

Abstract
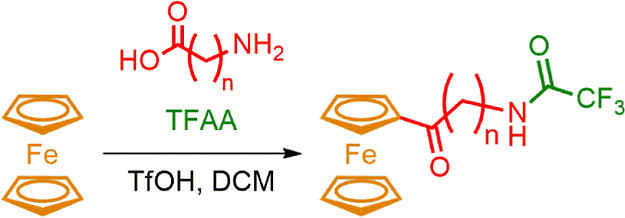
The Friedel–Crafts acylation of ferrocene with amino acids carried out under mild conditions (metal-free catalytic system, room temperature, and a short reaction time of 1 h) has been reported. The acylating agent is generated in situ by N-protection of the amino group of the amino acid, followed by formation of mixed anhydride. This one-pot triflic-acid-promoted reaction provides N-trifluoroacetyl-protected amidoketones in good to excellent yields. Moreover, the trifluoroacetyl group can be easily removed or replaced with another protecting group under mild conditions in a one-pot procedure.
1. Introduction
Aminoketones constitute an important group of bifunctional organic compounds.1,2 Representatives of this class of compounds have various applications. As the compounds possess biological activity, they are utilized in medicinal chemistry as drugs, active substances, and their precursors.3−12
For example, α-aminoketones, exhibiting a wide spectrum of regulatory properties, serve as the basis for antidepressant drugs (Bupropion),13 appetite suppressants (Amfepramone),14 ACE inhibitors for hypertension treatment (Keto-ACE),15,16 or antiplatelet agents (Prasugrel).17 Similarly, the β-aminoketone skeleton can be found in Proroxane (nonselective α-adrenergic blocker),18,19 Tolperisone (vasodilation),20 Oxyfedrine (coronary artery disease),21 or Sitagliptin (antidiabetic).22
On the other hand, the presence of both carbonyl and amino groups in their molecular structure makes them extremely useful building blocks employed in organic synthesis.23,24 As building blocks, α-aminoketones play an important role in the synthesis of heterocycles.25−28 Particularly significant is their role in the formation of nitrogen-containing ring systems such as pyrazines29,30 or pyrroles.31,32 Through the reduction of the carbonyl group, α-aminoketones yield 1,2-amino alcohols,33−35 which are useful ligands and chiral auxiliaries.36 In turn, β-aminoketones find application in the synthesis of 1,3-amino alcohols,37−42 1,3-diamines,43−45 and γ-aryl amines46−49 and the asymmetric synthesis of β-amino acids.50,51 In the Mannich reaction, β-aminoketones can undergo cyclization with aldehydes to form piperidones, which constitutes an important aspect of the asymmetric synthesis of these compounds.52
The importance of this group of compounds, also as useful synthetic intermediates, has led to the development of various synthesis methods for aminoketones. Mainly, these include the addition reactions of organolithium and organomagnesium compounds to α-amino acids53,54 and their derivatives such as esters55 or Weinreb amides56 or the direct amination of ketones in the presence of oxidants for the synthesis of α-aminoketones.57 For β-aminoketones, these are mainly various variants of Mannich reactions.2 Most of these reactions require, due to the presence of two active groups in aminoketone molecules, the use of blocking groups, the use of transition metal catalysis, or the use of organocatalysts with complex structures.1,2
This prompted us to develop a simple one-step synthesis enabling the production of aromatics α, β, γ, etc., aminoketones from aromatic compounds and unprotected amino acids through electrophilic substitution in the aromatic system. There are few known examples in the literature of using α-amino acid derivatives in the Friedel–Crafts intra- and intermolecular reactions with benzene and its derivatives.58−65 However, they require α-amino acid chlorides with blocked amino groups, which are unstable, and their synthesis requires a two-step process.
Herein, we report the efficient acylation of ferrocene with unprotected amino acids and trifluoroacetic anhydride (TFAA) which undergoes under mild conditions in the presence of triflic acid.
2. Results and Discussion
2.1. Synthesis
There are literature reports of the Friedel–Crafts acylation reactions of ferrocene using carboxylic acids with trifluoroacetic anhydride and trifluoromethanesulfonic acid. The carboxylic acids used in these reactions are either the simple ones (Scheme 1)66,67 but also substrates with more complicated structure like biotin can play this role efficiently.68,69
Scheme 1. Acylation of Ferrocene with the Carboxylic Acids.

In these reactions, carboxylic acid and a stoichiometric amount of TFAA form acyl trifluoroacetate in situ, which then, under protonation by trifluoromethanesulfonic acid (TfOH), generates the acylating agent. This allows one to avoid the use of AlCl3, which is moisture-sensitive, difficult to handle, and causes difficulties during workup (Scheme 1).
The above facts prompted us to try to apply this methodology for the use of unprotected amino acids as acylating agents. We assumed that using a 2-fold excess of TFAA, it would be possible to in situ protect the amino group of an amino acid with the trifluoroacetyl group and subsequently to generate acyl trifluoroacetate from such N-protected amino acid. The existing literature examples that the N-trifluoroacetyl group is stable against TfOH70−72 assured us about the chosen approach.
For our studies, we chose glycine 2a as the simplest amino acid as a model compound. Since glycine does not dissolve in dichloromethane, we mixed 1 mmol of glycine with 2 mmol of trifluoroacetic anhydride and stirred this mixture for 15 min in anhydrous conditions to protect the amino group and convert the carboxyl group into acyl trifluoroacetate. Then, we were adding anhydrous dichloromethane, followed by ferrocene 1 (1 mmol) and TfOH (1 mmol), and stirred the reaction mixture for 1 h at ambient temperature. The workup of the reaction afforded the corresponding N-trifluoroacetylaminoketone 3a, albeit with low yield, a significant amount of trifluoroacetylferrocene 4 as the main product, and also unreacted ferrocene (Scheme 2). The assumption that the N-trifluoroacetyl protecting group is too weak for the reaction conditions prompted us to try this reaction with N-protected glycine derivatives, namely, N-acetylglycine 5 and Fmoc-glycine 7. The rationale behind it was that 5 is known of its stability in various reaction conditions,73 and the Fmoc group present in compound 7 is stable toward acid conditions.74 We conducted both reactions with only one equivalent of TFAA and found that in the first case, we obtained only acetylferrocene 6, and in the second, no formation of the product was observed (Scheme 2).
Scheme 2. Attempts of the Acylation of Ferrocene with Glycine (2a) and Its Derivatives (5 and 7).

We attempted to optimize the reaction conditions of the reaction of unprotected glycine by extending the reaction time and increasing the amount of TFAA. We also tried to extend the time of generation of acyl trifluoroacetate to ensure that most of TFAA was consumed and therefore decrease the amount of byproduct 4. Both attempts yielded moderate success. We slightly improved the yield but still observed a significant amount of byproduct 4 as well as the unreacted ferrocene. The results are summarized in Table 1.
Table 1. Optimization of the Acylation of Ferrocene with Unprotected Glycine.
| entry | TFAA eq | acyl trifluoroacetate generation time min | reaction time h | 3a | 4 | % of reacting Fc-H |
|---|---|---|---|---|---|---|
| 1 | 2 | 15 | 1 | 17% | 46% | 97% |
| 2 | 2 | 30 | 1 | 21% | 45% | 94% |
| 3 | 2 | 30 | 4 | 25% | 45% | 96% |
| 4 | 3 | 30 | 1 | 26% | 44% | 94% |
The possible explanation is due to the proximity of the N-trifluoroacetyl group and the acyl trifluoroacetate group, and the proton from TfOH can be transferred from the latter to the first and lead to the formation of trifluoroacetyl carbocation, which is responsible for the formation of 4 (Scheme 3).
Scheme 3. Mechanism of Formation of the Products 3a and 4.

The formation of acetylferrocene has been also observed in the sole example of acylation of ferrocene with N-acetylamino acid (6-acetamidohexanoic acid) in similar conditions, but only as a small amount of byproduct.69
Next, we studied how the elongation of the hydrocarbon chain would affect the course of the reaction. We chose β-alanine 2b and reacted it under basic conditions with ferrocene. After a 1 h reaction, we obtained the corresponding N-trifluoroacetyl amidoketone 3b in excellent 91% yield, virtually without any byproducts, so the reaction workup required only extraction and crystallization from hexane to obtain the pure product. This showed that one more methylene group between the N-trifluoroacetyl group and the acyl trifluoroacetate group is sufficient to isolate them from each other enough to prevent the proton transfer and equilibrium which lead to the mixture of products. Therefore, with such an efficient reaction system in hand, we decided to extend the scope of our reaction to amino acids with a longer chain (Scheme 4).
Scheme 4. Acylation of Ferrocene with Amino Acids with Different Chain Lengths.

reaction time 4h.
reaction without TFAA.
reaction with 1 eq. H3PO4 instead of TfOH.
reaction with 1 eq. BF3xEt2O instead of TfOH.
The reactions of ferrocene with a series of amino acids, from 4-aminobutyric 2c acid to 6-aminohexanoic acid 2e, and two long-chain ones, 11-aminoundecanoic acid 2f and 12-aminododecanoic acid 2g, yielded the corresponding N-trifluoroacetamidoketones in good yields. In the case of products 3d and 3e, reactions proceed almost without byproducts and require only simple crystallization like in the case of 3b. The long-chain products require purification by flash chromatography due to the formation of polar byproducts during the course of the reactions. We also attempted to replace the triflic acid with other acids but with moderate success. We conducted the synthesis of products 3a (with the lowest yield) and 3b (with the highest yield) using the orthophosphoric acid and the boron trifluoride diethyl etherate. In the reactions with orthophosphoric acid, we did not observe the formation of the product. In the ones where the boron trifluoride diethyl etherate was used, the yield was lower compared to the triflic acid (Scheme 4).
This is the first example of using unblocked amino acids of different hydrocarbon chain lengths and the positions of amino and carboxyl groups as acylating agents in the Friedel–Crafts reaction of ferrocene. Earlier literature reports include one example of using N-acylated 6-aminohexanoic acid in this role69 and the use of β-lactam and its N-protected derivatives as acylating agents, which undergo ring opening under the influence of a strong acid, with this method being limited to the synthesis of β-aminoketones.75
2.2. Study on the Reaction Mechanism
To further investigate the formation of N-trifluoroacetylaminoacyl trifluoroacetates from amino acids under the reaction conditions, we conducted the following experiment. We charged the NMR test tube with appropriate amino acid, apply TFAA, and left the mixture for 15 min to generate mixed anhydride under conditions similar to the reaction conditions. Then, we dissolved the reaction mixture in deuterated dichloromethane (CD2Cl2) and recorded the 1H and 19F NMR spectra (for details, see the Experimental Section). We repeat this procedure for all amino acids in series 2a–g (Scheme 5).
Scheme 5. NMR Experiment to Investigate the Formation of N-Trifluoroacetylaminoacyl Trifluoroacetates (8a–g).

We obtain the series of 1H NMR spectra presented below (Figure 1).
Figure 1.

1H NMR spectra of acyl trifluoroacetates (8a–g) in CD2Cl2.
On each of them, we can observe the signal from the acidic proton from trifluoroacetic acid (TFA) around 10 ppm and the signal from the N-trifluoroacetylamide proton in the area between 6 and 7 ppm. On all spectra except 8c, we observed the integration ratio 2:1 between signals of TFA and amide protons, which is consistent with the stoichiometry presented in Scheme 5 and supports the mechanism involving the formation of N-trifluoroacetylaminoacyl trifluoroacetates. In the case of 8c, the integration ratio between the above-mentioned signals is 5:1, and two sets of signals are present in the alkyl region of the spectra, suggesting the formation of a mixture of products. The 19F NMR spectra support this hypothesis. Again on all of them, except 8c, we can observe four signals: one from TFA, second from TFAA, and the third and fourth from N-trifluoroacetylamide and acyl trifluoroacetate groups, respectively (Figure 2). On spectrum 8c, an additional fifth signal is present, at −73 ppm, which can originate from the second product, apart from the expected mixed anhydride.
Figure 2.

19F NMR spectra of acyl trifluoroacetates (8a–g) (TFA and TFAA for comparison) in CD2Cl2.
This second product could be 5-member N-trifluoroacetylated lactam 9. This lactam may form from part of in situ generated mixed anhydride 8c (Scheme 6).
Scheme 6. Possible Pathway of Formation of Lactam 9 from 4-Aminobutyric Acid 2c.

Such a compound will contribute to 19F NMR spectra with a single signal from the only CF3 group, which is consistent with the experiment. Lactam 9 also does not undergo acid-driven ring opening, so it would be inactive in reaction conditions.75 This explains the lower yield of product 3c (Scheme 4).
2.3. Deprotection–Reprotection Studies
The N-trifluoroacetyl group is labile under mild basic conditions,76,77 allowing its easy deprotection. We decided to try such deprotection on compound 3b to obtain its aminoketone derivative 10 (Scheme 7). To achieve this, we treated compound 3b with an excess of potassium carbonate in a mixture of methanol and water.
Scheme 7. Deprotection and Deprotection/Protection of 3b.

We found by comparative TLC that in the course of the reaction, the product is more polar than the substrate is formed, which suggested the formation of β-aminopropionyl ferrocene 10. However, it turned out that this compound undergoes slow (few days) decomposition, when exposed to air and room temperature. This decomposition undergoes probably due to the simultaneous presence of the carbonyl group and the free amino group in the molecule. To solve this problem, we modified the deprotection procedure to the deprotection and subsequent protection of the free aminoketone with the Boc group. Since such protection can be done in aqueous conditions,78 we were able to achieve it in a one-pot reaction. The only issue is to replace methanol with tetrahydrofuran between the deprotection and subsequent Boc protection to make the latter possible. The reaction proceeds with excellent yield, giving the bench-stable compound 11. Such Boc derivative can be easily deprotected79,80 before use in synthesis, which makes it a very useful building block.
2.4. Crystallographic Studies
Crystal structures for compounds 3a–3g and 11 were determined by means of single-crystal X-ray diffraction. Detailed information about data processing, structure solution, and refinement is shown in Table S1 in the Supporting Information. Atom and centroid labeling used for structure description is shown in Figure 3. In the case of 3a and 3e, there were, respectively, three and two molecules in the crystallographic asymmetric unit. They were denoted as molecules I–III or I–II depending on the number of the iron atoms.
Figure 3.

Numbering scheme common for all described structures.
Ferrocene conformation in almost all structures was typical, close to eclipsed, with relative rotation of cyclopentadienyl (Cp) rings in the range −7.18–6.74° (Table S4). Only for 3f, there was staggered conformation (relative rotation of −23.13°). Distance between Cp rings was in the range of 3.26–3.32 Å.
The C1–C11 bond was close to 1.46–1.48 Å, which suggests a proper single bond (Table S2.). As expected, the carbonyl group C11–O1 bound to cyclopentadienyl was almost in plane with the ring. The biggest out-of-plane bend was found in the structure of 3a (n = 1) and equaled 11.7(5)°. For the majority of structures, the beginning of the alkyl chain was almost in the cyclopentadienyl plane, with rotation around the first single C–C bond in the range of −160–178°. Only for 3f alkyl chain was bent at the Cp end (dihedral angle −75(2)°). Different behavior was visible at the amide end of the alkyl chain. It was bent for most structures and almost straight (rotated by less than 10°) for the structures with longest alkyl chain −3f and 3g (n = 10 and 11, Figure S3 in the Supporting Information).
Molecules in all structures were interacting by hydrogen bonds between the amide group and carbonyl group bonded to Cp (for 3a–g) or from other amide groups (for 3a and 11). The geometry of hydrogen bonds is shown in Table S1, and motifs are shown in Figure S2. In 3a, hydrogen bond interactions built a branched chain (Figure 4). The chain core (network C(5) in Etter notation81) consisted of molecules I, where the hydrogen atoms bonded to the amide group interacted with the oxygen atoms from the carbonyl group bonded to Cp (O1). Each second oxygen atom (O2–from amide moiety) of molecule I was an acceptor to the hydrogen bond from the amide group of molecule II. Similarly, molecule III was a donor of the hydrogen bond to the oxygen atom of the amide group in the molecule (Figure S2).
Figure 4.

Crystal packing in described structures with supramolecular motifs related to H-bond presence highlighted: 3a in the [100] direction, 3c in the [100] direction, 3g in the [010] direction, 3f in the [010] direction, and 11 in the [010] direction.
3b–d had different hydrogen bond motifs, which can be described as R(4 + n). Their molecules formed dimers, acting as a donor and an acceptor (Figures 4 and S2b). The longer aliphatic chain in 3e, 3f, and 3g and the Boc moiety in 11 resulted in chain formation by hydrogen bonds. Aliphatic chains were parallel in structure with even n value (3f, n = 10, Figure 4: 3f) and crossed when the n value was odd (3e and 3g, n = 5 and n = 11, Figure 4: 3g), in both cases, resulting in the C(4 + n) motif. The Boc derivative in hydrogen bond formation contributed only amide moiety (Figure S2e), creating the C(4) network.
There were no visible differences between the geometry of hydrogen bonds as a function of aliphatic chain length. All N···O distances in structures of 3a–g were in the range 2.83–2.93 Å, and N–H···O angles were 149–169°. The structure of 11, because of steric bulk due to the Boc moiety, was an outlier with N···O distance 3.43 Å.
Hydrogen bond motifs have an impact on crystal packing. In the structure of 3a, there were columns of branched chains in the [100] direction, creating a herringbone pattern on the (100) plane (Figure 4: 3a). There were no distinguishable layers. In structures of 3b–d, dimers were separated. Layers were formed in structures with longer aliphatic chains 3e–g or with Boc derivative 11. Nonparallelism in H-bond chain formation resulted in layers with crossed aliphatic chains (Figure 4: 3g). Crystal packing tended to maximize the number of Fc···Fc interactions.
3. Conclusions
In this study, we obtained a series of trifluoroacetamide ketones by the acylation of ferrocene with unprotected amino acids. We used trifluoroacetic anhydride to protect and activate the amino acids simultaneously and trifluoromethanesulfonic acid (TfOH) as a proton source. We proposed a mechanism for this reaction, involving the in situ creation of N-trifluoroacetylaminoacyl trifluoroacetate, which is subsequently protonated by TfOH to generate a carbocation. Our study demonstrated the possibility of using unprotected amino acids as acylating agents, allowing these reactions to be conducted under relatively mild conditions. This approach gradually expanded the scope of Friedel–Crafts reactions with mixed anhydrides. Additionally, the obtained ferrocene trifluoroacetamide ketones could easily undergo deprotection or a deprotection–protection procedure to yield β-aminopropyl ferrocene derivatives in good yields.
4. Experimental Section
4.1. General Information
All reactions were carried out in an Ar atmosphere using typical glassware. Commercially available reagents and solvents were used as received. Column chromatography was performed on silica gel 60 (230–400 mesh) purchased from Merck. Thin-layer chromatography was performed on aluminum sheets precoated with silica purchased from Silicycle. Elemental analysis were performed on an Elementar Vario Micro Cube. Infrared (IR) spectra were recorded in KBr on a Fourier Transform InfraRed (FTIR) NEXUS (Thermo Nicolet) spectrometer. Electrospray ionization mass spectrometry (ESI-MS) spectra were recorded on a Varian 500 MS LC ion trap spectrometer. High-resolution mass spectrometry (HRMS) measurements were performed using a Synapt G2 Si mass spectrometer (Waters) equipped with an ESI source and a quadrupole-time-of-flight mass analyzer. 1H and 13C{1H} NMR spectra were recorded at 300 K on a Bruker UltraShield Avance III 600 MHz spectrometer (600.26 MHz for 1H and 150.94 MHz for 13C) equipped with a BBFO probe and a Bruker Ascend Neo 600 MHz spectrometer (600.14 MHz for 1H and 150.90 MHz for 13C) equipped with a Prodigy cryoprobe. 19F NMR spectra were recorded at 300 K on a Bruker Ascend Neo 600 MHz spectrometer (564.70 MHz for 19F). Chemical shifts were calibrated on the residual solvent signals: CDCl3 7.26 ppm for 1H and 77.16 ppm for 13C, CD3OD 3.31 ppm for 1H and 49.00 ppm for 13C, and CD2Cl2 5.32 ppm for 1H and 53.84 ppm for 13C.82 Melting points were measured with a Stanford Research Systems DigiMelt MPA161.
4.2. Synthesis of 3a–g
Corresponding amino acid (1 mmol, 1 equiv) was stirred with TFAA (280 μL, 2 mmol, 2 equiv) in a 10 mL round-bottom flask equipped with a magnetic stirring bar until complete dissolution. Dry DCM (5 mL) was added, followed by ferrocene (186 mg, 1 mmol, 1 equiv) and triflic acid (90 μL, 1 mmol, 1 equiv). Reaction progress was monitored by TLC. After 1 h, the reaction was quenched by pouring into ice–water (50 mL) and then placing in a separatory funnel. The organic layer was washed with water (3 × 25 mL) and brine (1 × 25 mL) and dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the crude product was purified by crystallization or column chromatography on silica gel.
4.2.1. 3a
Purified by column chromatography using 1:1 DCM/hexane as an eluent. The product was crystallized from DCM/hexane. Orange–red crystals (71 mg, 21%), mp 98.6–100.3 °C. 1H NMR (600 MHz, CDCl3): δ 7.43 (s, 1H), 4.87 (t, J = 1.7 Hz, 2H), 4.65 (t, J = 1.7 Hz, 2H), 4.52 (d, J = 4.3 Hz, 2H), 4.25 (s, 5H). 13C{1H} NMR (151 MHz, CDCl3): δ 196.4, 76.9, 75.2, 73.5, 70.5, 69.2, 46.5. 19F NMR (565 MHz, CDCl3): δ −75.64. IR (KBr, cm–1): 3292, 3101, 2933, 1734, 1711, 1682, 1558, 1464, 1438, 1323, 1252, 1211, 1188, 1144, 1106, 1047, 818. MS (ESI) m/z: [M + Na]+ calcd for C14H12F3FeNO2Na+ 362.0; found, 362.1. Anal. Calcd for C14H12F3FeNO2: C, 49.59; H, 3.57; N, 4.13. Found: C, 49.55; H, 3.73; N, 4.38.
4.2.2. 3b
Purified by crystallization from DCM/hexane. Orange-red crystals (321 mg, 91%), mp 127.2–128.1 °C. 1H NMR (600 MHz, CDCl3): δ 7.25 (s, 1H), 4.79 (t, J = 1.9 Hz, 2H), 4.57 (t, J = 1.9 Hz, 2H), 4.19 (s, 5H), 3.74 (q, J = 5.8 Hz, 2H), 3.03 (t, J = 5.6 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3): δ 203.5, 78.0, 73.1, 70.1, 69.4, 38.1, 35.0. 19F NMR (565 MHz, CDCl3): δ −76.05. IR (KBr, cm-1): 3239, 3071, 2966, 2910, 1714, 1646, 1552, 1458, 1279, 1244, 1200, 1176, 1153, 1079, 1003, 824. MS (ESI) m/z: [M + H]+ calcd for C15H15F3FeNO2+ 354.0; found, 354.1. Anal. Calcd for C15H14F3FeNO2: C, 51.02; H, 4.00; N, 3.97. Found: C, 51.23; H, 4.02; N, 3.91.
4.2.3. 3c
Purified by column chromatography using DCM/MeOH 200:1 as an eluent. The product was crystallized from DCM/hexane. Orange-red crystals (141 mg, 38%), mp 86.5–87.4 °C. 1H NMR (600 MHz, CDCl3): δ 7.50 (s, 1H), 4.80 (t, J = 1.9 Hz, 2H), 4.55 (t, J = 1.9 Hz, 2H), 4.19 (s, 5H), 3.45 (q, J = 6.1 Hz, 2H), 2.89 (t, J = 6.4 Hz, 2H), 2.02 (quint, J = 6.4 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3): δ 205.1, 78.3, 72.9, 70.0, 69.6, 40.5, 37.6, 22.6. 19F NMR (565 MHz, CDCl3): δ −75.92. IR (KBr, cm–1): 3260, 3095, 2951, 2892, 1720, 1649, 1561, 1461, 1211, 1179, 1150, 820. MS (ESI) m/z: [M + H]+ calcd for C16H17F3FeNO2+ 368.1; found, 368.1. Anal. Calcd for C16H16F3FeNO2: C, 52.34; H, 4.39; N, 3.82. Found: C, 52.27; H, 4.53; N, 3.60.
4.2.4. 3d
Purified by crystallization from DCM/hexane. Orange-red crystals (240 mg, 63%), mp 99.8–101.6 °C. 1H NMR (600 MHz, CDCl3): δ 6.90 (br s, 1H), 4.79 (t, J = 1.9 Hz, 2H), 4.52 (t, J = 1.9 Hz, 2H), 4.20 (s, 5H), 3.40 (q, J = 6.4 Hz, 2H), 2.79 (t, J = 6.7 Hz, 2H), 1.77 (m, 2H), 1.68 (m, 2H). 13C{1H} NMR (151 MHz, CDCl3): δ 204.4, 78.7, 72.4, 69.85, 69.32, 39.5, 38.5, 28.4, 20.6. 19F NMR (565 MHz, CDCl3): δ −75.87. IR (KBr, cm–1): 3280, 3083, 2951, 1720, 1661, 1558, 1455, 1358, 1255, 1208, 1182, 1156, 1103, 818. MS (ESI) m/z: [M + H]+ calcd for C17H19F3FeNO2+ 382.1; found, 382.2. Anal. Calcd for C17H18F3FeNO2: C, 53.57; H, 4.76; N, 3.67. Found: C, 53.58; H, 4.80; N, 3.54.
4.2.5. 3e
Purified by crystallization from DCM/hexane. Orange-red crystals (254 mg, 64%), mp 71.8–73.3 °C. 1H NMR (600 MHz, CDCl3): δ 6.65 (br s, 1H), 4.78 (t, J = 1.9 Hz, 2H), 4.51 (t, J = 1.8 Hz, 2H), 4.19 (s, 5H), 3.43 (q, J = 6.7 Hz, 2H), 2.74 (t, J = 7.1 Hz, 2H), 1.74 (quint, J = 7.5 Hz, 2H), 1.65 (quint, J = 7.3 Hz, 2H), 1.43 (m, 2H). 13C{1H} NMR (151 MHz, CDCl3): δ 204.5, 79.1, 72.4, 69.92, 69.43, 39.6, 39.3, 28.7, 26.4, 23.4. 19F NMR (565 MHz, CDCl3): δ −75.85. IR (KBr, cm–1): 3289, 3080, 2945, 2866, 1711, 1658, 1555, 1458, 1255, 1197, 1182, 1150, 820. MS (ESI) m/z: [M + H]+ calcd for C18H21F3FeNO2+ 396.1; found, 396.2. Anal. Calcd for C18H20F3FeNO2: C, 54.71; H, 5.10; N, 3.54. Found: C, 54.57; H, 5.15; N, 3.68.
4.2.6. 3f
Purified by column chromatography using a DCM/MeOH 400:1 as an eluent. The product was crystallized from DCM/hexane. Orange powder (246 mg, 53%), mp 58.1–59.5 °C. 1H NMR (600 MHz, CDCl3): δ 6.33 (s, 1H), 4.78 (t, J = 1.9 Hz, 2H), 4.49 (t, J = 1.9 Hz, 2H), 4.19 (s, 5H), 3.36 (q, J = 6.8 Hz, 2H), 2.69 (t, J = 7.4 Hz, 2H), 1.70 (quint, J = 7.4 Hz, 2H), 1.58 (quint, J = 7.2 Hz, 2H), 1.35 (m, 12H). 13C{1H} NMR (151 MHz, CDCl3):δ 204.9, 79.3, 72.3, 69.88, 69.47, 40.1, 39.9, 29.58, 29.53, 29.40, 29.16, 29.04, 26.7, 24.7. 19F NMR (565 MHz, CDCl3): δ −75.91. IR (KBr, cm–1): 3301, 3083, 2939, 2916, 2851, 1705, 1646, 1552, 1458, 1376, 1200, 1164, 1108, 824. MS (ESI) m/z: [M + H]+ calcd for C23H31F3FeNO2+ 466.2; found, 466.4. Anal. Calcd for C23H30F3FeNO2: C, 59.37; H, 6.50; N, 3.01. Found: C, 59.52; H, 6.55; N, 2.91.
4.2.7. 3g
Purified by column chromatography using DCM/MeOH 400:1 as an eluent. The product was crystallized from DCM/hexane. Orange powder (246 mg, 51%), mp 79.4–80.9 °C. 1H NMR (600 MHz, CDCl3): δ 6.31 (s, 1H), 4.78 (t, J = 1.9 Hz, 2H), 4.49 (t, J = 1.9 Hz, 2H), 4.19 (s, 5H), 3.36 (q, J = 6.8 Hz, 2H), 2.69 (t, J = 7.4 Hz, 2H), 1.70 (quint, J = 7.4 Hz, 2H), 1.58 (quint, J = 7.4 Hz, 2H), 1.33 (m, 14H). 13C{1H} NMR (151 MHz, CDCl3):δ 204.9, 79.4, 72.2, 69.88, 69.49, 40.2, 39.9, 29.64, 29.57, 29.52, 29.46, 29.19, 29.08, 26.8, 24.8. 19F NMR (565 MHz, CDCl3): δ −75.93. IR (KBr, cm–1): 3321, 3083, 2916, 2851, 1714, 1655, 1549, 1473, 1455, 1385, 1270, 1202, 1182, 1153, 820. MS (ESI) m/z: [M + H]+ calcd for C24H33F3FeNO2+ 480.2; found, 480.4. Anal. Calcd for C24H32F3FeNO2: C, 60.13; H, 6.73; N, 2.92. Found: C, 60.20; H, 6.81; N, 2.84.
4.3. Synthesis of 10
Compound 3b (353 mg, 1 mmol) was suspended in MeOH/water 1:1 (10 mL) in a 25 mL round-bottom flask equipped with a magnetic stirring bar, and potassium carbonate (1.38 g, 10 mmol) was added to the reaction mixture. Reaction was stirred overnight when TLC indicated completion of deprotection. One M NaOH (3 mL) was added to the reaction mixture, and the product was extracted with an EtOAc/MeOH 20:1 solution. Combined organic extracts were washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the crude product was purified by column chromatography using DCM/MeOH 20:1 as an eluent. The compound slowly decomposes and should be stored in the dark under argon at −20 °C. Reddish oil (180 mg, 70%). 1H NMR (600 MHz, CD3OD): δ 4.85 (t, J = 1.7 Hz, 2H), 4.62 (t, J = 1.7 Hz, 2H), 4.26 (s, 5H), 3.06 (m, 4H). 13C{1H} NMR (151 MHz, CD3OD): δ 205.7, 79.7, 74.0, 71.1, 70.5, 41.2, 37.5. IR (KBr, cm–1): 3500, 3442, 3348, 3097, 2961, 2924, 1664, 1479, 1454, 1379, 1252, 1103, 821. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H16FeNO+ 258.0576; found, 258.0578.
4.4. Synthesis of 11
Compound 3b (353 mg, 1 mmol) was suspended in MeOH/water 1:1 (10 mL) in a 25 mL round-bottom flask equipped with a magnetic stirring bar, and potassium carbonate (1.38 g, 10 mmol) was added to the reaction mixture. The reaction was stirred overnight when TLC indicated completion of deprotection. MeOH was removed by evaporation and replaced with THF (5 mL), and Boc2O (436 mg, 2 mmol) was added to the reaction mixture. After 3 h, TLC indicated completion of the reaction. The reaction mixture was concentrated to remove THF and taken between water and DCM. The organic layer was washed with 1 M HCl and brine and dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the crude product was purified by column chromatography using DCM/MeOH 200:1 as an eluent. The product 11 was crystallized from DCM/hexane. Orange powder (324 mg, 91%), mp 93.1–94.6 °C. 1H NMR (600 MHz, CDCl3): δ 5.17 (br s, 1H), 4.78 (t, J = 1.7 Hz, 2H), 4.52 (t, J = 1.8 Hz, 2H), 4.20 (s, 5H), 3.50 (q, J = 5.7 Hz, 2H), 2.95 (t, J = 5.3 Hz, 2H), 1.43 (s, 9H). 13C{1H} NMR (151 MHz, CDCl3): δ 203.6, 156.1, 79.4, 78.9, 72.6, 70.0, 69.4, 39.6, 35.6, 28.6. IR (KBr, cm–1): 3424, 3377, 3359, 3130, 2972, 2936, 1702, 1655, 1499, 1452, 1367, 1288, 1247, 1164, 1085, 973, 824. MS (ESI) m/z: M+ calcd for C18H23FeNO3357.1; found, 357.3. Anal. Calcd for C18H23FeNO3: C, 60.52; H, 6.49; N, 3.92. Found: C, 60.64; H, 6.38; N, 3.87.
4.5. 1H and 19F NMR Experiments (8a–g and 9)
The NMR test tube was charged with an appropriate amino acid (0.07 mmol, 1 equiv), TFAA (30 μL, 0.22 mmol, 3.1 equiv) was added, and the tube was closed with a stopper and left for 15 min to generate N-trifluoroacetylaminoacyl trifluoroacetate. Then, CD2Cl2 (0.5 mL) was added to the tube, and 1H, 13C{1H}, and 19F NMR spectra were recorded. All compounds were generated in situ and have not been separated from the reaction mixture.
4.5.1. 8a
1H NMR (600 MHz, CD2Cl2): δ 7.09 (s, 1H), 4.44 (d, J = 5.8 Hz, 2H). 13C{1H} NMR (151 MHz, CD2Cl2): δ 161.8, 160.9, 160.6, 160.3, 160.0, 152.5, 152.2, 151.9, 151.6, 117.9, 117.1, 116.0, 115.18, 115.14, 114.2, 113.2, 112.3, 42.6. 19F NMR (565 MHz, CD2Cl2): δ −76.44, −76.74. HRMS (ESI-TOF) m/z: [M – H]− calcd for C6H2F6NO4– 265.9894; found, 265.9891.
4.5.2. 8b
1H NMR (600 MHz, CD2Cl2): δ 7.11 (s, 1H), 3.76 (q, J = 6.1 Hz, 2H), 3.00 (t, J = 6.0 Hz, 2H). 13C{1H} NMR (151 MHz, CD2Cl2):δ 165.4, 159.8, 159.5, 159.2, 159.0.153.0, 152.7, 152.4, 152.1, 117.9, 117.1, 116.0, 115.25, 115.20, 114.1, 113.3, 112,3, 35.5, 34.6. 19F NMR (565 MHz, CD2Cl2): δ −76.58, −76.85. HRMS (ESI-TOF) m/z: [M – H]− calcd for C7H4F6NO4– 279.0204; found, 279.0204.
4.5.3. 8c and 9 (mixture) 8c
1H NMR (600 MHz, CD2Cl2): δ 6.87 (s, 1H),3.52 (q, J = 6.7 Hz, 2H), 2.73 (m, 2H), 2.04 (quint, J = 7.0 Hz, 2H). 13C{1H} NMR (151 MHz, CD2Cl2): δ 166.0, 159.7, 159.4, 159.2, 153.3, 153.0, 152.7, 152.4, 117.8, 117.2, 116.0, 115.3, 115.2, 114.1, 113.3, 112.2, 32.6, 30.8, 23.4, 18.2. 19F NMR (565 MHz, CD2Cl2): δ −76.61, −76.78. HRMS (ESI-TOF) m/z: [M – H]− calcd for C8H6F6NO4– 294.0207; found, 294.0203. 9(83)1H NMR (600 MHz, CD2Cl2): δ 3.93 (t, J = 7.2 Hz, 2H), 2.72 (m, 2H), 2.19 (m, 2H). 13C{1H} NMR (151 MHz, CD2Cl2): δ 160.2, 159.9, 159.6, 159.3, 116.7, 114.8, 47.3, 45.3, 39.7. 19F NMR (565 MHz, CD2Cl2): δ −73.34. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C6H7F3NO2+ 182.0423; found, 182.0425.
4.5.4. 8d
1H NMR (600 MHz, CD2Cl2): δ 6.79 (s, 1H), 3.45 (q, J = 6.6 Hz, 2H), 2.70 (t, J = 6.9 Hz, 2H), 1.74 (m, 4H). 13C{1H} NMR (151 MHz, CD2Cl2): δ 166.1, 159.4, 159.2, 158.9, 158.7, 153.5, 153.2, 152.9, 152.6, 117.9, 117.2, 116.0, 115.3, 115.2, 114.1, 113.3, 112.2, 40.4, 34.7, 28.1, 21.2. 19F NMR (565 MHz, CD2Cl2): δ −76.67, −76.81. HRMS (ESI-TOF) m/z: [M – H]– calcd for C9H8F6NO4– 308.0363; found, 308.0357.
4.5.5. 8e
1H NMR (600 MHz, CD2Cl2): δ 6.72 (s, 1H), 3.42 (q, J = 6.8 Hz, 2H), 2.65 (t, J = 7.2 Hz, 2H), 1.75 (m, 2H), 1.65 (m, 2H), 1.45 (m, 2H). 13C{1H} NMR (151 MHz, CD2Cl2): δ 166.2, 159.8, 159.5, 159.2, 158.9, 153.6, 153.3, 153.0, 152.7, 117.9, 117.3, 116.0, 115.4, 115.3, 114.1, 113.4, 111.5, 40.7, 35.2, 28.7, 26.1, 23.7. 19F NMR (565 MHz, CD2Cl2): δ −76.70, −76.84. HRMS (ESI-TOF) m/z: [M – H]− calcd for C10H10F6NO4– 322.0520; found, 322.0523.
4.5.6. 8f
1H NMR (600 MHz, CD2Cl2): δ 6.66 (s, 1H), 3.39 (q, J = 6.8 Hz, 2H), 2.62 (t, J = 7.4 Hz, 2H), 1.70 (quint, J = 7.4 Hz, 2H), 1.60 (quint, J = 7.2 Hz, 2H), 1.35 (m, 12H). 13C{1H} NMR (151 MHz, CD2Cl2): δ 166.4, 159.2, 158.9, 158.7, 158.4, 153.7, 153.4, 153.1, 152.8, 119.2, 117.4, 116.0, 115.4, 115.3, 114.1, 113.4, 111.6, 41.1, 35.5, 29.8, 29.7, 29.52, 29.50, 29.12, 29.08, 27.1, 24.3. 19F NMR (565 MHz, CD2Cl2): δ −76.74, −76.89. HRMS (ESI-TOF) m/z: [M – H]− calcd for C15H20F6NO4– 392.1302; found, 392.1302.
4.5.7. 8g
1H NMR (600 MHz, CD2Cl2): δ 6.65 (s, 1H), 3.39 (q, J = 6.8 Hz, 2H), 2.62 (t, J = 7.4 Hz, 2H), 1.70 (quint, J = 7.4 Hz, 2H), 1.60 (quint, J = 7.3 Hz, 2H), 1.35 (m, 14H). 13C{1H} NMR (151 MHz, CD2Cl2): δ 166.4, 159.2, 159.0, 158.7, 158.5, 153.8, 153.5, 153.2, 152.9, 119.3, 117.4, 166.0, 115.5, 115.4, 114.2, 113.6, 113.5, 41.2, 35.5, 29.9, 29.8, 29.60, 29.57, 29.18, 29.14, 27.1, 24.4. 19F NMR (565 MHz, CD2Cl2): δ −76.75, −76.90. HRMS (ESI-TOF) m/z: [M – H]− calcd for C16H22F6NO4– 406.1459; found, 406.1452.
4.6. X-ray Diffraction Experiments
Crystals of 3a–g and 11 were obtained by n-pentane vapor diffusion into a dichloromethane solution in NMR-like thin glass sample tubes. Data for 3a were collected using a SuperNova diffractometer with a microfocus sealed source of MoKα X-ray radiation (λ = 0.71073 Å) and a CCD Eos detector. Data for the remaining data sets were collected using a SuperNova diffractometer with a microfocus sealed source of CuKα X-ray radiation (λ = 1.54184 Å) and a Hybrid Pixel Array detector. Low temperature was achieved and maintained by keeping crystals in a cold nitrogen stream using an Oxford Cryosystems liquid nitrogen device. Data reduction was performed with CrysAlisPro.81 Gaussian absorption correction was applied using spherical harmonics with the SCALE3 ABSPACK algorithm. Crystal fae indexing was performed for all data sets but 3c, 3d, and 3f. The structure was solved with SHELXT and refined with SHELXL in Olex2.84−86 Detailed information about data processing, structure solution, and refinement is shown in Table 1. Crystal structures were deposited with the Cambridge Crystallographic Data Centre (3a—deposition number 2370728, 3b—deposition number 2370783, 3c—deposition number 2370787, 3d—deposition number 2370786, 3e—deposition number 2370729, 3f—deposition number 2370784, 3g—deposition number 2370785, and 11—deposition number 2370730).
Acknowledgments
This work was financially supported by the Faculty of Chemistry, University of Lodz (grant No. IDUB 38/2021). AM acknowledges the support of the National Science Centre, Poland (grant No. DEC-2021/41/B/ST4/02760). MP gratefully acknowledges MSc Karolina Koprowska (University of Lodz, Faculty of Chemistry, Department of Organic Chemistry, Poland) for conducting the IR measurements.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information The crystallographic data are openly available in CCDC at https://www.ccdc.cam.ac.uk/structures/.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.4c02717.
1H, 13C{1H}, 19F NMR, and IR spectra for compounds 3a–g; 1H, 13C{1H} NMR, and IR spectra for compounds 10 and 11; 1H, 13C{1H}, and 19F NMR spectra for experiments 8a–g and 9; HRMS spectra for experiments 8a–g, 9, and 10; and X-ray diffraction data for compounds 3a–g and 11 (PDF)
Author Contributions
MP: (organic synthesis) conceptualization, investigation, characterization, supervision (NM), validation, and writing—original draft; NM: (organic synthesis) investigation and characterization; RD: (crystallography) investigation, validation, visualization, and writing—part of original draft; AM: (crystallography) investigation, validation, and supervision (RJ); and BR: (organic synthesis) writing—review and editing.
The authors declare no competing financial interest.
This paper was published ASAP on February 18, 2025. The footnote was not included for Scheme 4. The corrected version was reposted on February 19, 2025.
Supplementary Material
References
- Allen L. A. T.; Raclea R.-C.; Natho P.; Parsons P. J. Recent advances in the synthesis of α-amino ketones. Org. Biomol. Chem. 2021, 19, 498–513. 10.1039/D0OB02098B. [DOI] [PubMed] [Google Scholar]
- Hammouda M. M.; Elattar K. M. Recent progress in the chemistry of β-aminoketones. RSC Adv. 2022, 12, 24681–24712. 10.1039/D2RA03864A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blough B. E.; Landavazo A.; Partilla J. S.; Baumann M. H.; Decker A. M.; Page K. M.; Rothman R. B. Hybrid Dopamine Uptake Blocker–Serotonin Releaser Ligands: A New Twist on Transporter-Focused Therapeutics. ACS Med. Chem. Lett. 2014, 5 (6), 623–627. 10.1021/ml500113s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer P. C.; Butler D.; Deschamps J. R.; Madras B. K. 1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (Pyrovalerone) Analogues: A Promising Class of Monoamine Uptake Inhibitors. J. Med. Chem. 2006, 49 (4), 1420–1432. 10.1021/jm050797a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers M. C.; Wang J.; Iera J. A.; Bang J.; Hara T.; Saito S.; Zambetti G. P.; Appella D. H. A New Family of Small Molecules To Probe the Reactivation of Mutant p53. J. Am. Chem. Soc. 2005, 127 (17), 6152–6153. 10.1021/ja045752y. [DOI] [PubMed] [Google Scholar]
- Foley K. F.; Cozzi N. V. Novel aminopropiophenones as potential antidepressants. Drug Dev. Res. 2003, 60 (4), 252–260. 10.1002/ddr.10297. [DOI] [Google Scholar]
- Kolanos R.; Partilla J. S.; Baumann M. H.; Hutsell B. A.; Banks M. L.; Negus S. S.; Glennon R. A. Stereoselective Actions of Methylenedioxypyrovalerone (MDPV) To Inhibit Dopamine and Norepinephrine Transporters and Facilitate Intracranial Self-Stimulation in Rats. ACS Chem. Neurosci. 2015, 6, 771–777. 10.1021/acschemneuro.5b00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F. F.; Zhang X.; Zhang G. H.; Xie D.; Chen P. Y.; Zhang W. R.; Jiang J. P.; Liang M.; Wang G. B.; Liu Z. R.; et al. Efficacy and Safety of Benazepril for Advanced Chronic Renal Insufficiency. N. Engl. J. Med. 2006, 354 (2), 131–140. 10.1056/NEJMoa053107. [DOI] [PubMed] [Google Scholar]
- Sham H. L.; Kempf D. J.; Molla A.; Marsh K. C.; Kumar G. N.; Chen C. M.; Kati W.; Stewart K.; Lal R.; Hsu A.; et al. ABT-378, a Highly Potent Inhibitor of the Human Immunodeficiency Virus Protease. Antimicrob. Agents Chemother. 1998, 42 (12), 3218–3224. 10.1128/aac.42.12.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y.; Li Q.; Xiong B.; Hui X.; Wang X.; Feng Y.; Meng T.; Hu D.; Zhang D.; Wang M. Aromatic β-amino-ketone derivatives as novel selective non-steroidal progesterone receptor antagonists. Bioorg. Med. Chem. 2010, 18 (12), 4255–4268. 10.1016/j.bmc.2010.04.092. [DOI] [PubMed] [Google Scholar]
- Makarova N.; Boreko E.; Moiseev I.; Pavlova N.; Zemtsova M.; Nikolaeva S.; Vladyko G. Antiviral Activity of Adamantyl-Containing β-Aminoketones, Enaminoketones, and Related Compounds. Pharm. Chem. J. 2001, 35 (9), 480–484. 10.1023/A:1014086507352. [DOI] [Google Scholar]
- Altmeyer M.; Amtmann E.; Heyl C.; Marschner A.; Scheidig A. J.; Klein C. D. Beta-aminoketones as prodrugs for selective irreversible inhibitors of type-1 methionine aminopeptidases. Bioorg. Med. Chem. Lett. 2014, 24, 5310–5314. 10.1016/j.bmcl.2014.09.047. [DOI] [PubMed] [Google Scholar]
- Perrine D. M.; Ross J. T.; Nervi S. J.; Zimmerman R. H. A Short, One-Pot Synthesis of Bupropion (Zyban, Wellbutrin). J. Chem. Educ. 2000, 77 (11), 1479. 10.1021/ed077p1479. [DOI] [Google Scholar]
- Silverstone T. Appetite Suppressants. Drugs 1992, 43, 820–836. 10.2165/00003495-199243060-00003. [DOI] [PubMed] [Google Scholar]
- Nchinda T.; Chibale K.; Redelinghuys P.; Sturrock E. D. Synthesis of novel keto-ACE analogues as domain-selective angiotensin I-converting enzyme inhibitors. Bioorg. Med. Chem. Lett. 2006, 16 (17), 4612–4615. 10.1016/j.bmcl.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Almquist R. G.; Chao W.-R.; Ellis M. E.; Johnson H. L. Synthesis and biological activity of a ketomethylene analog of a tripeptide inhibitor of angiotensin converting enzyme. J. Med. Chem. 1980, 23 (12), 1392–1398. 10.1021/jm00186a020. [DOI] [PubMed] [Google Scholar]
- Shan J.; Sun H. The discovery and development of prasugrel. Expert Opin. Drug Discovery 2013, 8 (7), 897–905. 10.1517/17460441.2013.793668. [DOI] [PubMed] [Google Scholar]
- Krechetov S.; Nifontova G.; Dolotova O.; Veselov M. Solubility and Stability of Proroxan at Various PH Values. Pharm. Chem. J. 2018, 52, 236–240. 10.1007/s11094-018-1798-1. [DOI] [Google Scholar]
- Nifontova G.; Krechetov S.; Dolotova O.; Buyukli S.; Akhmetzyanova A.; Krasnyuk I. Granulation of Effervescent Ingredients for Optimization of Gastroretentive Properties of Floating Proroxan Prolonged-Release Tablets. Pharm. Chem. J. 2018, 52, 361–365. 10.1007/s11094-018-1822-5. [DOI] [Google Scholar]
- Hofer D.; Lohberger B.; Steinecker B.; Schmidt K.; Quasthoff S.; Schreibmayer W. A. A comparative study of the action of tolperisone on seven different voltage dependent sodium channel isoforms. Eur. J. Pharmacol. 2006, 538, 5–14. 10.1016/j.ejphar.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Kaski J. C.; Araujo L.; Maseri A. Effects of oxyfedrine on regional myocardial blood flow in patients with coronary artery disease. Cardiovasc. Drugs Ther. 1991, 5, 991–996. 10.1007/BF00143526. [DOI] [PubMed] [Google Scholar]
- Concellón J. M.; Rodríguez-Solla H. Synthesis and Synthetic Applications of α-Amino Ketones Derived from Natural α-Amino Acids. Curr. Org. Chem. 2008, 12 (7), 524–543. 10.2174/138527208784245996. [DOI] [Google Scholar]
- Nguyen N. H.; Hughes A. B.; Sleebs B. E. Stereoselective Synthesis and Application of β-Amino Ketones. Curr. Org. Chem. 2014, 18 (2), 260–289. 10.2174/138527281802140129104218. [DOI] [Google Scholar]
- Deacon C. F. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 642–653. 10.1038/s41574-020-0399-8. [DOI] [PubMed] [Google Scholar]
- Langer P.; Bodtke A. Sequential cyclizations of 2-isothiocyanatobenzonitrile and 2-isocyanatobenzonitrile with α-aminoketones. Tetrahedron Lett. 2003, 44 (32), 5965–5967. 10.1016/S0040-4039(03)01489-8. [DOI] [Google Scholar]
- Sorrell T. N.; Allen W. E. A regiospecific synthesis of 1,4-disubstituted imidazoles. J. Org. Chem. 1994, 59 (6), 1589–1590. 10.1021/jo00085a056. [DOI] [Google Scholar]
- Frantz D. E.; Morency L.; Soheili A.; Murry J. A.; Grabowski E. J. J.; Tillyer R. D. Synthesis of Substituted Imidazoles via Organocatalysis. Org. Lett. 2004, 6 (5), 843–846. 10.1021/ol0498803. [DOI] [PubMed] [Google Scholar]
- Huang J.; Luo L.; Xing N.; Gu L.; Li C.; Han Q.; Zheng S.; He L. Novel synthesis of divergent aryl imidazoles from ketones involving copper-catalyzed α-amination and oxidative C–C bond cleavage. RSC Adv. 2020, 10, 13815–13819. 10.1039/D0RA01408G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida H.; Hayashida K.; Yamada M.; Takahashi K.; Yamada K. Synthesis of 2,5-Dimethylpyrazine-3,6-Dicarboxylic Acid Derivatives. Synth. Commun. 1973, 3 (3), 225–230. 10.1080/00397917308062041. [DOI] [Google Scholar]
- Chiba T.; Sakagami H.; Murata M.; Okimoto M. Electrolytic Oxidation of Ketones in Ammoniacal Methanol in the Presence of Catalytic Amounts of KI. J. Org. Chem. 1995, 60 (21), 6764–6770. 10.1021/jo00126a027. [DOI] [Google Scholar]
- Knorr L. Synthese von Pyrrolderivaten. Ber. Dtsch. Chem. Ges. 1884, 17 (2), 1635–1642. 10.1002/cber.18840170220. [DOI] [Google Scholar]
- Knorr L. Synthetische Versuche mit dem Acetessigester. II. Mittheilung: Ueberführung des Diacetbernsteinsäureesters und des Acetessigesters in Pyrrolderivate. Adv. Cycloaddit. 1886, 236 (3), 290–332. 10.1002/jlac.18862360303. [DOI] [Google Scholar]
- Klingler F. D. Asymmetric Hydrogenation of Prochiral Amino Ketones to Amino Alcohols for Pharmaceutical Use. Acc. Chem. Res. 2007, 40 (12), 1367–1376. 10.1021/ar700100e. [DOI] [PubMed] [Google Scholar]
- Gediya S. K.; Clarkson G. J.; Wills M. Asymmetric Transfer Hydrogenation: Dynamic Kinetic Resolution of α-Amino Ketones. J. Org. Chem. 2020, 85 (17), 11309–11330. 10.1021/acs.joc.0c01438. [DOI] [PubMed] [Google Scholar]
- Wu W.; You C.; Yin C.; Liu Y.; Dong X. Q.; Zhang X. Enantioselective and Diastereoselective Construction of Chiral Amino Alcohols by Iridium–f-Amphox-Catalyzed Asymmetric Hydrogenation via Dynamic Kinetic Resolution. Org. Lett. 2017, 19 (10), 2548–2551. 10.1021/acs.orglett.7b00844. [DOI] [PubMed] [Google Scholar]
- Ager D. J.; Prakash I.; Schaad D. R. 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev. 1996, 96 (2), 835–876. 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]
- Bartoli G.; Cimarelli C.; Palmieri G. Convenient procedure for the reduction of β-enamino ketones: synthesis of γ-amino alcohols and tetrahydro-1,3-oxazines. J. Chem. Soc., Perkin Trans. 1994, 1 (5), 537–543. 10.1039/P19940000537. [DOI] [Google Scholar]
- Bates R. W.; Sa-Ei K. Syntheses of the sedum and related alkaloids. Tetrahedron 2002, 58 (30), 5957–5978. 10.1016/S0040-4020(02)00584-7. [DOI] [Google Scholar]
- Davis F. A.; Prasad K. R.; Nolt M. B.; Wu Y. N-Sulfinyl β-Amino Weinreb Amides: Synthesis of Enantiopure β-Amino Carbonyl Compounds. Asymmetric Synthesis of (+)-Sedridine and (−)-Allosedridine. Org. Lett. 2003, 5 (6), 925–927. 10.1021/ol034119z. [DOI] [PubMed] [Google Scholar]
- Jefford C. W.; Wang J. B. An enantiospecific synthesis of solenopsin A. Tetrahedron Lett. 1993, 34 (18), 2911–2914. 10.1016/S0040-4039(00)60479-3. [DOI] [Google Scholar]
- Keck G. E.; Truong A. P. Directed Reduction of β-Amino Ketones to Syn or Anti 1,3-Amino Alcohol Derivatives. Org. Lett. 2002, 4 (18), 3131–3134. 10.1021/ol026456y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner K. T.; Höfner G. Chelat- und nicht-chelat-kontrollierte reduktionen von β-amido-ketonen: Synthese nicht-racemischer 1,3-aminoalkohole mit pyrrolidinstruktur. Tetrahedron 1991, 47 (10), 1895–1910. 10.1016/S0040-4020(01)96102-2. [DOI] [Google Scholar]
- Barluenga J.; Tomas M.; Kouznetsov V.; Jardon J.; Rubio E. Reduction of the Pyrimidine Ring: Regio- and Stereoselective Synthesis of 1,3-Diamine Derivatives. Synlett 1991, 1991 (11), 821–823. 10.1055/s-1991-20890. [DOI] [Google Scholar]
- Couty F.; David O.; Durrat F.; Evano G.; Lakhdar S.; Marrot J.; Vargas-Sanchez M. Nucleophilic Ring-Opening of Azetidinium Ions: Insights into Regioselectivity. Eur. J. Org Chem. 2006, 2006 (15), 3479–3490. 10.1002/ejoc.200600200. [DOI] [Google Scholar]
- Shustov G.; Denisenko S.; Chervin I.; Asfandiarov N.; Kostyanovsky R. Asymmetric nitrogen—41: Stereochemistry of bicyclic 1,2-cis-diaziridines. Tetrahedron 1985, 41 (23), 5719–5731. 10.1016/S0040-4020(01)91378-X. [DOI] [Google Scholar]
- Colyer J. T.; Andersen N. G.; Tedrow J. S.; Soukup T. S.; Faul M. M. Reversal of Diastereofacial Selectivity in Hydride Reductions of N-tert-Butanesulfinyl Imines. J. Org. Chem. 2006, 71 (18), 6859–6862. 10.1021/jo0609834. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Weber T.; Piotrowski D. W. Organocerium additions to SAMP-hydrazones: general synthesis of chiral amines. J. Am. Chem. Soc. 1987, 109 (7), 2224–2225. 10.1021/ja00241a073. [DOI] [Google Scholar]
- González-Sabín J.; Gotor V.; Rebolledo F. CAL-B-catalyzed resolution of some pharmacologically interesting β–substituted isopropylamines. Tetrahedron: Asymmetry 2002, 13 (12), 1315–1320. 10.1016/S0957-4166(02)00336-1. [DOI] [Google Scholar]
- Nechab M.; Azzi N.; Vanthuyne N.; Bertrand M.; Gastaldi S.; Gil G. Highly Selective Enzymatic Kinetic Resolution of Primary Amines at 80 °C: A Comparative Study of Carboxylic Acids and Their Ethyl Esters as Acyl Donors. J. Org. Chem. 2007, 72 (18), 6918–6923. 10.1021/jo071069t. [DOI] [PubMed] [Google Scholar]
- C Cole D. Recent stereoselective synthetic approaches to β-amino acids. Tetrahedron 1994, 50 (32), 9517–9582. 10.1016/S0040-4020(01)85527-7. [DOI] [Google Scholar]
- Liu M.; Sibi M. P. Recent advances in the stereoselective synthesis of β-amino acids. Tetrahedron 2002, 58 (40), 7991–8035. 10.1016/S0040-4020(02)00991-2. [DOI] [Google Scholar]
- Davis F. A.; Song M.; Qiu H.; Chai J. Total synthesis of (5R,6R,8R,9S)-(−)-5,9Z-indolizidine 221T using sulfinimine-derived N-sulfinyl β-amino ketones. Org. Biomol. Chem. 2009, 7 (24), 5067–5073. 10.1039/b915796d. [DOI] [PubMed] [Google Scholar]
- Klix R. C.; Chamberlin S. A.; Bhatia A. V.; Davis D. A.; Hayes T. K.; Rojas F. G.; Koops R. W. A practical, large-scale procedure for the preparation of N-protected α-amino ketones from α-amino acids. Tetrahedron Lett. 1995, 36 (11), 1791–1794. 10.1016/0040-4039(95)00144-2. [DOI] [Google Scholar]
- Florjancic A.; Sheppard G. S. A Practical Synthesis of α-Amino Ketones via Aryllithium Addition to N-Boc-α-Amino Acids. Synthesis 2003, 11, 1653–1656. 10.1055/s-2003-40875. [DOI] [Google Scholar]
- De Luca L.; Giacomelli G.; Porcheddu A. A. Simple Preparation of Ketones. N-Protected α-Amino Ketones from α-Amino Acids. Org. Lett. 2001, 3, 1519–1521. 10.1021/ol015840c. [DOI] [PubMed] [Google Scholar]
- Zhou Z. H.; Tang Y. L.; Li K. Y.; Liu B.; Tang C. C. Synthesis of optically active N-protected α-aminoketones and α-amino alcohols. Heteroat. Chem. 2003, 14, 603–606. 10.1002/hc.10195. [DOI] [Google Scholar]
- Lv Y.; Li Y.; Xiong T.; Lu Y.; Liu Q.; Zhang Q. nBu4NI-Catalyzed oxidative imidation of ketones with imides: synthesis of α-amino ketones. Chem. Commun. 2014, 50, 2367–2369. 10.1039/c3cc48887j. [DOI] [PubMed] [Google Scholar]
- Buckley T. F. III; Rapoport H. .alpha.-Amino acids as chiral educts for asymmetric products. Amino acylation with N-acylamino acids. J. Am. Chem. Soc. 1981, 103 (20), 6157–6163. 10.1021/ja00410a030. [DOI] [Google Scholar]
- McClure D. E.; Arison B. H.; Jones J. H.; Baldwin J. J. Chiral.alpha.-amino ketones from the Friedel-Crafts reaction of protected amino acids. J. Org. Chem. 1981, 46 (11), 2431–2433. 10.1021/jo00324a057. [DOI] [Google Scholar]
- McClure D. E.; Lumma P. K.; Arison B. H.; Jones J. H.; Baldwin J. J. 1,4-Oxazines via intramolecular ring closure of.beta.-hydroxydiazoacetamides: phenylalanine to tetrahydroindeno[1,2-b]-1,4-oxazin-3(2H)-ones. J. Org. Chem. 1983, 48 (16), 2675–2679. 10.1021/jo00164a009. [DOI] [Google Scholar]
- Nordlander J. E.; Payne M. J.; Njoroge F. G.; Balk M. A.; Laikos G. D.; Vishwanath V. M. Friedel-Crafts acylation with N-(trifluoroacetyl)-.alpha.-amino acid chlorides. Application to the preparation of.beta.-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines. J. Org. Chem. 1984, 49 (22), 4107–4111. 10.1021/jo00196a001. [DOI] [Google Scholar]
- Nordlander J. E.; Njoroge F. G.; Payne M. J.; Warman D. N-(Trifluoroacetyl)-.alpha.-amino acid chlorides as chiral reagents for Friedel-Crafts synthesis. J. Org. Chem. 1985, 50 (19), 3481–3484. 10.1021/jo00219a012. [DOI] [Google Scholar]
- Itoh O.; Honnami T.; Amano A.; Murata K.; Koichi Y.; Sugita T. Friedel-Crafts.alpha.-aminoacylation of alkylbenzene with a chiral N-carboxy-.alpha.-amino acid anhydride without loss of chirality. J. Org. Chem. 1992, 57 (26), 7334–7338. 10.1021/jo00052a059. [DOI] [Google Scholar]
- Di Gioia M. L.; Leggio A.; Liguori A.; Napoli A.; Siciliano C.; Sindona G. Facile Approach to Enantiomerically Pure α-Amino Ketones by Friedel–Crafts Aminoacylation and Their Conversion into Peptidyl Ketones. J. Org. Chem. 2001, 66 (21), 7002–7007. 10.1021/jo010414q. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Jiang R.; Suzuki K. N-Tfa- and N-Fmoc-(α-aminoacyl)benzotriazoles as Chiral C-Acylating Reagents under Friedel–Crafts Reaction Conditions. J. Org. Chem. 2005, 70 (13), 4993–5000. 10.1021/jo050226q. [DOI] [PubMed] [Google Scholar]
- Plażuk D.; Zakrzewski J. Acylation of Ferrocene and a 1,1′-Diphosphaferrocene with Acyl Trifluoroacetates in the Presence of Trifluoromethanesulfonic (Triflic) Acid or Some Metal Triflates. Synth. Commun. 2004, 34 (1), 99–107. 10.1081/SCC-120027243. [DOI] [Google Scholar]
- Plażuk D.; Zakrzewski J. Friedel–Crafts acylation of ferrocene with alkynoic acids. J. Organomet. Chem. 2009, 694 (12), 1802–1806. 10.1016/j.jorganchem.2009.01.007. [DOI] [Google Scholar]
- Plażuk D.; Zakrzewski J.; Salmain M. Biotin as acylating agent in the Friedel–Crafts reaction. Avidin affinity of biotinyl derivatives of ferrocene, ruthenocene and pyrene and fluorescence properties of 1-biotinylpyrene. Org. Biomol. Chem. 2011, 9, 408–417. 10.1039/C0OB00319K. [DOI] [PubMed] [Google Scholar]
- Plażuk D.; Zakrzewski J.; Salmain M.; Błauż A.; Rychlik B.; Strzelczyk P.; Bujacz A.; Bujacz G. Ferrocene–Biotin Conjugates Targeting Cancer Cells: Synthesis, Interaction with Avidin, Cytotoxic Properties and the Crystal Structure of the Complex of Avidin with a Biotin–Linker–Ferrocene Conjugate. Organometallics 2013, 32 (20), 5774–5783. 10.1021/om4003126. [DOI] [Google Scholar]
- Murai Y.; Wang L.; Muto Y.; Sakihama Y.; Hashidoko Y.; Hatanaka Y.; Hashimoto M. Simple and Stereocontrolled Preparation of Benzoylated Phenylalanine Using Friedel–Crafts Reaction in Trifluoromethanesulfonic Acid for Photoaffinity Labeling. Heterocycles 2013, 87 (10), 2119–2126. 10.3987/COM-13-12815. [DOI] [Google Scholar]
- Murashige R.; Hayashi Y.; Ohmori S.; Torii A.; Aizu Y.; Muto Y.; Murai Y.; Oda Y.; Hashimoto M. Comparisons of O-acylation and Friedel–Crafts acylation of phenols and acyl chlorides and Fries rearrangement of phenyl esters in trifluoromethanesulfonic acid: effective synthesis of optically active homotyrosines. Tetrahedron 2011, 67 (3), 641–649. 10.1016/j.tet.2010.11.047. [DOI] [Google Scholar]
- Murai Y.; Hashidoko Y.; Hashimoto M. Novel Synthesis of Optically Active Bishomotyrosine Derivatives Using the Friedel-Crafts Reaction in Triflic Acid. Biosci., Biotechnol., Biochem. 2011, 75 (2), 352–354. 10.1271/bbb.100595. [DOI] [PubMed] [Google Scholar]
- Wuts P. G. M.Greene’s Protective Groups in Organic Synthesis, 5th ed.; John Wiley & Sons, Inc., Hoboken, NJ, 2014; pp 993–994 [Google Scholar]
- Carpino L. A. The 9-fluorenylmethyloxycarbonyl family of base-sensitive amino-protecting groups. Acc. Chem. Res. 1987, 20 (11), 401–407. 10.1021/ar00143a003. [DOI] [Google Scholar]
- Anderson K. W.; Tepe J. J. Trifluoromethanesulfonic acid catalyzed Friedel–Crafts acylation of aromatics with β-lactams. Tetrahedron 2002, 58 (42), 8475–8481. 10.1016/S0040-4020(02)01026-8. [DOI] [Google Scholar]
- Newman H. Trifluoroacetyl as a Protecting Group for 1-Halo Sugars. J. Org. Chem. 1965, 30 (4), 1287–1288. 10.1021/jo01015a533. [DOI] [Google Scholar]
- Boger D. L.; Yohannes D. Total synthesis of K-13. J. Org. Chem. 1989, 54 (11), 2498–2502. 10.1021/jo00272a003. [DOI] [Google Scholar]
- Tarbell D. S.; Yamamoto Y.; Pope B. M. New Method to Prepare N-t-Butoxycarbonyl Derivatives and the Corresponding Sulfur Analogs from di-t-Butyl Dicarbonate or di-t-Butyl Dithiol Dicarbonates and Amino Acids. Proc. Natl. Acad. Sci. U.S.A. 1972, 69 (3), 730–732. 10.1073/pnas.69.3.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl G. L.; Walter R.; Smith C. W. General procedure for the synthesis of mono-N-acylated 1,6-diaminohexanes. J. Org. Chem. 1978, 43 (11), 2285–2286. 10.1021/jo00405a045. [DOI] [Google Scholar]
- Martin C. L.; Nakamura S.; Otte R.; Overman L. E. Total Synthesis of (+)-Condylocarpine, (+)-Isocondylocarpine, and (+)-Tubotaiwine. Org. Lett. 2011, 13 (1), 138–141. 10.1021/ol102709s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter M. C.; MacDonald J. C.; Bernstein J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. 1990, 46, 256–262. 10.1107/S0108768189012929. [DOI] [PubMed] [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29 (9), 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- Dales N.; Zhang Z.; Kamboj R.; Fu J.; Sun S.; Pokrovskaia N.; Sviridov S.. Heterocyclic organic compounds. WO 2008036715 A1, 2008.
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. SHELXT – Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information The crystallographic data are openly available in CCDC at https://www.ccdc.cam.ac.uk/structures/.