

Abstract
Background: DNA microarray technology has permitted the analysis of global gene expression profiles for several diseases, including cancer. However, standard hybridisation and detection protocols require micrograms of mRNA for microarray analysis, limiting broader application of this technology to small excisional biopsies, needle biopsies, and/or microdissected tissue samples. Therefore, linear amplification protocols to increase the amount of RNA have been developed. The correlation between the results of microarray experiments derived from non-amplified RNA and amplified samples needs to be evaluated in detail.
Methods: Total RNA was amplified and replicate hybridisation experiments were performed with linearly amplified (aRNA) and non-amplified mRNA from tonsillar B cells and the SUDHL-6 cell line using cDNA microarrays containing approximately 4500 genes. The results of microarray differential expression using either source of RNA (mRNA or aRNA) were also compared with those found using real time quantitative reverse transcription polymerase chain reaction (QRT-PCR).
Results: Microarray experiments using aRNA generated reproducible data displaying only small differences to data obtained from non-amplified mRNA. The quality of the starting total RNA template and the concentration of the promoter primer used to synthesise cDNA were crucial components of the linear amplification reaction. Approximately 80% of selected upregulated and downregulated genes identified by microarray analysis using linearly amplified RNA were confirmed by QRT-PCR using non-amplified mRNA as the starting template.
Conclusions: Linear RNA amplification methods can be used to generate high fidelity microarray expression data of comparable quality to data generated by microarray methods that use non-amplified mRNA samples.
Keywords: amplified RNA, in vitro transcription, microarray, expression profiling, real time PCR
 The development of cDNA microarray technology has made it possible to carry out large scale parallel analyses of gene expression, allowing the simultaneous comparison of the levels of expression of several thousand genes in different cell types.1,2 This approach has been used to identify distinct gene expression patterns in several distinct tumour types, tumour cell lines, and disease states.3–10 However, broader application of this technology has been limited by the requirement for relatively large amounts of RNA. To expand the use of this technology, there is a great need for robust methods that can amplify small amounts of RNA without greatly altering the information content of the original RNA samples. This is particularly true of needle biopsies and laser capture microdissection specimens, where limited amounts of RNA require the use of RNA amplification methods to obtain sufficient RNA for microarray experiments.
The development of cDNA microarray technology has made it possible to carry out large scale parallel analyses of gene expression, allowing the simultaneous comparison of the levels of expression of several thousand genes in different cell types.1,2 This approach has been used to identify distinct gene expression patterns in several distinct tumour types, tumour cell lines, and disease states.3–10 However, broader application of this technology has been limited by the requirement for relatively large amounts of RNA. To expand the use of this technology, there is a great need for robust methods that can amplify small amounts of RNA without greatly altering the information content of the original RNA samples. This is particularly true of needle biopsies and laser capture microdissection specimens, where limited amounts of RNA require the use of RNA amplification methods to obtain sufficient RNA for microarray experiments.
In general, two major strategies have been used to obtain sufficient quantities of RNA for subsequent cDNA microarray analysis when RNA quantities are limiting. Polymerase chain reaction (PCR) based methods, although effective for the generation of abundant amplified RNA,11 are hindered by a propensity to skew expression data as a result of preferential amplification of specific transcripts.12 Additional approaches involve linear amplification of RNA using in vitro transcription based protocols.13,14 RNA amplification by in vitro transcription of cDNA14 has been shown to maintain relative mRNA expression levels with as little as 1 μg of mRNA12 or 10 μg of total RNA.15 Furthermore, amplified RNA (aRNA) samples from much smaller amounts of starting material have been shown to generate reproducible microarray data, in addition to similar results to hybridisations using non-amplified RNA.16–20 Nevertheless, more studies assessing the validity of microarray expression data obtained from linearly amplified RNA using hybridisation independent methods, such as real time quantitative reverse transcriptase (RT) PCR, are warranted.
“There is a great need for robust methods that can amplify small amounts of RNA without greatly altering the information content of the original RNA samples”
In our study, we examined linear amplification protocols and compared cDNA expression profiles of mRNA and linearly amplified total RNA. Systematic analysis of various primer and template concentrations allowed us to define the optimal parameters for obtaining the best aRNA product yields for microarray studies. We also sought to determine the relation between microarray expression data obtained from linearly amplified RNA and non-amplified RNA samples through the analysis of replicate hybridisation experiments using cDNA microarrays consisting of approximately 4500 genes. Using twofold differences in expression as the threshold for scoring differential gene expression, we directly compared the differentially expressed genes identified by linearly amplified and non-amplified RNA microarray hybridisations and found that a large number of these genes were identified in both types of RNA samples. Differentially expressed genes identified by microarray analysis of linearly amplified RNA were validated by real time quantitative fluorescence RT-PCR. Our results indicate that RNA amplification methods can be used to generate microarray data of comparable quality to standard microarray protocols that do not involve RNA amplification, and that these methods can also be used successfully to identify authentic differentially expressed genes that are verifiable by hybridisation independent methods.
MATERIALS AND METHODS
Tonsillar B cells
The isolation of phenotypically enriched tonsillar B cells was performed as described previously.21 Briefly, a routine tonsillectomy sample was obtained from a single patient (with informed consent) who underwent surgical removal for chronic tonsillitis. A single specimen was processed to minimise the heterogeneity of the purified B cell population. Tonsillar tissue was finely minced and the resulting cell suspension was depleted of non-B cells by plastic adherence and rosetting with sheep erythrocytes. This method routinely yields tonsillar B cells with approximately 98% purity, as determined by immunophenotypic analysis with CD3, CD19, CD20, and CD45 antibodies.
RNA preparation
RNA was extracted from the diffuse large B cell lymphoma cell line (DLBCL) SUDHL-6 (kindly provided by Dr L Sorbara, National Cancer Institute, Bethesda, Maryland, USA) and purified tonsillar B cells using TRIzol reagent (Life Technologies, Rockville, Maryland, USA), essentially as described by the manufacturer. The RNA concentration was determined by absorbance at 260 nm and the quality of the RNA was assessed by electrophoresis in 2% agarose gels.
mRNA was isolated from total RNA using the Qiagen Oligotex mRNA purification system, according to the manufacturer’s instructions (Qiagen Inc, Valencia, California, USA).
RNA amplification
Total cellular RNA and purified mRNA samples from the SUDHL-6 cell line and purified tonsillar B cells were subjected to linear amplification using a procedure adapted from that described by Wang et al.17
Total RNA was diluted to 0.1 ng/μl, 1 ng/μl, 10 ng/μl, 100 ng/μl, and 1000 ng/μl, and purified mRNA from these samples was diluted to 1 ng/μl, 10 ng/μl, and 100 ng/μl. The samples were subjected to two rounds of amplification as described by Wang et al.17
DNA microarray analysis
Microarray analysis was performed in the Huntsman Cancer Institute Microarray Core Facility at the University of Utah using Molecular Dynamics/Amersham Pharmacia Biotech (Piscataway, New Jersey, USA) instrumentation to print and scan microarray slides containing 4364 sequence verified clones obtained from Research Genetics (Huntsville, Alabama, USA), as described previously.22 Control tonsillar B cell RNA was labelled with Cy3-dCTP, and experimental SUDHL-6 RNA was labelled with Cy5-dCTP. Manipulation of raw fluorescence data was accomplished using GeneSpring software (Redwood City, California, USA). In accordance with the recommendations of Lee et al,23 gene expression patterns were considered reproducible only if they were maintained in at least three replicate hybridisations (from a total of four hybridisations).
Quantitative fluorescence RT-PCR analysis
First strand cDNA synthesis was performed using 1.0 μg of total RNA and SuperScript II RNase H RT (Life Technologies), according to the manufacturer’s instructions. Fluorescence PCR amplifications were performed in triplicate using the LightCycler™ (Roche Molecular Biochemicals, Indianapolis, Indiana, USA) and SYBR Green I™ (Molecular Probes, Eugene, Oregon, USA), essentially as described previously.24 A 1.0 μl aliquot of each first strand cDNA reaction was amplified by primer pairs specific for the glyceraldehyde phosphate dehydrogenase (GAPDH), potassium voltage gated channel shaker related subfamily β member 2 (KCNAB2), KIAA0246, DNA (cytosine-5)-methyltransferase 3α (DNMT3A), major histocompatibility complex (MHC) class II DMα (HLA-DMA), MHC class II DPβ1 (HLA-DPB1), junD, fosB, and CD79A antigen (immunoglobulin associated α) genes, and selected expressed sequence tags in a 10 μl reaction containing 1× PCR buffer (50mM Tris, pH 8.3, 250 μg/ml bovine serum albumin, 2% sucrose, 3.0mM MgCl2), dNTPs at 200 μmol each, 0.5 μmol of each primer, 0.4 units of Taq DNA polymerase (Promega, Madison, Wisconsin, USA), 8.8 ng/μl TaqStart antibody (Clontech, Palo Alto, California, USA), and the double stranded DNA binding dye SYBR Green I™ (1/30 000 dilution). Amplification reactions consisted of 45 cycles of denaturation at 94°C (0 seconds), annealing at 55°C (0 seconds), and extension at 72°C (15 seconds). Fluorescence signals were obtained once in each cycle by sequential fluorescence monitoring of each sample tube at the end of extension. A fractional cycle number or crossing threshold (CT) was determined from the exponential phase of the fluorescence amplification profiles using the second derivative maximum function of the Roche LightCycler™ software. These CT values serve as indirect indicators of gene expression so that samples with high expression of a given gene will exhibit lower CTs than samples showing low level gene expression. Average CT values and standard deviations were calculated for each gene. Expression of the housekeeping gene GAPDH was used to control for input cDNA in each LightCycler™ amplification reaction. Once the CT for the GAPDH gene was determined for each cDNA sample, it was used to normalise all other genes tested in the same cDNA sample. Determination of fold increase or decrease in expression for selected genes in the SUDHL-6 cell line relative to levels of expression in tonsillar B cells was accomplished using the following formula, as described previously22,25:
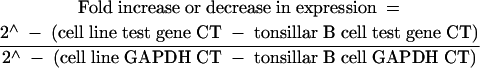 |
This formula permits an accurate estimation of the abundance of specific transcripts relative to a reference sample (phenotypically purified tonsillar B cells) using a relatively stable transcript (GAPDH) for normalisation of input cDNA.
RESULTS
Linear RNA amplification
Most commonly used labelling procedures for microarray experiments require relatively large amounts of total RNA (50–200 μg) or mRNA (2–5 μg).26 In our study, we sought to determine whether microarray expression data obtained using aRNA synthesised from small amounts (nanogram quantities) of total RNA were comparable to microarray data obtained using conventional methods. For this analysis, various amounts of total RNA isolated from the DLBCL derived cell line, SUDHL-6, were subjected to two rounds of linear amplification (fig 1A ▶). Using 1000 ng of starting total RNA template, we consistently observed an approximately 15-fold amplification after the first round of amplification (based upon absorbance at 260 nm) and an average of approximately 190-fold amplification after the second round (fig 1B ▶). By way of comparison, using the Arcturus RiboAmp™ RNA amplification kit, one round of amplification yielded a 49-fold increase and two rounds of amplification yielded a 121-fold increase (Arcturus, Mountain View, California, USA). However, after two rounds of amplification the average transcript size decreased as evaluated by gel electrophoresis. The average range of product sizes for one round of amplification was 50 to 800 bases and for two rounds of amplification 50 to 600 bases. The most consistent results were obtained when 500 ng or more of total RNA were used for the amplification reactions, as demonstrated by agarose gel electrophoresis (fig 1A ▶). We also found that the quality of the aRNA was directly dependent upon the quality of the starting total RNA (fig 1C ▶). For example, if partially degraded RNA was used for the linear amplification reaction, little amplification was observed, and the amplified product consisted of primarily short fragments of 100 bp or less (fig 1C ▶). The concentration of the oligo-dT(15)-T7 promoter primer used to make cDNA was also found to be a crucial component of the amplification reaction. In conditions of high primer concentrations, template independent (primer dimer) products were produced at the expense of RNA template products (fig 1D ▶). The best results were obtained when the primer to starting template ratio was approximately 1:10 (fig 1D ▶).
Figure 1.

Linear RNA amplification. (A) Various amounts of total RNA isolated from the SUDHL-6 cell line were used for two rounds of linear amplification. With decreasing template concentration below approximately 500 ng, the amplified RNA product shows a significant decline in both quantity and quality (size). Lane 1, RNA ladder; lane 2, 1000 ng starting RNA template; lane 3, 500 ng starting RNA template; lane 4, 100 ng starting RNA template; lane 5, 10 ng starting RNA template; lane 6, 1 ng starting RNA template; lane 7, 0.1 ng starting RNA template; lane 8, DNA ladder. (B) Aliquots of 1000 ng of total RNA from the SUDHL-6 cell line were subjected to two rounds of RNA amplification. One round of amplification resulted in approximately 15-fold amplification and two rounds showed approximately 190-fold amplification. Lane 1, RNA ladder; lane 2, two rounds of amplification; lane 3, one round of amplification; lane 4, DNA ladder. (C) The quality and quantity of the amplified RNA is directly dependent upon the quality of the starting template RNA. Partially degraded RNA results in much lower amounts of amplified product and smaller product size after two rounds of linear amplication compared with amplified product obtained from intact starting material. Lane 1, RNA ladder; lane 2, two rounds of amplification starting from 1000 ng of intact total RNA; lane 3, two rounds of amplification starting from 1000 ng of partially degraded total RNA; lane 4, DNA ladder. (D) The concentration of the oligo-dT(15)-T7 promoter primer used to make the cDNA is a crucial component of the RNA amplification reaction. Template independent products (primer dimers) are produced when higher concentrations of oligo-dT(15)-T7 primer are used in the amplification reaction. Lane 1, RNA ladder; lane 2, 1000 ng oligo-dT(15)-T7 primer; lane 3, 500 ng oligo-dT(15)-T7 primer; lane 4, 100 ng oligo-dT(15)-T7 primer; lane 5, DNA ladder. (E) One round of amplification produced amplified RNA (aRNA) from SUDHL-1 (lanes 1 and 2) and Karpas 299 (lanes 3 and 4) starting from 2 μg total RNA template using the RiboAMP kit; lane 5, DNA ladder.
cDNA microarrays
To assess the usefulness of our linear amplification procedure for microarray expression analysis, we performed replicate hybridisation experiments with linearly amplified and non-amplified RNA samples using cDNA microarrays consisting of approximately 4500 genes. When Cy5 labelled aRNA prepared from SUDHL-6 total RNA was compared with Cy3-labelled aRNA prepared from the same sample of SUDHL-6 total RNA in replicate cDNA microarray hybridisations (n = 4), the range of the correlation coefficients was quite high (0.9781 to 0.9803), as would be expected in comparisons of identical RNA samples (fig 2A ▶). However, when aRNA and non-amplified mRNA samples were compared with each other in replicate microarray hybridisations, the range of the correlation coefficients decreased to 0.8080–0.8242 (n = 3), indicating that these samples differed slightly from one another (fig 2B ▶).
Figure 2.

Representative scatter plots of log2 transformed data from cDNA microarray hybridisations. Equal amounts (2 μg) of either mRNA or amplified RNA (aRNA) were used for each comparison. (A) Scatter plots representing averaged cDNA microarray hybridisations comparing SUDHL-6 aRNA samples; r = 0.9904. (B) Scatter plots representing averaged cDNA microarray hybridisations comparing SUDHL-6 aRNA and mRNA samples; r = 0.9038. (C) Scatter plots representing averaged cDNA microarray hybridisations comparing SUDHL-6 mRNA to purified tonsillar B cell mRNA; r = 0.8783. (D) Scatter plots representing averaged cDNA microarray hybridisations comparing SUDHL-6 aRNA to purified tonsillar B cell aRNA; r = 0.9763.
To determine the extent of similarity between the aRNA and non-amplified RNA samples, we compared the microarray expression profiles of amplified and non-amplified SUDHL-6 samples with the genetic profile of phenotypically purified B cells obtained from hyperplastic tonsils. The range of the correlation coefficients for replicate cDNA microarray hybridisations (n = 4) using SUDHL-6 mRNA and tonsillar B cell mRNA was 0.7088 to 0.7982 (fig 2C ▶), whereas the correlation coefficients for replicate cDNA microarray hybridisations (n = 4) using SUDHL-6 aRNA and tonsillar B cell aRNA was 0.9484 to 0.9583 (fig 2D ▶). All raw data were log2 transformed before analysis. These results suggested that linear amplification caused “compression” of the microarray expression data (slightly reduced dynamic range), resulting in the identification of fewer differentially regulated genes at a given threshold of expression than non-amplified RNA samples.
This phenomenon of data “compression” was illustrated more clearly when specific differentially expressed genes were identified and compared in the mRNA and aRNA generated microarrays. Genes that were found to be either twofold overexpressed or underexpressed relative to tonsillar B cells in mRNA generated microarrays were compared with similarly overexpressed and underexpressed genes in aRNA generated microarrays. In mRNA generated microarrays, 88 genes were found to be overexpressed and 78 genes were underexpressed twofold or more relative to tonsillar B cells, whereas aRNA generated microarrays showed that 12 genes were overexpressed and 22 were underexpressed twofold or more. Using a twofold threshold of differential expression relative to purified tonsillar B cells, six genes were overexpressed and 20 genes were underexpressed in both the mRNA and aRNA generated microarrays. However, when a 1.5-fold threshold of differential expression relative to purified tonsillar B cells was used for the aRNA generated microarrays, the number of genes that were overexpressed and underexpressed in both the mRNA and aRNA generated microarrays increased to 10 and 43, respectively.
Validation of cDNA microarray differential expression data by quantitative fluorescence RT-PCR analysis
To confirm the differential expression of genes identified by cDNA microarray analysis of mRNA and aRNA samples isolated from the SUDHL-6 DLBCL cell line, we analysed several upregulated and downregulated genes by real time quantitative fluorescence RT-PCR (fig 3 ▶). Using GAPDH expression as a control for input cDNA, we directly compared levels of gene expression obtained with the use of both mRNA and aRNA samples in microarray and RT-PCR analyses. We were able to confirm most of the differentially expressed genes identified by microarray analysis of linearly amplified and non-amplified RNA samples, with the exception of the KIAA0246 gene, the aRNA PCR reaction for the DNMT3A gene, the aRNA reaction for EST1, and the mRNA reaction for EST2 (fig 3 ▶). Overall agreement of the quantitative RT-PCR data (using both aRNA and mRNA samples) with the mRNA generated microarray data was approximately 75%. Furthermore, the aRNA generated microarray data could be confirmed by quantitative RT-PCR using non-amplified RNA as the starting template in approximately 80% of the tested genes. These results are consistent with a recent study by Rajeevan et al,27 in which they were able to verify the expression levels of approximately 70% of differentially expressed genes identified by microarray analysis using a similar RT-PCR approach that also used the LightCycler™. In general, the trends of differential expression were maintained in both the microarray and quantitative RT-PCR analyses of mRNA and aRNA samples, but we often found a greater dynamic range of expression in quantitative RT-PCR analyses relative to the corresponding microarray expression data (fig 3 ▶). Although a direct comparison may not reflect the precise magnitude of differential expression, the analysis of the trends observed using RT-PCR and microarray is valid. We realise that the normalisation schemes for the microarray analysis and the quantitative real time RT-PCR are quite different, particularly in the number of genes used as calibrators. However, we must state again that the trends documented in the microarray experiments were also shown by quantitative real time RT-PCR.
Figure 3.

Comparison of cDNA microarray expression data and quantitative fluorescence reverse transcriptase polymerase chain reaction (RT-PCR) data for selected differentially expressed genes identified by microarray analysis of mRNA and amplified RNA (aRNA) samples obtained from the SUDHL-6 cell line. RT-PCR results were calculated as described in the Materials and Methods. We consistently found a larger dynamic range of expression for our RT-PCR data relative to the corresponding cDNA microarray expression data. Results are included for the potassium voltage gated channel shaker related subfamily β member 2 (KCNAB2), KIAA0246, DNA (cytosine-5)-methyltransferase 3α (DNMT3A), major histocompatibility complex (MHC) class II DMα (HLA-DMA), MHC class II DPβ1 (HLA-DPB1), junD, fosB, and CD79A antigen (immunoglobulin associated α) genes, and selected expressed sequence tags (EST1 and EST2).
DISCUSSION
In our study, we compared the differential expression identified by cDNA microarray using mRNA and linearly amplified total RNA, with that observed by real time quantitative RT-PCR. We found that the quality of the aRNA produced was directly dependent upon the quality of the starting total RNA. When partially degraded RNA was used for the linear amplification reaction, little amplification was observed, and primarily short fragments of 100 bp or less were produced. This suggests that small tissue biopsies, needle biopsies, and/or microdissection samples should be processed carefully to avoid possible RNA degradation if these samples are to be subjected to linear amplification and subsequent microarray analysis. Ideally, tissue samples should be immersed in RNA preservation medium as soon as possible to protect RNA integrity (for example, RNAlater™; Ambion, Austin, Texas, USA). The concentration of the promoter primer used to make cDNA was also found to be a crucial component of the amplification reaction. High primer concentrations resulted in the production of abundant template independent (primer dimer) product. Statistical analysis of replicate hybridisation experiments performed with linearly amplified RNA samples using cDNA microarrays consisting of approximately 4500 genes indicated that the amplified RNA obtained by this method was of sufficient quality to produce highly reproducible microarray expression data (mean correlation coefficient, 0.9904).
To determine the extent of similarity between amplified and non-amplified RNA samples, we compared the microarray expression profiles of amplified and non-amplified samples to that of phenotypically purified B cells. These data indicated that two rounds of linear amplification caused a slight reduction in the dynamic range of the expression data, resulting in the identification of fewer differentially expressed genes at a given expression threshold (twofold) than non-amplified RNA samples. Furthermore, the decreases in the dynamic range of expression in the aRNA generated microarrays appeared to occur more frequently among the overexpressed genes than the underexpressed genes. However, we found that by decreasing the screening threshold for the aRNA generated microarrays from twofold to 1.5-fold overexpression or underexpression relative to purified tonsillar B cells, we could increase the number of differentially expressed genes common to both the mRNA and aRNA generated microarrays. Thus, when using linearly amplified RNA for microarray studies, we recommend using lower screening thresholds to identify the maximum number of differentially regulated genes for further analysis. However, we do not recommend using statistical tests on each gene to identify differentially expressed genes. Given the large number of spots on a slide, this practice would be labour intensive and not conducive to genome wide analyses.
“Small tissue biopsies, needle biopsies, and/or microdissection samples should be processed carefully to avoid possible RNA degradation if these samples are to be subjected to linear amplification and subsequent microarray analysis”
Confirmation of the usefulness of linear amplification based methods was demonstrated by the fact that we were able to validate approximately 80% of the gene expression trends observed by amplified RNA generated microarrays by real time quantitative fluorescence RT-PCR using non-amplified RNA samples. Comparisons of the results of quantitative RT-PCR analysis of samples derived from mRNA and aRNA showed strong overall concordance of differential expression trends when either RNA species was used as a template. Interestingly, the dynamic range of differential expression seen with quantitative RT-PCR analysis was often much greater than that seen with microarray analysis.
Take home messages.
Linear RNA amplification can be used to amplify nanogram quantities of unselected total RNA samples, so that it can be used in cases where only small amounts of sample are available for microarray analysis
The amplified RNA generated high fidelity microarray expression data of comparable quality to that generated by microarray methods using non-amplified mRNA samples
The quality of the starting total RNA template and the concentration of the promoter primer used to synthesise cDNA were crucial components of the linear amplification reaction
In conclusion, linear amplification methods such as the protocol described here can be used to amplify nanogram quantities of unselected total RNA samples, making it applicable for use in cases where only small amounts of sample are available for microarray analysis. Because clinicians are using smaller and smaller tissue samples for diagnostic purposes, the use of this and other linear amplification methods may pave the way towards the routine use of small tissue biopsies, fine needle aspirates, and microdissection samples for microarray analysis.
Acknowledgments
This study was supported by grant CA83984-01 from the National Institutes of Health (NIH) to KSJE-J and the ARUP Institute for Clinical and Experimental Pathology. RSR receives partial support from a NIH Haematology Training Grant (T32 DK07115-24) and a College of American Pathologists Foundation Research Fellowship.
Abbreviations
aRNA, amplified RNA
CT, crossing threshold
DLBCL, diffuse large B cell lymphoma
GAPDH, glyceraldehyde phosphate dehydrogenase
KCNAB2, potassium voltage gated channel shaker related subfamily β member 2
DNMT3A, DNA (cytosine-5)-methyltransferase 3α
MHC, major histocompatibility complex
PCR, polymerase chain reaction
RT, reverse transcriptase
REFERENCES
- 1.Schena M, Shalon D, Davis RW, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995;270:467–70. [DOI] [PubMed] [Google Scholar]
- 2.Schena M, Shalon D, Heller R, et al. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci U S A 1996;93:10614–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeRisi J, Penland L, Brown PO, et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet 1996;14:457–60. [DOI] [PubMed] [Google Scholar]
- 4.Heller RA, Schena M, Chai A, et al. Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci U S A 1997;94:2150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ross DT, Scherf U, Eisen MB, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet 2000;24:227–35. [DOI] [PubMed] [Google Scholar]
- 6.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52. [DOI] [PubMed] [Google Scholar]
- 7.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 8.Welsh JB, Zarrinkar PP, Sapinoso LM, et al. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc Natl Acad Sci U S A 2001;98:1176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Ross DT, Kadin ME, et al. Comparative genome-scale analysis of gene expression profiles in T cell lymphoma cells during malignant progression using a complementary DNA microarray. Am J Pathol 2001;158:1231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young AN, Amin MB, Moreno CS, et al. Expression profiling of renal epithelial neoplasms: a method for tumor classification and discovery of diagnostic molecular markers. Am J Pathol 2001;158:1639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jena PK, Liu AH, Smith DS, et al. Amplification of genes, single transcripts and cDNA libraries from one cell and direct sequence analysis of amplified products derived from one molecule. J Immunol Methods 1996;190:199–213. [DOI] [PubMed] [Google Scholar]
- 12.Lockhart DJ, Dong H, Byrne MC, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol 1996;14:1675–80. [DOI] [PubMed] [Google Scholar]
- 13.Eberwine J, Yeh H, Miyashiro K, et al. Analysis of gene expression in single live neurons. Proc Natl Acad Sci U S A 1992;89:3010–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Gelder RN, von Zastrow ME, Yool A, et al. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci U S A 1990;87:1663–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahadevappa M, Warrington JA. A high-density probe array sample preparation method using 10- to 100-fold fewer cells. Nat Biotechnol 1999;17:1134–6. [DOI] [PubMed] [Google Scholar]
- 16.Luo L, Salunga RC, Guo H, et al. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat Med 1999;5:117–22. [DOI] [PubMed] [Google Scholar]
- 17.Wang E, Miller LD, Ohnmacht GA, et al. High-fidelity mRNA amplification for gene profiling. Nat Biotechnol 2000;18:457–9. [DOI] [PubMed] [Google Scholar]
- 18.Luzzi V, Holtschlag V, Watson MA. Expression profiling of ductal carcinoma in situ by laser capture microdissection and high-density oligonucleotide arrays. Am J Pathol 2001;158:2005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pabon C, Modrusan Z, Ruvolo MV, et al. Optimized T7 amplification system for microarray analysis. Biotechniques 2001;31:874–9. [DOI] [PubMed] [Google Scholar]
- 20.Baugh LR, Hill AA, Brown EL, et al. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res 2001;29:E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan JW, Reddy GS, Uskokovic MR, et al. Functional block for 1 alpha,25-dihydroxyvitamin D3-mediated gene regulation in human B lymphocytes. J Biol Chem 1994;269:13437–43. [PubMed] [Google Scholar]
- 22.Robetorye RS, Bohling SD, Morgan JW, et al. Microarray analysis of B-cell lymphoma cell lines with the t (14; 18). J Mol Diagn 2002;4:123–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee ML, Kuo FC, Whitmore GA, et al. Importance of replication in microarray gene expression studies: statistical methods and evidence from repetitive cDNA hybridizations. Proc Natl Acad Sci U S A 2000;97:9834–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison TB, Weis JJ, Wittwer CT. Quantification of low-copy transcripts by continuous SYBR Green I monitoring during amplification. Biotechniques 1998;24:954–8, 960962. [PubMed] [Google Scholar]
- 25.Elenitoba-Johnson KS, Bohling SD, Jenson SD, et al. Fluorescence PCR quantification of cyclin D1 expression. J Mol Diagn 2002;4:90–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duggan DJ, Bittner M, Chen Y, et al. Expression profiling using cDNA microarrays. Nat Genet 1999;21 (suppl 1):10–14. [DOI] [PubMed] [Google Scholar]
- 27.Rajeevan MS, Vernon SD, Taysavang N, et al. Validation of array-based gene expression profiles by real-time (kinetic) RT-PCR. J Mol Diagn 2001;3:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]