

Abstract
In the present work, an endopin-like elastase inhibitor was purified for the first time from bovine muscle. A three-step chromatography procedure was developed including successively SP-Sepharose, Q-Sepharose and EMD-DEAE 650. This procedure provides about 300 μg of highly pure inhibitor from 500 g of bovine diaphragm muscle. The N-terminal sequence of the muscle elastase inhibitor, together with the sequence of a trypsin-generated peptide, showed 100% similarity with the cDNA deduced sequence of chromaffin cell endopin 1. Hence, the muscle inhibitor was designated muscle endopin 1 (mEndopin 1). mEndopin 1 had a molecular mass of 70 kDa, as assessed by both gel filtration and SDS/PAGE. According to the association rates determined, mEndopin 1 is a potent inhibitor of elastase (kass=2.41×107 M−1·s−1) and trypsin (kass=3.92×106 M−1·s−1), whereas plasmin (kass=1.78×103 M−1·s−1) and chymotrypsin (kass=1.0×102 M−1·s−1) were only moderately inhibited. By contrast, no inhibition was detected against several other selected serine proteinases, as well as against cysteine proteinases of the papain family. The cellular location of mEndopin in muscle tissue and its tissue distribution were investigated using a highly specific rabbit antiserum. The results obtained demonstrate an intracellular location and a wide distribution in bovine tissues.
Keywords: bovine muscle, elastase inhibitor, muscle endopin 1, serine proteinase, serpin
Abbreviations: CBZ, benzyloxycarbonyl; HLE, human leucocyte neutrophil elastase; MALDI–TOF, matrix-assisted laser-desorption ionization–time-of-flight; MCE, 2-mercaptoethanol; mEndopin 1, muscle endopin 1; MeO, methoxy; NHMec, aminomethyl coumarylamide; RCL, reactive centre loop; Suc, succinyl
INTRODUCTION
The serine proteinase inhibitors (serpins) are a large family of functionally diverse proteins (approx. 700 to date) which are essential regulators of a wide variety of biological processes and are associated with numerous familial disorders [1]. Most serpins are inhibitory, although their proteinase targets are not restricted to the serine proteinases. In particular, several serpins have been identified that inhibit papain-like cysteine proteinases. In addition to the inhibitory serpins, some members have evolved to perform non-inhibitory roles. The inhibitory function of the serpins involves a marked conformational transition, but this inherent molecular flexibility also renders the serpins susceptible to form abnormal intermolecular linkages. The effects of such protein aggregation are cumulative, with a progressive loss of cellular function that results in diseases as diverse as cirrhosis or emphysema. The insidious nature of protein aggregation and accumulation affecting individuals in middle- or old-age are now known to be a feature of many of the neurodegenerative diseases, notably of Alzheimer's and Parkinson's diseases and spongiform encephalopathies. These particular features of the serpins explain why these protein inhibitors are so extensively studied in human and mammals in general, but not exclusively (reviewed in [1]).
Despite the fact that most of the body's proteins are concentrated in muscles, this tissue has been poorly investigated in this context. On the other hand, one important characteristic of muscle tissue is its high functional plasticity and adaptability [2]. Hence, the absolute requirement for a continuous adaptation of fibres in response to various types of stress (biological, physiological, nutritional, physical injury, etc.) under normal and pathological conditions contribute to maintain a relatively intense and permanent metabolic activity in this tissue throughout life. Such changes are obviously ensured by a series of strictly regulated proteolytic enzymes, including, for example, the well-known calpains, proteasomes, cathepsins and collagenases. Undoubtedly the least investigated group of enzymes in muscle tissue, namely the serine proteinases, is also likely to contribute to muscle differentiation, remodelling and regeneration processes [3–7]. The few studies on muscle serpins were indeed carried out either because of their possible implication in different types of pathologies or because they allow a better understanding of the serpins function in the complex biological processes ensuring muscle differentiation and homoeostasis. Several serpins have been thus described in muscle, including protease nexin 1, a thrombin inhibitor [8], plasminogen activator inhibitor [9], and kallistatin, a kallikrein-binding protein [10]. Muscle cells also express α-1-antichymotrypsin and β-amyloid precursor protein [6]. To fill in this gap, investigations with the goal of obtaining more knowledge of muscle serine proteinase inhibitors, including serpins, are presently being carried out in our laboratory.
The present report describes the purification and characterization of a 70 kDa serpin which strongly inhibits elastase and trypsin with Kd values of 2.4×107 and 3.9×106 M−1·s−1 respectively. Plasmin, another basic-residue-cleaving proteinase, and chymotrypsin were moderately inhibited. By contrast, no inhibitory activity was observed against cysteine proteinases, including papain and cathepsins B and L, as well as against the set of other serine proteinases tested. This widely distributed elastase inhibitor was essentially intracellular. Partial sequencing and peptide map analysis by MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight) MS clearly indicate that this inhibitor is the same protein as endopin 1, a chromaffin cell serpin which has been identified at the cDNA level and further characterized using Escherichia coli-expressed recombinant protein [11]. Hence, the present elastase inhibitor will be subsequently designated muscle endopin 1 (mEndopin 1). It must be further emphasized that endopin 1 has never been purified to date from any mammalian tissue, and all data available were obtained with the non-mature recombinant protein expressed in E. coli.
EXPERIMENTAL
Materials
The trypsin/elastase inhibitor (mEndopin 1) was purified from bovine diaphragm muscle, obtained from the local slaughterhouse of the INRA Research Centre. Thrombin from bovine plasma (EC 3.4.21.5) was from Roche diagnostics (Meylan, France). Human leucocyte elastase (EC 3.4.21.37) and cathepsin G (EC 3.4.21.20) were from Calbiochem (Meudon, France). Bovine pancreatic trypsin (EC 3.4.21.4), chymotrypsin (EC 3.4.21.1) and plasmin (EC 3.4.21.7) were from bovine plasma, urokinase from human kidney cells (EC 3.4.21.73), porcine pancreatic kallikrein (EC 3.4.21.35), tissue plasminogen activator, N-CBZ-Phe-Arg-NHMec, Suc-Ala-Ala-Ala-NHMec, CBZ-Gly-Gly-Arg-NHMec, Suc-Ala-Phe-Lys-NHMec, MeO-Suc-Ala-Ala-Pro-Val-NHMec (where: CBZ, benzyloxycarbonyl; NHMec, aminomethyl coumarylamide; Suc, succinyl; MeO, methoxy) and 4-nitrophenyl-guanidinobenzoate were from Sigma Chemical Co. (St Quentin-Fallavier, France). SP-Sepharose, Q-Sepharose and Superose 12 HR 10/30 were purchased from Amersham Pharmacia Biotech (Orsay, France). Fractogel EMD-DEAE-650 was from Merck (Nogent-sur-Marne, France). Goat anti-rabbit IgG conjugated with either alkaline phosphatase or FITC were from Jackson Immunoresearch Laboratories (Baltimore, MD, U.S.A.).
Purification of mEndopin 1
The purification procedure included three to four chromatography steps and starts from a crude extract prepared as described previously [12] from bovine diaphragm muscle obtained within 1 h after killing. Briefly, a 40–70% (NH4)2SO4 precipitate was dissolved in 50 ml of 50 mM sodium acetate buffer, pH 4.5 (buffer A), dialysed overnight, clarified by centrifugation at 20000 g for 15 min and applied to a SP-Sepharose (5 cm×10 cm) column equilibrated with buffer A. The bound proteins were eluted at 3 ml/min with a 0–0.2 M linear NaCl gradient and fractions of 4.5 ml were collected. Fractions of the S-1 peak (see Figure 1a) were pooled, dialysed against 30 mM Tris/HCl buffer, pH 8.0 (buffer B), and loaded on to a Q-Sepharose column (2.5 cm×10 cm) which had been previously equilibrated with buffer B. The bound proteins were eluted at 3 ml/min with a linear 0–0.25 M NaCl gradient and fractions of 4.5 ml were collected. The pooled trypsin-inhibiting peak was dialysed against buffer B and then run on a Fractogel EMD-DEAE-650 column (1 cm×8 cm) equilibrated with the same buffer. Proteins were eluted at 1 ml/min with a linear 0–0.25 M NaCl gradient and fractions of 1.5 ml were collected. After dialysis of the eluted active fractions against buffer B containing 0.3 M NaCl, the sample was loaded on to a Superose 12 HR 10/30 column, and proteins were eluted at 0.5 ml/min and collected in 1 ml fractions. Active fractions were pooled, desalted by dialysis against buffer B and stored at 0–4 °C until use.
Figure 1. Major chromatography steps used for the purification of mEndopin 1.

(a) Elution profile of the muscle crude extract at pH 4.5 from a SP-Sepharose column (5 cm×10 cm). Only the S-1 trypsin inhibitory peak will be subsequently considered. (b) Elution profile of the S-1 from a Q-Sepharose column (2.5 cm×10 cm) at pH 8.0. (c) Gel filtration profile of the Fractogel EMD-DEAE-650 pooled active fractions run on a Superose 12 HR 10/10 column at pH 8.0. The protein peak was eluted just before BSA (arrow). Calibration of the gel filtration column was performed using ferritin (440 kDa), catalase (232 kDa), aldolase (150 kDa), BSA (67 kDa), β-lactoglobulin (35 kDa) and α-lactalbumin (14 kDa). In all profiles, the continuous line is the absorbance at 280 nm, the broken line corresponds to the NaCl gradient and open circles indicate the trypsin inhibitory activity.
Monitoring of the trypsin inhibitory activity in collected fractions
Aliquots of each fraction (30 μl) were mixed with 750 μl of a trypsin solution (40 μg/ml) in 50 mM Tris/HCl buffer, pH 8.0, containing 10 mM CaCl2, and then incubated at 37 °C for 20 min before addition of 250 μl of a 40 μM substrate stock solution (N-CBZ-Phe-Arg-NHMec). After 20 min, the reaction was stopped by adding 3 ml of a stopping solution containing 100 mM chloroacetate, 70 mM acetic acid and 30 mM sodium acetate, pH 4.3. Fluorescence was then measured using a PerkinElmer LS 50B spectrofluorimeter (λexcitation at 360 nm−λemission 440 nm). Unless otherwise indicated, all activity measurements were performed at 37 °C.
PAGE and Western blotting
SDS/PAGE was performed as described previously [13] under reducing conditions on a 12.5% slab gel. Molecular masses were estimated by using the Pharmacia low Mr calibration kit. Proteins were revealed using either Coomassie Brilliant blue R250 or silver staining. Electrophoresis in non-denaturing conditions was carried out as above on 10% slab gel, but in non-reducing conditions and in the absence of SDS. Immunoblot analysis were carried out as described previously [14].
pH and heat stability of the mEndopin 1
pH stability of the purified inhibitor was tested in the pH range 2 to 12. The trypsin inhibitor (approx. 10 nM) was incubated for 1 h at room temperature in the following (buffer) solutions: 0.2 M KCl/HCl (pH 2), 0.1 M formic acid/NaOH (pH 4), 0.1 M Bis/Tris/HCl (pH 6), 0.1 M Tris/HCl (pH 8), 0.1 M sodium bicarbonate/NaOH (pH 10) and 0.15 M KCl/NaOH (pH 12). The residual trypsin inhibiting activity was then measured as described above. Heat stability of the trypsin inhibitor was determined by heating the purified inhibitor (approx. 10 nM) in Tris/HCl buffer, pH 8.0, for 15 min at different temperatures ranging from 40° to 100 °C and residual activity measured as described above.
Enzyme titration
Titration of bovine pancreatic trypsin and bovine plasmin were carried out as in [15] using 4-nitrophenyl p-guanidinobenzoate. HLE (human leucocyte neutrophil elastase) was tritrated as described in [16] using acetyl-Ala-Ala-AzaAla-p-nitrophenylester. Chymotrypsin and cathepsin G were titrated as described in [17] using N-trans-cinnamoyl-imidazole. All cysteine proteinases were titrated as described in [18] using E-64 [trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane], a common irreversible inhibitor. Concentration of the purified muscle inhibitor was measured by titration with previously titrated trypsin.
Stoichiometry of the enzyme–inibitor interaction
The defined concentrations of enzyme, i.e. trypsin or HLE, were incubated with increasing amounts of mEndopin 1, and the residual activity was measured as described above.
Measurement of the association rate constants
Second order association rate constant (kass) of the muscle endopin 1 with trypsin and elastase were determined as described in [19]. The purified inhibitor (5 nM) was pre-incubated with equimolar amounts (5 nM) of enzyme in 50 mM Tris/HCl buffer, pH 8.0, containing 10 mM CaCl2 for given periods of time. 100 μl of 100 μM substrate stock solutions (N-CBZ-Phe-Arg-NHMec and MeO-Suc-Ala-Pro-Val-NHMec for trypsin and elastase respectively) were then added and the mixture incubated for 20 min before measuring residual activity. The association rate with plasmin and chymotrypsin was determined under pseudo-first-order conditions by pre-incubating the enzymes (15 nM) with a 10-fold molar excess of inhibitor in 50 mM Tris/HCl buffer containing 100 mM NaCl, pH 7.5, in a total volume of 200 μl. After different pre-incubation times, the substrates (Suc-Ala-Phe-Lys-NHMec and Suc-Ala-Ala-Pro-Phe-NHMec for plasmin and chymotrypsin respectively) were added at a final concentration of 100 μM and incubated for 20 min. The reaction was stopped by addition of the stopping buffer and the fluorescence measured as indicated above.
Peptide preparation and sequence analysis
After SDS/PAGE and Coomassie Blue staining, the band of gel corresponding to mEndopin 1 was cut out and partially dehydrated in a SpeedVac. The gel sample was then subjected to hydrolysis for 18 h at 30 °C by addition of 0.1 μg of pig trypsin (Sigma) in 200 μl of Tris/HCl buffer, pH 8.6, containing 0.01% Tween 20. Peptides were extracted, fractionated by HPLC on a DEAE-C18 column (1 mm in diameter) and eluted with a 2–70% linear acetonitrile gradient in 0.1% TFA (trifluoroacetic acid). These peptides were then subjected to N-terminal sequencing. Sequence analysis by the Edman method was performed using an Applied Biosystems 477A pulsed liquid sequencer (Perseptive Biosystems, Framingham, MA, U.S.A.). Phenylthiohydantoin derivatives were identified with an on-line model 120A analyser.
N-terminal sequencing of mEndopin 1
After SDS/PAGE, proteins were transferred on to a PVDF membrane by electroblotting and revealed by Coomassie Blue staining. The bands corresponding to the inhibitor and its dimer were cut out and subjected to Edman sequence analysis as described above.
MS peptide map of mEndopin 1
Peptide map of the trypsin digest was performed as previously described [14], using a MALDI–TOF mass spectrometer (Voyager DE-Pro, Applied BioSystems, Courtaboeuf, France) in the reflectron mode for peptide-mass fingerprinting.
Preparation of the antiserums and Western blot analysis
Preparation of the antiserums and Western blot analysis were carried out as previously described [20]. The antiserum directed against mEndopin 1 was used at a dilution of 1/500. The rabbit anti-trypsin polyclonal antibody was similarly prepared and used at a dilution of 1/500. The anti-HLE polyclonal antibody was purchased from BioTrends (Köln, Germany) and used at a dilution of 1/150.
Immunofluorescence localization of mEndopin 1
Muscle strips (3 mm×10 mm) isolated from fresh cuts of bovine longissimus muscle, perpendicular to the fibre axis were immersed in 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.4, for 45 min at room temperature. Samples were then bathed in 30% sucrose in PBS buffer up to equilibrium. Blocks were mounted in a Reichert Frigocut 2800 (Leika, Heidelberg, Germany) and frozen in situ at −20 °C. Thick transverse sections of about 10 μm were cut and treated with non-immune goat serum (diluted to 1/20 in PBS) for 15 min followed by three 5 min successive washes in PBS. The sections were then incubated with the anti-mEndopin 1 polyclonal antibody (diluted to 1/50 in PBS) for 1 h at room temperature. After three 5 min washes in PBS, the sections were treated with the goat FITC-labelled anti-rabbit IgG (diluted to 1/500 in PBS) for 1 h at room temperature, rinsed for 30 min in PBS and then mounted in glycerol on glass plates. Sections were then examined using an Axioplan 2E light microscope (Zeiss, Lyon, France).
Assessment of tissue distribution by double immunodiffusion
Double immunodiffusion was performed as described in [21]. Crude tissue extracts were obtained by homogenization of 1 g of fresh tissue, previously sliced into small pieces with a sterile blade, in 2 ml of cold 50 mM Tris/HCl buffer, pH 7.6, containing 150 mM KCl and 4 mM EDTA, and further clarified by centrifugation at 20000 g for 20 min. In addition to plasma, different bovine tissues were tested, including spleen, liver, kidney, thymus and diaphragm muscle. Immunodiffusion was performed using a 2.5-mm thick agar plate containing 1.2% Noble Agar in 0.05 M veronal buffer, pH 7.3 (Buffer V). Circular wells (3 mm in diameter) were punched out of the agar gel and filled with 20 μl of each crude tissue extract. The central well was filled in with 20 μl of the antiserum diluted to 1/8 in Buffer V. After diffusion for 24 h at 37 °C in a humidified chamber, the plate was examined using dark-field oblique illumination. Picture was taken using a video printer (Sony UP 1200EPM, Manganelly, Clermont Ferrand, France) coupled to a magnifying video camera (Sony XCOO3P).
Protein determination
Protein concentrations were measured as described in [22] with rabbit Ig as the standard.
RESULTS
Purification of the mEndopin 1
When applied to an ESP-Sepharose column, the ammonium sulphate precipitate was separated into three trypsin-inhibiting peaks designated S-1, S-2 and S-3 (Figure 1a). The S-1 fraction was pooled, loaded on to a Q-Sepharose column and the trypsin-inhibiting activity eluted in the gradient at approx. 0.1 M NaCl (Figure 1b). After overnight dialysis against 30 mM Tris/HCl buffer, pH 8.0, the active fractions were applied on to a Fractogel EMD-DEAE 650 column equilibrated with the same buffer. Elution with a 0–0.25 M NaCl smooth linear gradient gave one active peak (results not shown). Generally, the preparation was pure enough at this stage and the last step was then omitted. Otherwise, the purification was ended by chromatography of the active fraction on a Superose 12 HR 10/30 column (Figure 1c) from which the serpin was eluted as a single peak, coming out just before BSA (Mr 67000), with a molecular mass of 70 kDa. When analysed by SDS/PAGE, one or two bands, with Mrs of 140000 and 70000 respectively, were detected depending on the amount of protein loaded into the wells (Figure 2a, lanes 1 and 2). In contrast and irrespective of the amount of proteins loaded, only one band was obtained when run in non-denaturing conditions (Figure 2b, lanes 3 to 5).
Figure 2. SDS/PAGE and Western blot analysis of the purified preparation (40 μg/ml).

(a) The inhibitor was run in denaturing conditions on a 12.5% polyacrylamide slab gel and revealed by silver staining. The volume of the preparation loaded in the wells were 1 μl (lane 1) and 3 μl (lane 2). (b) Inhibitor run in non-denaturing conditions and revealed by silver staining. The volume of the preparation loaded in the wells were 1 μl (lane 3), 3 μl (lane 4) and 5 μl (lane 5). (c) Analysis of the dimer formation by immunoblotting using the mEndopin 1 antibody. Sample volume loaded into the wells was from left to right: 1, 3, 5, 7, 9, 11, 15, 17 and 20 μl.
Characterization of the 140 and 70 kDa bands
Unexpectedly and even after gel filtration, SDS/PAGE analysis of the purified mEndopin 1 revealed two bands with Mr of 70000 and 140000, a finding in contrast with those obtained by gel filtration and electrophoresis under non-denaturing conditions, which suggested the presence of only one protein in the purified preparation. Upon gel filtration, the whole inhibitory activity was indeed eluted in a single peak just before BSA (Mr 67000) and not two, as would have been expected if the 140 kDa band corresponded to a contaminant protein. Interestingly, in denaturing conditions the upper band was not detectable when 1 μl of the serpin preparation was loaded on to the gel (Figure 2c, left-hand lane), whereas its concentration increased as the amount of protein loaded was raised from 3 μl (Figure 2c, second lane from the left-hand side) to 20 μl (Figure 2c, right-hand lane). It is worthy to note that the dimer/monomer ratio seemed to be constant as the amount of protein loaded on to the gel increased. The upper band starts to be detectable at a total protein concentration of about 100 nM (Figure 2c, second lane from the left-hand side) and the proportion remained quite constant for all other concentrations. As assessed by densitometry the amount of dimer corresponds to approx. one-third of the total protein in the denatured sample (28.7±1.5%, as estimated from five different preparations).
When analysed by Western blotting, both bands were labelled by the specific antibody raised against the muscle serpin, suggesting large structural similarities between them (Figure 2c).
Primary sequence analysis using the Edman procedure showed a common N-terminal amino-acid sequence (indicated in bold) for both bands: bovine 140 kDa band, L1PENVVVKDQHRRV14; bovine mEndopin 1, L1PENVVVKDQHRRV14; bovine chromaffin endopin 1, L1PENVVVKDQHRRVDGHTLA20.
Screening of the protein databases with this sequence suggested that this serpin corresponds very likely to the bovine chromaffin cell endopin 1 (Swiss-Prot and TrEMBL ID, Q9TTE1). Similarly, the use of an internal peptide sequence isolated from the trypsin digest of mEndopin 1 extracted again only one protein from the databanks, i.e. the chromaffin endopin 1: bovine mEndopin 1 peptide, — SNYELNDILSQLGIR —; bovine chromaffin endopin 1, –S313NYELNDILSQLGIR327—.
Analysis of both bands by MALDI–TOF MS further gave a similar trypsin peptide map (results not shown) confirming that the 140 kDa band is a dimer of the muscle serpin [23–25].
Identity of mEndopin 1
Similarity searches of databanks with the N-terminal sequence were carried out using BlastP2 (Bork Group, EMBL, Heidelberg). This sequence showed 100% similarity with bovine endopin 1 (ID Q9TTE1), a serpin first identified at the cDNA level in adrenal medulla neuroendocrine chromaffin cells [11], which strongly inhibits trypsin, but not elastase or chymotrypsin. Several other serine protease inhibitors (plasma bovine trypsin inhibitor or serpin TI, ID Q9TS73 [26]; goat contrapsin, ID S54330 [27] and sheep plasma alpha-1-proteinase inhibitor, ID A60812 [28]), for which partial N-terminal sequences are available in the databases, exhibited lower similarity scores. A similarity search performed with the sequence of a trypsin-generated peptide containing 15 amino acids (S1NYELNDILSQLGIR15) extracted only one protein from the databases showing a full identity of this sequence with an internal peptide (residues 313–327 of the precursor protein) of chromaffin cell endopin 1. To confirm the identity of the present muscle serpin, the protein was subjected to trypsin digestion and the mixture then analysed by MALDI–TOF MS. As shown in Figure 3(a), the peptides generated matched extensively with the mature protein sequence. Of these, four matched with the C-terminal region of the protein, including a large part of the RCL (reactive centre loop) and the suspected scissile bond (between Arg350 and Thr351), indicated in Figure 3(b) by two opposite arrow heads within the underlined sequence ‘SMERTI’ [11].
Figure 3. mEndopin 1 identity with chromaffin cell endopin.

(a) mEndopin 1 peptide map of the trypsin digest as assessed by MALDI–TOF MS. The protein sequence taken in reference corresponds to the mature chromaffin cell endopin 1 without the signal peptide. (b) C-terminal sequence of chromaffin cell endopin 1. Alignment of peptides matching with the sequence. (Chromaffin cells endopin 1 sequence was from Swiss-Prot database, ID Q9TTE1).
pH and heat stability of mEndopin 1
The muscle endopin 1 is highly stable at pH values ranging 4.0 to 12. In this pH range, activity was not affected after 1 h incubation and decreased by less than 5% after 24 h (Figure 4a). At more acidic pH values (<4.0), the activity was not detectable even after 1 h.
Figure 4. pH (a) and heat (b) stability of the mEndopin 1.

(a) Residual activity (percentage of the control) of mEndopin 1 (10 nM) pre-incubated for 1 h (○) or 24 h (●) at different pH ranging from 2 to 12 (for buffer composition see the Experimental section). (b) Residual activity of mEndopin (10 nM) heated for 15 min at different temperatures ranging from 40° to 100 °C.
Upon heating for 15 min to various temperatures ranging from 40° to 100 °C, the mEndopin 1 activity was unchanged between 40° and 70 °C and decreased rapidly for higher temperature values (Figure 4b). No activity was thus detectable at 80 °C and above. These results argued for a relative heat stability of this serpin compared with others for which total inactivation occurs at temperatures of 60 °C or lower [29–31].
Stoichiometry for trypsin and elastase inhibition
To assess the stoichiometry of interaction, trypsin (20 nM) and HLE (50 nM) were titrated with mEndopin 1 in a total volume of 200 μl. As shown in Figure 5, complete trypsin inhibition was achieved for an mEndopin 1 concentration of 20 nM (mean±S.D. for 5 independent determinations, 20.0±0.5 nM), indicating a 1:1 trypsin/mEndopin 1 ratio of interaction. Total elastase inhibition was achieved for an mEndopin 1 concentration of 50 nM (mean±S.D. for 5 independent determinations, 50.2±0.8 nM), suggesting a 1:1 elastase/mEndopin 1 ratio of interaction. To ascertain these findings, we tested the ability of mEndopin 1 to form SDS-stable complexes with each of these proteases. Trypsin (20 nM) and elastase (20 nM) were pre-incubated at 37 °C with 50 nM of mEndopin 1 for 20 min in a total volume of 100 μl and the mixture then heated in the presence of SDS/MCE (2-mercaptoethanol) buffer (50 μl) for subsequent SDS/PAGE analysis. After electrotransfer on to the membrane, proteins were revealed selectively using polyclonal antibodies directed against mEndopin 1, trypsin and HLE antiserums (Figure 6). The HLE polyclonal antibody labelled a first band (Figure 6a, lane 4) corresponding to free elastase and a second one with a Mr of approx. 95000, very likely to be the elastase–mEndopin 1 complex. Two bands were also revealed upon treatment of the membrane with the anti-mEndopin 1 antibody with Mr of 70000 (free monomeric form of mEndopin) and 95000 respectively (Figure 6a, lane 2). As the 95 kDa band was labelled by both anti-elastase and anti-mEndopin 1 antibodies, this band corresponds undoubtedly to the elastase–mEndopin 1 complex. These results confirm those previously obtained by titration. Similar findings were obtained upon analysis of the trypsin–mEndopin 1 mixture, since a 93-kDa band corresponding to the complex was recognized by both anti-trypsin and anti-mEndopin 1 antibodies (Figure 6b, lanes 2 and 4). Taken together and according to the Mr of the enzyme–inhibitor complexes, these results stressed that inhibition of both HLE and trypsin by mEndopin 1 occurs through equimolar interaction.
Figure 5. Stoichiometry of interaction of mEndopin 1 with trypsin (○) and HLE (●).

Regression analysis of the linear part of the curves cross the x-axis at 20 nM and 50 nM. These concentrations correspond to the amount of mEndopin 1 needed for a total inhibition of trypsin (20 nM) and elastase (50 nM), indicative of a 1:1 enzyme/inhibitor ratio.
Figure 6. Stability under SDS/PAGE of the enzyme–inhibitor complexes obtained with elastase (a) and trypsin (b) as revealed by Western blotting using specific inhibitors directed against trypsin (Tryp-Ab), elastase (HLE-Ab) and mEndopin 1 (mEndo-Ab).

(a) Lane 1, purified mEndopin 1; lane 2, elastase incubated with mEndopin 1; lane 3, elastase control; lane 4, elastase incubated with mEndopin 1; proteins were revealed with mEndo-Ab (lanes 1 and 2) and HLE-Ab (lanes 3 and 4). (b) Lane 1, trypsin control; lane 2, trypsin incubated with mEndopin 1; lane 3, purified mEndopin 1; lane 4, trypsin incubated with mEndopin 1; proteins were revealed with Tryp-Ab (lanes 1 and 2) and mEndo-Ab (lanes 3 and 4). The 140 kDa band revealed with the mEndo-Ab antibody corresponds to the mEndopin 1 dimer. E, elastase; T, trypsin bands.
Inhibitory pattern of mEndopin 1 and association rate constant determination
The inhibitory activity of the purified inhibitor against various serine and cysteine proteinases was determined (Table 1). No inhibitory activity was detected with any cysteine proteinase tested, including papain, cathepsin B and cathepsin L. Amongst the set of serine proteinases only trypsin, plasmin, elastase and chymotrypsin were inhibited, to various extents, by mEndopin 1. By contrast, the activities of thrombin, urokinase, kallikrein, tissue plasminogen activator or cathepsin G were not affected at all. According to the experimentally determined association rate constants (kass), muscle endopin 1 is a strong inhibitor of HLE (kass=2.41×107 M−1·s−1) and trypsin (kass=3.92×106 M−1·s−1), whereas plasmin (kass=1.78×103 M−1·s−1) and chymotrypsin (kass=1.0×102 M−1·s−1) were only moderately affected.
Table 1. Association rate constants for inhibition of serine and cysteine proteinases.
N.I., no inhibition.
| Proteinase | kass (M−1·s−1) |
|---|---|
| Serine | |
| Trypsin | 3.92×106 |
| Chymotrypsin | 1.0×102 |
| Plasmin | 1.78×103 |
| Elastase | 2.41×107 |
| Cathepsin G | N.I. |
| Thrombin | N.I. |
| Kallikrein | N.I. |
| Urokinase | N.I. |
| Plasminogen activator | N.I. |
| Cysteine | |
| Papain | N.I. |
| Cathepsin B | N.I. |
| Cathepsin L | N.I. |
Immunolocalization of the inhibitor and tissue distribution
Immunolocalization of mEndopin 1 was performed on transverse sections of freshly excised adult bovine longissimus muscle using the polyclonal rabbit antiserum described above. As depicted in Figure 7(B), the muscle endopin 1 is highly concentrated between the plasma membrane and the myofibrils, whereas a lower fluorescence intensity can be seen within the myofibrils in the centre of the muscle fibres, indicating that muscle endopin 1 is exclusively intracellular with a preferential peripheral localization. No fluorescence was detected in the control sample for which the primary antibody was omitted (Figure 7A).
Figure 7. Immunolocalization of the mEndopin in bovine longissimus muscle.

Immunolocalization was revealed using a specific rabbit antiserum diluted to 1/50 revealed by a FITC-labelled goat anti-rabbit IgG (dilution 1/500). (A) Control sample incubated with the secondary antibody alone. (B) Sample treated with the primary and the secondary antibodies (magnification, ×1200).
Tissue distribution of mEndopin 1 was assessed using the double immunodiffusion technique. Different bovine tissues and fluids were tested, including plasma, spleen, liver, kidney, thymus and muscle. According to the results presented in Figure 8, a continuous precipitating line was obtained, suggesting that the antibody recognized the same protein in all tissues and fluids and that mEndopin 1 is widely distributed in the body. In addition and with respect to the intensity of the precipitate line, mEndopin 1 was more abundant in liver, spleen and kidney than in the other tissues and fluids, whereas the lowest levels were in muscle tissue.
Figure 8. Tissue distribution of mEndopin as assessed by double immunodiffusion.

The central well contains the mEndopin rabbit antiserum (dilution 1/8; 20 μl); well 1, spleen; well 2, bovine liver; well 3, bovine plasma (20 μl); well 4, bovine kidney; well 5, bovine thymus; well 6, bovine diaphragm muscle (20 μl of each crude extract was loaded into the wells).
DISCUSSION
In the present work, we developed a rapid protocol enabling the purification, to homogeneity, of a potent elastase inhibitor from bovine muscle using a three-step procedure, including chromatography with S-Sepharose, Q-Sepharose, EMD-DEAE 650 and, occasionally, Superose 12. On average, about 300 μg of purified inhibitor were obtained from 500 g of freshly excised bovine diaphragm muscle (approx. 0.6 μg/g of wet muscle). The purified inhibitor exhibited a Mr of 70000, as assessed by both gel filtration and SDS/PAGE.
When analysed by SDS/PAGE, the purified inhibitor gives two bands with Mr of 140000 and 70000 respectively. Using different experimental approaches, including Western blotting, N-terminal sequence analysis and MALDI–TOF MS peptide mapping, we confirmed that the 140 kDa form is a dimer of mEndopin 1. The fact that serpins are able to form SDS-resistant dimers is not new. Dimer formation was shown to be catalysed by heating and denaturing agents, such as acidic pH [23–25]. As observed here and according to a series of previous reports [23–25], in non-denaturing conditions (no heating or denaturing components, i.e. SDS and MCE), only one band must be expected irrespective of the amount of protein loaded into the well. By contrast, in denaturing conditions, the samples are heated in the presence of both SDS and MCE, a set of conditions necessary and sufficient to induce the formation of the dimer, heating very likely being the primary cause of this process. However, SDS also contributes to the distortion of the serpin molecule by increasing the space between β-strands where the RCL is inserted. As shown in the present work, the dimer level increases with the protein concentration in the sample, a finding already mentioned in a series of previous reports (reviewed in [25]). According to these authors, serpin polymerization might proceed through formation of a protodimer stabilized by subsequent structural rearrangement and thus resistant to the action of SDS [25–26]. This protodimer is very likely the structure observed in SDS/PAGE.
Preliminary similarity searches performed with the partial N-terminal sequence and the sequence of an internal peptide suggested that the muscle serpin is very likely a chromaffin endopin 1-like serpin. To ascertain this finding, the monomeric form was subjected to trypsin digestion and the peptides generated analysed by MALTI–TOF MS. The peptides obtained map a large part of the chromaffin endopin 1 sequence. The analysis further shows some peptides matching well with the sequence of the RCL region of mEndopin 1, where serpin specificities are encoded, and containing the ‘SMERTI’ sequence characterizing chromaffin cell endopin 1 [11]. Hence, we concluded that the muscle serpin is undoubtedly the same protein as the chromaffin cell endopin 1. To distinguish it from the chromaffin cells serpin, the elastase inhibitor purified from bovine skeletal muscle was designated mEndopin 1. It must be emphasized that this is the first time that endopin 1 has been purified from mammalian tissue, since all the data available so far concern a recombinant endopin 1 expressed in E. coli [11].
As in chromaffin cells, the cellular localization of mEndopin 1 in muscle tissue was found to be essentially intracellular. In addition, it shows a wide distribution in bovine tissue, since it was found in all tissues and fluids examined including plasma, liver, spleen, kidney and bovine thymus. As suspected from the double immunodiffusion analysis, its concentration is highly variable from one tissue to another. Preliminary quantification of mEndopin 1 by ELISA indicates a concentration of 1 mg/ml in plasma, 14 μg/g of wet tissue in liver, 2 μg/g in kidney and 1 μg/g in diaphragm muscle. Of these tissues, skeletal muscle exhibited the lowest concentration of mEndopin 1. Note, however, that this concentration varies from one muscle to another (results not shown).
In their native state, serpins are generally known to be unstable [31,32]. Most of them exhibited a melting temperature (Tm) of about 60 °C [29–33]. The melting temperature of mEndopin 1 was found to be about 70 °C. This value is 10 °C above those reported for many native serpins, suggesting that it could be in a latent form. However, before the present work, a latent form of mEndopin 1 has never been detected and this was substantiated by the ability of this inhibitor to form SDS-stable complexes with trypsin in the initial crude muscle extract (results not shown). As serpins are mostly glycoproteins [34], one possible reason for that would be the extensive glycosylation of endopin 1. Glycosylation is, indeed, known to affect the protein folding and structure and generally stabilizes the conformation of the mature glycoproteins [35–37]. Conversely, induction of glycosylation in a non-glycosylated protein might have deleterious effects on its functionality [38]. Chromaffin cell endopin 1 was shown to be extensively glycosylated, and at least five potential N-glycosylation sites were predicted from the cDNA-deduced amino-acid sequence [11]. The particular high glycosylation of mEndopin 1 affected its conformation, a finding supported by the large over-estimation of its Mr by gel filtration as compared with the value deduced from its amino acid sequence. This could explain its higher stability to heating and also its stability in a wide pH range from pH 4 to 12.
With regard to the inhibitory pattern, mEndopin 1 strongly inhibited elastase (kass=2.41×107 M−1·s−1) and trypsin (kass=3.92×106 M−1·s−1) and, although to a much lesser extent, plasmin (kass=1.78×103 M−1·s−1) and chymotrypsin (kass=1.0×102 M−1·s−1). By contrast, no inhibitory activity was detected against a series of other serine proteinases (thrombin, cathepsin G, kallikrein, urokinase and plasminogen activator) or against cysteine proteinases of the papain family (papain and cathepsins B and L). Chromaffin cell recombinant endopin 1 showed a different specificity pattern, since only trypsin and plasmin, two basic residue-cleaving proteinases, were inhibited, whereas no inhibition was detected against HLE and chymotrypsin [11].
Attempts to deduce the N-terminal sequence of the C-terminal peptide released upon formation of the enzyme–inhibitor complex and to identify the scissile bond for both trypsin and HLE were unsuccessful. Although not clearly proved, the predicted scissile bond in chromaffin endopin 1, established by analogy with the RCL sequence of trypsin inhibiting serpins, was assumed to be between Arg374 and Thr375 in the following sequence [11]:
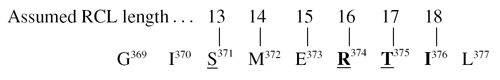 |
Examination of that sequence indicates that several bonds are susceptible to cleavage by HLE, which cleaves preferentially peptide bonds on the COOH-side of hydrophobic uncharged residues at P1 position [39]. However, as emphasized by Gettins [1], the length of the RCL is essential for the serpin to efficiently inhibit their target protease. The length of the RCL up to the scissile bond was almost always 17 residues which reduced extensively the potential P1 residue identity for inhibition of elastase and chymotrypsin. This RCL length invariance is directly related to the inhibition mechanism, which requires that the proteinase be fully translocated to the distal pole of the serpin from the initial interaction site, and that in such a location there be sufficient compression on the proteinase, resulting from it being held by the end of the RCL against the body of the serpin, for the active site to be distorted effectively enough to create the kinetic trap. Cleavage at Arg374 involves a 16-residue RCL length which, though not common, is found in some serpins [1]. Much more common is a 17-residue length to the scissile bond which could make Thr375 the P1 residue for elastase inhibition. In addition, chymotrypsin might utilize Ile376, giving an RCL length one residue longer. Such assumptions are rather speculative and would need a more accurate identification of the target scissile bond for all sensitive peptidases studied in the present work.
To conclude, endopin 1 was purified for the first time from a mammalian tissue and this was all the more original in that it was from bovine muscle, a tissue poorly investigated with regard to serpins and their target proteinases. mEndopin 1 is widely distributed in bovine tissues and fluids, and exhibited an intracellular location in muscle, as well as in liver (results not shown). It is a potent inhibitor of neutrophil elastase, but whether this enzyme is its natural and real target proteinase in muscle cells remains to be elucidated. Identification of its target enzyme(s) will be indeed be a prerequisite for a better understanding of its biological function in muscle cells.
References
- 1.Gettins P. G. W. Serpin structure, mechanism, and function. Chem. Rev. 2002;102:4751–4803. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 2.Pette D., Staron R. S. Cellular and molecular diversities of mammalian skeletal muscle fibers. Rev. Physiol. Biochem. Pharmacol. 1990;116:1–76. doi: 10.1007/3540528806_3. [DOI] [PubMed] [Google Scholar]
- 3.Festoff B. W., Reddy R. B., VanBecelaere M., Smirnova I., Chao J. Activation of serpins and their cognate proteases in muscle after crush injury. J. Cell. Physiol. 1994;159:11–18. doi: 10.1002/jcp.1041590103. [DOI] [PubMed] [Google Scholar]
- 4.Parra M., Carmeliet P., Munoz-Canoves P. Plasmin activity is required for myogenesis in vitro and skeletal muscle regeneration in vivo. Blood. 2002;99:2835–2844. doi: 10.1182/blood.v99.8.2835. [DOI] [PubMed] [Google Scholar]
- 5.Businaro R., Nori S. L., Toesca A., Evangelisti E., De Renzis G., Fumagalli L. Altered balance of proteinase inhibitors in atrophic muscle after denervation. Ital. J. Anat. Embryol. 2001;106(suppl. 1):159–165. [PubMed] [Google Scholar]
- 6.Akaaboune M., Verdiere-Sahuque M., Lachkar S., Festoff B. W., Hantai D. Serine proteinase inhibitors in human skeletal muscle: expression of beta-amyloid protein precursor and alpha-1-antichymotrypsin in vivo and during myogenesis in vitro. J. Cell. Physiol. 1995;165:503–511. doi: 10.1002/jcp.1041650308. [DOI] [PubMed] [Google Scholar]
- 7.Festoff B. W., Patterson M. R., Romstedt K. Plasminogen activator: the major secreted neutral protease of cultured skeletal muscle cells. J. Cell. Physiol. 1982;110:190–195. doi: 10.1002/jcp.1041100213. [DOI] [PubMed] [Google Scholar]
- 8.Verdiere-Sahuque M., Akaaboune M., Lachkar S., Festo B. W., Jandrot-Perrus M., Garcia L., Barlovatz-Meimon G., Hantai D. Myoblast fusion promotes the appearance of active protease nexin I on human muscle cell surfaces. Exp. Cell Res. 1996;222:70–76. doi: 10.1006/excr.1996.0009. [DOI] [PubMed] [Google Scholar]
- 9.Fibbi G., Barletta E., Dini G., Del Rosso A., Pucci M., Cerletti M., Del Rosso M. Cell invasion is affected by differential expression of the urokinase plasminogen activator/urokinase plasminogen activator receptor system in muscle satellite cells from normal and dystrophic patients. Lab Invest. 2001;81:27–39. doi: 10.1038/labinvest.3780209. [DOI] [PubMed] [Google Scholar]
- 10.Richards G. P., Chao L., Chao J. Distribution of tissue kallikreins in lower vertebrates: potential physiological roles for fish kallikreins. Comp. Biochem. Physiol., Part C: Pharmacol., Toxicol. Endocrinol. 1997;118:49–58. doi: 10.1016/s0742-8413(97)00031-5. [DOI] [PubMed] [Google Scholar]
- 11.Hwang S. R., Steineckert B., Yasothornsrikul S., Sei C. A., Tonef T., Rattan J., Hook V. Y. H. Molecular cloning of endopin 1, a novel serpin localized to neurosecretory vesicles of chromaffin cells. Inhibition of basic residue-cleaving proteases by endopin 1. J. Biol. Chem. 1999;274:34164–34173. doi: 10.1074/jbc.274.48.34164. [DOI] [PubMed] [Google Scholar]
- 12.Bige L., Ouali A., Valin C. Purification and characterization of a low molecular weight cysteine proteinase inhibitor from bovine muscle. Biochim Biophys. Acta. 1985;843:269–275. doi: 10.1016/0304-4165(85)90148-5. [DOI] [PubMed] [Google Scholar]
- 13.Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 14.Sentandreu M. A., Aubry L., Ouali A. Purification of bovine cathepsin B: proteomic characterization of the different forms and production of specific antibodies. Biochem. Cell Biol. 2003;81:317–326. doi: 10.1139/o03-060. [DOI] [PubMed] [Google Scholar]
- 15.Chase T., Shaw E. p-nitrophenyl-p′-guanidinobenzoate HCl: a new active site titrant for trypsin. Biochem. Biophys. Res. Comm. 1967;29:508–514. doi: 10.1016/0006-291x(67)90513-x. [DOI] [PubMed] [Google Scholar]
- 16.Powers J. C., Boon R. E., Carroll D. L., Gupton B. F., Kam C. M., Nishino N., Sakamoto M., Tuhy P. M. Reaction of azapeptides with human leukocyte elastase and porcine pancreatic elastase. New inhibitors and active site titrants. J. Biol. Chem. 1984;259:4288–4294. [PubMed] [Google Scholar]
- 17.Kezdy F. J., Kaiser E. T. Principles of active site titration of proteolytic enzymes. Methods Enzymol. 1969;29:3–27. doi: 10.1016/s0076-6879(76)45003-6. [DOI] [PubMed] [Google Scholar]
- 18.Barrett A. J., Kembhavi A. A., Brown M. A., Kirschke H., Knight C. G., Tamai M., Hanada K. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 1982;201:189–198. doi: 10.1042/bj2010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beatty K., Bieth J., Travis J. Kinetics of association of serine proteinases with native and oxidized α-proteinase inhibitor and α-1-antichymotrypsin. J. Biol. Chem. 1980;255:3931–3934. [PubMed] [Google Scholar]
- 20.Dutaud D., Aubry L., Henry L., Levieux D., Hendil K. B., Kueh L. N., Bureau J. P., Ouali A. Development and evaluation of a sandwich ELISA for quantification of the 20 S proteasome in human plasma. J. Immunol. Methods. 2002;260:183–193. doi: 10.1016/s0022-1759(01)00555-5. [DOI] [PubMed] [Google Scholar]
- 21.Ouchterlony O. Antigen–antibody reactions in gels. Acta Path. 1948;26:507–514. doi: 10.1111/j.1699-0463.1949.tb00751.x. [DOI] [PubMed] [Google Scholar]
- 22.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 23.Patston P. A., Hauert J., Michaud M., Schapira M. Formation and properties of C1-inhibitor polymers. FEBS Lett. 1995;368:401–404. doi: 10.1016/0014-5793(95)00694-5. [DOI] [PubMed] [Google Scholar]
- 24.Devlin G. L., Chow M. K., Howlett G. J., Bottomley S. P. Acid denaturation of α1-antitrypsin: characterization of a novel mechanism of serpin polymerization. J. Mol. Biol. 2002;324:859–870. doi: 10.1016/s0022-2836(02)01088-4. [DOI] [PubMed] [Google Scholar]
- 25.Chow M. K., Lomas D. A., Bottomley S. P. Promiscuous β-strand interactions and the conformational diseases. Curr. Med. Chem. 2004;11:491–499. doi: 10.2174/0929867043455936. [DOI] [PubMed] [Google Scholar]
- 26.Christensen S., Sottrup-Jensen L. Characterization of two serpins from bovine plasma and milk. Biochem. J. 1994;303:383–390. doi: 10.1042/bj3030383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Potempa J., Enghild J. J., Travis J. The primary elastase inhibitor (elastasin) and trypsin inhibitor (contrapsin) in the goat are serpins related to human α1-antichymotrypsin. Biochem. J. 1995;306:191–197. doi: 10.1042/bj3060191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinha U., Sinha S., Janoff A. Characterization of sheep alpha-1-proteinase inhibitor. Important differences from the human protein. Am. Rev. Respir. Dis. 1988;137:558–563. doi: 10.1164/ajrccm/137.3.558. [DOI] [PubMed] [Google Scholar]
- 29.Busby T. F., Atha D. H., Ingham K. C. Thermal denaturation of antithrombin III. Stabilization by heparin and lyotropic anions. J. Biol. Chem. 1981;256:12140–12147. [PubMed] [Google Scholar]
- 30.Carrell R. W., Lomas D., Stein P., Whisstock J. Dysfunctional variants and the structural biology of the serpins. In: Church F. C., Cunningham D. D., Ginsburg D., Hoffman M., Stone S. R., Hoffman M., editors. Chemistry and Biology of Serpins. New York: Plenum Press; 1997. pp. 207–222. [DOI] [PubMed] [Google Scholar]
- 31.Hekman C. M., Loskutoff D. J. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J. Biol. Chem. 1985;260:11581–11587. [PubMed] [Google Scholar]
- 32.Gettins P. G. W., Patston P. A., Olson S. T. Austin: RG Landes Company; 1996. Serpins: Structure, Function and Biology. [Google Scholar]
- 33.Kaslik G., Kardos J., Szabo E., Szilagyi L., Zavodszky P., Westler W. M., Markley J. L., Graf L. Effects of serpin binding on the target proteinase: global stabilization, localized increased structural flexibility, and conserved hydrogen bonding at the active site. Biochemistry. 1997;36:5455–5464. doi: 10.1021/bi962931m. [DOI] [PubMed] [Google Scholar]
- 34.Marshall C. J. Evolutionary relationships among the serpins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1993;342:101–119. doi: 10.1098/rstb.1993.0141. [DOI] [PubMed] [Google Scholar]
- 35.Helenius A., Markus A. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 36.Kern G., Kern D., Jaenicke R., Seckler R. Kinetics of folding and association of differently glycosylated variants of invertase from Saccharomyces cerevisiae. Protein Sci. 1993;2:1862–1868. doi: 10.1002/pro.5560021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imperiali B., O'Connor S. E. Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 1999;3:643–649. doi: 10.1016/s1367-5931(99)00021-6. [DOI] [PubMed] [Google Scholar]
- 38.Das T., Sen A., Kempf T., Pramanik S. R., Mandal C., Mandal C. Induction of glycosylation in human C-reactive protein under different pathological conditions. Biochem. J. 2003;373:345–355. doi: 10.1042/BJ20021701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blow D. M. Structure and mechanism of chymotrypsin. Acc. Chem. Res. 1976;9:145–152. [Google Scholar]