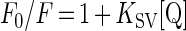 |
3 |
In this relationship, F0 is the fluorescence in the absence of quencher, F is the fluorescence in the presence of quencher, [Q] is the concentration of quencher, and KSV, the Stern–Volmer quenching constant, is an approximate measure of the degree of exposure of tryptophan residues to the solvent.
Steady-state anisotropy measurements were made using the fast filter component of the PerkinElmer LS 50 B spectrometer. Anisotropy values (R) for each sample, in 1 cm path-length cuvettes, recorded with λexcitation=295 nm and λemission=350 nm, were calculated from the equations
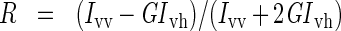 |
4 |
 |
5 |
in which Ivv, Ivh, Ihv and Ihh are the emission intensities (I) when the excitation and emission polarizers are in the vertical (v) and horizontal (h) orientations, and G is the g-factor of the emission monochromator.
Release of catalytic zinc
The presence of free zinc ions was measured by using the zinc chelator PAR [4-(2-pyridylazo)resorcinol]. A calibration curve at pH 7.8 yielded an molar absorption coefficient for the Zn–PAR complex of 60000 M−1·cm−1 at 500 nm [21]. Calibration curves were also determined at each urea concentration tested. Variation in the molar absorption coefficient was found to be negligible. PAR (final concentration 0.10 mM) was added to protein samples (approx. 2.0 μM) immediately before measurement of absorbance. The concentration of Zn(II) released by the enzyme in various concentrations of urea was calculated using the determined molar absorption coefficient at 500 nm and then converted into the percentage of Zn(II) released based on the protein concentration.
RESULTS
Urea-induced unfolding of TOP
Figures 2(A) and 2(B) show the changes in the intrinsic emission spectrum of TOP as a result of chemically induced unfolding by urea. Two distinct changes are apparent. An increase in intensity occurs at low urea concentrations (Figure 2A), followed by a decrease in intensity and red-shift in the emission maximum at high urea concentrations (Figure 2B). The resulting changes in emission at 326 nm and 377 nm as a function of urea are shown in Figure 3(A), along with the urea-dependent changes in the fluorescence anisotropy. The continuous lines in Figure 3(A) represent the fit of the data to double hyperbolic functions 1 and 2. In each case, the unfolding of TOP by urea at physiological pH was revealed to occur in two co-operative steps. The first transition begins below 1 M urea and reaches a plateau between 2–3 M urea. This transition accounts for approx. 15% of the intrinsic protein fluorescence, and is characterized by an increase in emission intensity at 326 and 377 nm and a small decrease in the fluorescence anisotropy. Also consistent among the plots is a second transition that occurs with equilibrium urea concentrations of 5.5 M and plateau at 7.0 M urea. In this case, the emission intensity decreases at 326 nm and increases at 377 nm, as expected from the red shift in the peak maximum in Figure 3(A) at this concentration of urea. Concurrent with these changes in fluorescence is a substantial decrease in the fluorescence anisotropy.
Figure 2. Change in the fluorescence emission spectra of TOP from (A) 0–3 M urea and (B) 3–9 M urea.

Arrows show the direction of change with increasing urea concentrations.
Figure 3. Effect of urea on the structure and stability of TOP.

(A) Unfolding of TOP monitored by intrinsic fluorescence of tryptophan at 326 nm (○) and 377 nm (●) upon excitation at 295 nm and fluorescence anisotropy (▲). All data were normalized to the maximum intensity. Lines represent the fit of the data to double sigmoidal equations (1) and (2) in the Experimental section. (B) The effect of urea on the activity of TOP. The kinetic parameter kcat/Km for MCA (▲) and mca-Bk (□), normalized to 100% activity at 0 M urea, is plotted as a function of increasing urea concentration. (C) Zn(II) loss from the enzyme as a function of urea monitored by the change in absorption of PAR upon binding Zn. The data were normalized to the percentage of Zn(II) lost at minimum and maximum absorption of PAR at 500 nm.
To probe further the nature of the different conformational states of the enzyme, collisional quenching with I− was performed at 0 M, 3.5 M and 7 M urea, corresponding to the three plateau regions in the unfolding curves shown in Figure 3(A). The resulting Stern–Volmer plots are shown in Figure 4. An increase in the Stern–Volmer constant KSV, represented by slope of the plots, is an indication of increased accessibility of tryptophan residues to the quencher [20]. In comparison with the native enzyme at 0 M urea, there is a small increase in the slope of the Stern–Volmer plot at 3.5 M urea and a large increase in the slope at 7 M urea.
Figure 4. Stern–Volmer plots for the quenching (λexcitation, 295 nm) of tryptophan emission by I− at 0 M urea (△), 3.5 M urea (◆) and 7.0 M urea (◇).

These concentrations of urea represent the three plateau regions represented by the change in emission at 377 nm shown in Figure 2(A).
Kinetic studies
The differential change in enzyme activity toward the two peptide substrates MCA and mca-Bk is shown in Figure 3(B) and kinetic constants are summarized in Table 1. Activity of TOP towards the small (5-residue) synthetic substrate MCA dramatically decreased in the presence of urea. As shown in Table 1, this is primarily due to a change in kcat, which decreases over 6-fold by 1.5 M urea and over 40-fold by 2.5 M urea. There was relatively smaller corresponding increase in Km.
Table 1. Kinetic constants for activity of TOP.
The results are expressed as the means±S.E.M., obtained from curve fitting a hyperbolic function to the initial rates at 8–9 different concentrations of substrate. Each individual substrate concentration was carried out in at least triplicate.
| MCA | mca-Bk | |||
|---|---|---|---|---|
| [Urea] (M) | kcat (s−1) | Km (μM) | kcat (s−1) | Km (μM) |
| 0 | 0.42±0.02 | 6.4±0.7 | 0.30±0.01 | 0.057±0.005 |
| 1.5 | 0.066±0.01 | 13.7±2.6 | 0.72±0.01 | 0.13±0.01 |
| 2.5 | 0.010±0.001 | 9.4±1.1 | 0.91±0.01 | 0.48±0.02 |
| 3.5 | – | – | 1.07±0.06 | 1.27±0.16 |
In contrast, TOP activity toward the somewhat larger, more polar, mca-Bk substrate (nine residues) was increased by low levels of urea (Figure 3B and Table 1). As can be seen in Table 1, kcat for the mca-Bk substrate increased 2.5-fold from 0 to 1.5 M urea, and 3.5-fold by 3.5 M urea. Similar to the results with MCA, Km increased slightly as a function of urea.
Zinc release
In order to monitor Zn(II) release from the enzyme as a function of urea concentration, the enzyme in various concentrations of urea was incubated with the Zn(II) chelator, PAR. PAR shows a distinct UV–visual spectral change from yellow (maximum at 412 nm) to orange (maximum at 500 nm) upon binding Zn(II). Unfolding of the active-site domain causes Zn(II) to released from the enzyme and taken up by PAR. Figure 3(C) shows the loss of zinc from TOP as a function of increasing urea concentration at pH 7.8. Release of zinc occurred at urea concentrations corresponding to the second transition of TOP's unfolding (compare with Figure 3A), whereas at the first transition state the zinc was still present.
Thermal unfolding of TOP
The thermal behavior of TOP was monitored by DSC. Thermal unfolding of TOP in 25 mM Tris/HCl at pH 7.8 and glycerol (Figure 5) revealed a two-step process in which a major endotherm occurs with a Tm of 64 °C and minor endotherm occurs with a Tm of 55 °C. Heating beyond the major peak resulted in aggregation and irreversible denaturation of the enzyme, as indicated by the exothermic transition after 70 °C.
Figure 5. Thermal unfolding of TOP monitored by DSC.

Note: 1 kcal=4.184 kJ.
HPLC analysis
To determine if the change in activity towards the mca-Bk substrate was due to a change in substrate recognition and cleavage site, the products obtained at 0 M and 2 M urea were analysed by HPLC. Examination of the elution products by UV–visual detection at 330 nm revealed two products, eluting at 11.86 min and 12.11 min (results not shown), suggesting that there is a single cleavage site in the mca-Bk substrate. Identical results concerning the position of the cleavage site were obtained in the presence of 2 M urea, indicating that urea does not alter the alignment of the substrate in the binding site.
DISCUSSION
TOP is likely to play a role in signalling and regulation of a variety of normal physiological processes, as well as in some disease states [22]. This range of functions is a result of its wide cellular distribution and its ability to cleave numerous peptides with no significant cleavage site or subsite specificity. TOP has been isolated as a secreted and cytosolic form, as a soluble and membrane-associated form and from various subcellular compartments [1]. TOP is known to cleave neurotensin, Bk, angiotensin I, somatostatin and gonadotropin-releasing hormone [8–13]. It is also involved in the degradation of MHC class I peptides produced by the proteosome for antigen presentation [23–25]. In this role subserved by TOP in regulating antigen presentation on the cell surface, there are hundreds of different peptides that will be bound and degraded by the enzyme. TOP plays an indirect role in the degradation of the Aβ peptides, associated with plaque formation in Alzheimer's disease, possibly by activating an Aβ-degrading serine protease [26,27]. On the basis of the importance of the multitude of roles, and large number of potential substrates of TOP, it is relevant to understand the structural mechanism by which it can accommodate its various substrates.
As revealed by structural studies, TOP comprises two domains (see Figure 1), divided by a deep channel that forms the substrate-binding pocket [3]. The two domains are connected by loops at four regions of the enzyme. Particular attention has been given to residues 599–611, which form a loop structure that is close enough to the active site to interact with even the smallest TOP substrates. This segment contains several glycine residues and should therefore be relatively flexible. On the basis of the inherent flexibility of this loop and on differences in this region between the crystal structures of TOP and its homologue neurolysin (60% sequence identity), it has been proposed that conformational changes in this segment, and in other loop and coil structures that connect the two domains, lead to changes in subsite specificity necessary to accommodate the numerous peptide substrates hydrolysed by TOP [3]. Oliveira et al. [28] have tried to build on this proposal by conducting activity assays under varying conditions of salt concentrations and temperature. In the study [28], increases in ionic strength resulted in displacement of the cleavage site of a series of quenched fluorescent peptides of sequence Abz-GGFLRRXQ-EDDnp (where Abz is o-aminobenzoic acid and EDDnp is ethylenediamine 2,4-dinitrophenyl) from the Leu–Arg to the Arg–Arg bond, and led to an increased preference for small non-polar amino acids at position X. However, no conformational change was detected by CD spectroscopy over the range of salt concentrations used. The net change detectable by CD spectroscopy is approx. 3%, which would account for a secondary change of approx. 20 residues.
In the present study, fluorescence spectroscopy has been used to characterize structural changes in TOP. At pH 7.8 TOP unfolds in two co-operative stages with increasing concentrations of urea. The two-step process is revealed in the biphasic nature of the change in emission intensity at 326 and 377 nm and the fluorescence anisotropy monitored as a function of urea (Figure 3A). A two-step unfolding process is also supported by DSC measurements. Changes in the heat capacity as a function of increasing temperature shows two endothermic peaks (Figure 5), indicative of two distinct steps in the thermal unfolding of TOP. The second unfolding step, represented by the major endothermic peak with a Tm of 64 °C (Figure 5) and the increase in emission intensity at 377 nm from 4–7 M urea (Figures 2B and 3A), undoubtedly represents complete unfolding of the enzyme. This is supported by the significant red shift in the TOP emission spectrum that occurs between 4–7 M urea (Figure 2B) and by anisotropy and quenching studies. A large decrease in fluorescence anisotropy (Figure 3A) occurs concomitantly with the red shift in emission, which is in agreement with the increased translational motion of tryptophan residues expected in a transition to a completely unfolded state of the enzyme. Quenching studies also showed an increase in solvent-accessible tryptophan residues for TOP in 7 M urea compared with TOP in buffer alone. The Stern–Volmer slope (KSV) for fluorescence quenching of tryptophan residues by I− is 2.85 in 7 M urea, compared with a negligible value of KSV (0.11) in 0 M urea (Figure 4).
The first unfolding transition leads to the formation of a stable intermediate between 2 M and 4 M urea. This transition, corresponding to the increase in emission intensity at low (0–4 M) urea concentrations (Figures 2A and 3A) and the DSC endotherm with a Tm of 55 °C (Figure 5), is more subtle than the second, but also more interesting, due to the changes in activity associated with the formation of this intermediate (Figure 3B).
Several possible explanations could account for the first transition and the structure of the intermediate. The transition could represent dimerization of the enzyme. TOP is known to form dimers in solution, linked via disulphide bridges between surface cysteine residues [15,29]. However, the formation of dimers is unlikely, since the transition at 0–4 M urea leads to a decrease in anisotropy. There is little change in the maximum emission wavelength during this transition, and most of the change in anisotropy in dilute solution can be attributed to changes in the rotational rate of tryptophan residues in the enzyme [19]. Dimerization would therefore lead to an increase in the anisotropy. Conversely, low concentrations of urea could promote the disruption of dimers under reducing conditions, but the activity data do not support this conclusion. Dimer formation is known to reduce kcat for both MCA and mca-Bk. If the intermediate could be accounted for by the disruption of dimers, an increase in kcat would be expected for both substrates in the 0–4 M urea range. Table 1 shows that although kcat increases for mca-Bk, it substantially decreases for the MCA substrate, thus ruling out this possibility.
It might be argued that the observed switch in specificity of the enzyme with increased urea concentration is due to inactivation of TOP (leading to lower activity towards MCA) coupled with changes in the structure of mca-Bk that make it more susceptible to hydrolysis. The results in Figure 3(B), which shows the percentage activity of the enzyme as a function of urea, argue against the first assumption. If the amount of activity towards MCA represented the remaining amount of active enzyme, then at 1.5 M urea the active enzyme would be only about 4% and at 2.5 M urea close to zero. If that were the case, it would be very unlikely to see the increase in activity towards mca-Bk at 1.5 M urea, and essentially impossible to retain the 40% of activity towards mca-Bk that is observed at 2.5 M urea. Furthermore, Bk itself has been shown to exist in an extended and disordered state in solution [30–32], making it more susceptible to peptidases than are peptides with discernible secondary structure [33].
On the basis of the present data, disruption of domain I (the non-catalytic domain) most reasonably accounts for the observed spectroscopic and activity changes in the 0–4 M urea range (Figures 2A, 3A and 3B). Whereas domain II contains all the residues responsible for binding Zn(II), domain I does not contain any of the residues comprising the active site, but instead is proposed to limit substrate access to the active site and possibly limit the size of substrates that can be accepted by the enzyme [3,14]. Therefore, a structural change in domain I would leave the active site intact, but could markedly change activity due to interactions with substrates. Figure 3(C) shows the change in Zn(II)-binding by PAR when this chelator is added to solutions of TOP containing increasing concentrations of urea. As Zn(II) is lost from the enzyme it is taken up by PAR, which subsequently undergoes a colour change that can be detected at 500 nm [21]. This change is expressed as a percentage of Zn(II) lost from the enzyme. From these data it is clear that domain II and the elements responsible for catalysis remain intact during formation of the intermediate from 0–4 M urea. An increase in absorbance [expressed as % Zn(II) loss] does occur when PAR is added to TOP in the presence of 4–7 M urea, supporting the hypothesis that the second unfolding event leads to complete disruption of the enzyme structure and loss of the active site. Further evidence that disruption of domain I accounts for the intermediate comes from the intrinsic fluorescence data. Figure 2(A) shows that some quenching is attenuated from 0–4 M urea, causing an increase in the total emission intensity with little to no shift (approx. 0.5 nm) shift in the λemission maximum (results not shown). This intensity increase accounts for only about 15% of the total emission change at 377 nm (Figure 3A). Domain I contains only two of the seven tryptophan residues in TOP, one of which (Trp613) is about 20% solvent-exposed within the large crevice between the domains, and the other (Trp334) is in a very polar environment in the folded enzyme, within 5 Å of Arg236, Arg237, Glu240, His332, Asp335 and Thr588. Thus changes in the structure of domain I are likely to contribute a small portion to the total fluorescence intensity, and result in minimal changes in the λemission max upon unfolding. Disruption of domain I is also supported by the decrease in anisotropy between 0–4 M urea and by the differential effect on activity of the MCA and mca-Bk substrates. Whether the transition leading to the intermediate is the result of complete unfolding or a more subtle conformational change of domain I cannot be definitively distinguished by our data, but either scenario would likely affect the flexible loop structures that connect the two domains at the base of the substrate-binding cleft.
In summary, the present work demonstrates that TOP undergoes a two-step unfolding process, as characterized by fluorescence studies and DSC. Based on the intrinsic tryptophan emission, anisotropy and quenching studies, the second step in the unfolding process that occurs at high urea concentrations leads to a complete unfolding of the enzyme structure. This is supported by evidence of the loss of Zn(II) from the active site. Importantly, a clear conformational change in TOP is apparent at lower urea concentrations (0–4 M) that can be connected to differential changes in kcat for two unrelated peptide substrates. This study provides the first evidence for a conformational change in TOP that leads to discrimination between different substrates and helps explain the wide array of peptide sequences recognized by this enzyme.
Acknowledgments
We thank Ms. Amanda Pabon for technical assistance. This study was supported by a Howard Hughes Medical Institute Undergraduate Science Education Program Grant and a National Science Foundation (NSF) Research Experiences for Undergraduate Award to Wellesley College, National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke grants NS 39892 and ADD S10-RR01935 (M.J.G.), and a Camille and Henry Dreyfus Scholar/Fellow Award (A.J.W.).
References
- 1.Shrimpton C. N., Smith A. I., Lew R. A. Soluble metalloendopeptidases and neuroendocrine signaling. Endocrinol. Rev. 2002;23:647–664. doi: 10.1210/er.2001-0032. [DOI] [PubMed] [Google Scholar]
- 2.Chu T. G., Orlowski M. Soluble metalloendopeptidase from rat brain: action on enkephalin-containing peptides and other bioactive peptides. Endocrinology (Baltimore) 1985;116:1418–1425. doi: 10.1210/endo-116-4-1418. [DOI] [PubMed] [Google Scholar]
- 3.Ray K., Hines C. S., Coll-Rodriguez J., Rodgers D. W. Crystal structure of human thimet oligopeptidase provides insight into substrate recognition, regulation, and localization. J. Biol. Chem. 2004;279:20480–20489. doi: 10.1074/jbc.M400795200. [DOI] [PubMed] [Google Scholar]
- 4.Ray K., Hines C. S., Rodgers D. W. Mapping sequence differences between thimet oligopeptidase and neurolysin implicates key residues in substrate recognition. Protein Sci. 2002;11:2237–2246. doi: 10.1110/ps.0216302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigman J. A., Edwards S. R., Pabon A., Glucksman M. J., Wolfson A. W. pH dependence studies provide insight into the structure and mechanism of thimet oligopeptidase (EC 3.4.24.15) FEBS Lett. 2003;545:224–228. doi: 10.1016/s0014-5793(03)00548-9. [DOI] [PubMed] [Google Scholar]
- 6.Oliveira V., Araujo M. C., Rioli V., de Camargo A. C., Tersariol I. L., Juliano M. A., Juliano L., Ferro E. S. A structure-based site-directed mutagenesis study on the neurolysin (EC 3.4.24.16) and thimet oligopeptidase (EC 3.4.24.15) catalysis. FEBS Lett. 2003;541:89–92. doi: 10.1016/s0014-5793(03)00310-7. [DOI] [PubMed] [Google Scholar]
- 7.Dando P. M., Brown M. A., Barrett A. J. Human thimet oligopeptidase. Biochem. J. 1993;294:451–457. doi: 10.1042/bj2940451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horsthemke B., Bauer K. Characterization of a nonchymotrypsin-like endopeptidase from anterior pituitary that hydrolyzes luteining hormone-releasing hormone at the tyrosyl-glycine and histidyl-tryptophan bonds. Biochemistry. 1980;19:2867–2873. doi: 10.1021/bi00554a008. [DOI] [PubMed] [Google Scholar]
- 9.Orlowski M., Reznik S., Ayala J., Pierotti A. R. Endopeptidase 24.15 from rat testes. Isolation of the enzyme and its specificity toward synthetic and natural peptides, including enkephalin-containing peptides. Biochem. J. 1989;261:951–958. doi: 10.1042/bj2610951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahms P., Mentlein R. Purification of the main somatostatin-degrading proteases from rat and pig brains, their action on other neuropeptides, and their identification as endopeptidases 24.15 and 24.16. Eur. J. Biochem. 1992;208:145–154. doi: 10.1111/j.1432-1033.1992.tb17168.x. [DOI] [PubMed] [Google Scholar]
- 11.Lew R. A., Hey N. J., Tetaz T. J., Glucksman M. J., Roberts J. L., Smith A. I. Substrate specificity differences between recombinant rat testes endopeptidase EC 3.4.24.15 and the native brain enzyme. Biochem. Biophys. Res. Commun. 1995;209:788–795. doi: 10.1006/bbrc.1995.1569. [DOI] [PubMed] [Google Scholar]
- 12.Vincent B., Jiracek J., Noble F., Loog M., Roques B., Dive V., Vincent J.-P., Checler F. Contribution of endopeptidase 3.4.24.15 to central neurotensin inactivation. Eur. J. Pharmacol. 1997;334:49–53. doi: 10.1016/s0014-2999(97)01209-0. [DOI] [PubMed] [Google Scholar]
- 13.Orlowski M., Michaud C., Chu T. G. A soluble metalloendopeptidase from rat brain. Purification of the enzyme and determination of specificity with synthetic and natural peptides. Eur. J. Biochem. 1983;135:81–88. doi: 10.1111/j.1432-1033.1983.tb07620.x. [DOI] [PubMed] [Google Scholar]
- 14.Brown C. K., Madauss K., Lian W., Beck M. R., Tolbert W. D., Rodgers D. W. Structure of neurolysin reveals a deep channel that limits substrate access. Proc. Natl. Acad. Sci. U.S.A. 2001;98:3127–3132. doi: 10.1073/pnas.051633198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shrimpton C. N., Glucksman M. J., Lew R. A., Tullai J. W., Margulies E. H., Roberts J. L., Smith A. I. Thiol activation of endopeptidase EC 3.4.24.15. A novel mechanism for the regulation of catalytic activity. J. Biol. Chem. 1997;272:17395–17399. doi: 10.1074/jbc.272.28.17395. [DOI] [PubMed] [Google Scholar]
- 16.Gill S. C., Von Hippel P. H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 17.Wolfson A. J., Shrimpton C. N., Lew R. A., Smith A. I. Differential activation of endopeptidase EC 3.4.24.15 toward natural and synthetic substrates by metal ions. Biochem. Biophys. Res. Commun. 1996;229:341–348. doi: 10.1006/bbrc.1996.1803. [DOI] [PubMed] [Google Scholar]
- 18.Pace C., Scholtz J. M. Measuring the conformational stability of a protein. In: Creighton C. E., editor. Protein Structure: A Practical Approach. Oxford University Press; 1997. pp. 299–321. [Google Scholar]
- 19.Lakowicz J. R. New York: Kluwer Academic/Plenum Publishers; 1999. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 20.Szabo A. G. Fluorescence principles and measurement. In: Gore M. G., editor. Spectrophotometry and Spectrofluorimetry: A Practical Approach. Oxford University Press; 2000. pp. 62–66. [Google Scholar]
- 21.Daiber A., Frein D., Namgaladze D., Ullrich V. Oxidation and nitrosation in the nitrogen monoxide/superoxide system. J. Biol. Chem. 2002;277:11882–11888. doi: 10.1074/jbc.M111988200. [DOI] [PubMed] [Google Scholar]
- 22.Kim S. I., Grum-Tokars V., Swanson T. A., Cotter E. J., Cahill P. A., Roberts J. L., Cummins P. M., Glucksman M. J. Novel roles of neuropeptide processing enzymes: EC3.4.24.15 in the neurome. J. Neurosci. Res. 2003;74:456–467. doi: 10.1002/jnr.10779. [DOI] [PubMed] [Google Scholar]
- 23.York I. A., Mo A. X., Lemerise K., Zeng W., Shen Y., Abraham C. R., Saric T., Goldberg A. L., Rock K. L. The cytosolic endopeptidase, thimet oligopeptidase, destroys antigenic peptides and limits the extent of MHC class I antigen presentation. Immunity. 2003;18:429–440. doi: 10.1016/s1074-7613(03)00058-x. [DOI] [PubMed] [Google Scholar]
- 24.Silva C. L., Portaro F. C., Bonato V. L., de Camargo A. C., Ferro E. S. Thimet oligopeptidase (EC 3.4.24.15), a novel protein on the route of MHC class I antigen presentation. Biochem. Biophys. Res. Commun. 1999;255:591–595. doi: 10.1006/bbrc.1999.0250. [DOI] [PubMed] [Google Scholar]
- 25.Kim S. I., Pabon A., Swanson T. A., Glucksman M. J. Regulation of cell-surface major histocompatibility complex class I expression by the endopeptidase EC 3.4.24.15 (thimet oligopeptidase) Biochem. J. 2003;375:111–120. doi: 10.1042/BJ20030490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamin R., Malgeri E. G., Sloane J. A., McGraw W. T., Abraham C. R. Metalloendopeptidase EC 3.4.24.15 is necessary for Alzheimer's amyloid-β peptide degradation. J. Biol. Chem. 1999;274:18777–18784. doi: 10.1074/jbc.274.26.18777. [DOI] [PubMed] [Google Scholar]
- 27.Koike H., Seki H., Kouchi Z., Ito M., Kinouchi T., Tomioka S., Sorimachi H., Saido T. C., Maruyama K., Suzuki K., Ishiura S. Thimet oligopeptidase cleaves the full-length Alzheimer amyloid precursor protein at a β-secretase cleavage site in COS cells. J. Biochem. (Tokyo) 1999;126:235–242. doi: 10.1093/oxfordjournals.jbchem.a022428. [DOI] [PubMed] [Google Scholar]
- 28.Oliveira V., Gatti R., Rioli V., Ferro E. S., Spisni A., Camargo A. C., Juliano M. A., Juliano L. Temperature and salts effects on the peptidase activities of the recombinant metallooligopeptidases neurolysin and thimet oligopeptidase. Eur. J. Biochem. 2002;269:4326–4334. doi: 10.1046/j.1432-1033.2002.03129.x. [DOI] [PubMed] [Google Scholar]
- 29.Sigman J. A., Sharky M. L., Walsh S. T., Pabon A., Glucksman M. J., Wolfson A. J. Involvement of surface cysteines in activity and multimer formation of thimet oligopeptidase. Protein Eng. 2003;16:623–628. doi: 10.1093/protein/gzg073. [DOI] [PubMed] [Google Scholar]
- 30.London R. E., Stewart J. E., Cann J. R., Matwiyoff N. A. 13C and 1H nuclear magnetic resonance studies of bradykinin and selected peptide fragments. Biochemistry. 1978;17:2270–2277. doi: 10.1021/bi00605a003. [DOI] [PubMed] [Google Scholar]
- 31.Denys L., Boythner-By A. A., Fisher G. H., Ryan J. W. Conformational diversity of bradykinin in aqueous solution. Biochemistry. 1982;21:6531–6536. doi: 10.1021/bi00268a032. [DOI] [PubMed] [Google Scholar]
- 32.Gaggelli E., D'Amelio N., Maccotta A., Valensin G. Calcium-binding properties and molecular organization of bradykinin A solution 1H-NMR study. Eur. J. Biochem. 1999;262:268–276. doi: 10.1046/j.1432-1327.1999.00420.x. [DOI] [PubMed] [Google Scholar]
- 33.Desai P., Coutinho E., Srivastava S. Conformational diversity of T-kinin in DMSO, water, and HFA. Eur. J. Med. Chem. 2002;37:135–146. doi: 10.1016/s0223-5234(01)01323-x. [DOI] [PubMed] [Google Scholar]