

Abstract
Polyethylene terephthalate (PET) is one of the most used polymers in the packaging industry; enzymatic recycling is emerging as a sustainable strategy to deal with waste PET, producing the virgin monomers terephthalic acid and ethylene glycol (EG). These monomers can be feedstocks for further microbial transformations. While EG metabolism has been uncovered in bacteria, in yeast the pathway for the oxidation to glycolic acid (GA) has only been proposed, but never experimentally elucidated. In this work, we investigated in Saccharomyces cerevisiae the potential contribution to this metabolism of two endogenous genes, YLL056C (a putative alcohol dehydrogenase) and GOR1 (glyoxylate reductase). Secondly, the possible role of alcohol dehydrogenases (ADHs) was considered, too. Finally, two heterologous genes (gox0313 from Gluconobacter oxydans and AOX1 from Komagataella phaffii) were expressed with the intent to push EG oxidation toward GA. Our main findings revealed that (i) Gor1, Yll056c, and ADHs are not involved in EG oxidation and (ii) the bottleneck of the catabolism is the first step in the pathway, due to the endogenous mechanisms for aldehyde detoxification. Multiomics studies are required to completely elucidate the pathway for EG catabolism, while further engineering directed toward relieving the bottleneck is needed to fully unleash the potential of yeasts for the upcycling of EG to GA.
Keywords: Saccharomyces cerevisiae, glycolic acid, ethylene glycol, metabolic engineering, synthetic biology, glycolaldehyde
This study aimed at exploring ethylene glycol metabolism in Saccharomyces cerevisiae and to improve its conversion into glycolic acid by metabolic engineering, uncovering the first reaction as the bottleneck of the pathway.
Introduction
After polyethylene (LDPE, LLDPE, and HDPE) and polypropylene, polyethylene terephthalate (PET) is the most used polymer in the packaging sector, this being also its major application (Nisticò 2020): PET is mostly used for the production of bottle for beverages, films, and in the food industry in general. As plastic packaging is generally single-use or for short-term usage, huge amounts of PET waste are generated daily, contributing to 8% by weight (12% by volume) of the world’s solid waste (Conroy and Zhang 2024).
Because of its properties, PET is—and will remain—one of the main polymers employed in the packaging industry (Nisticò 2020), thus PET waste management is fundamental to deal with these issues and to make the industry more sustainable. While PET has a relatively higher recycling rate with respect to other plastics, the main recycling techniques suffer from limitations: mechanical recycling—the most common one—yields a lower quality end product, and relies heavily on the quality of the waste material; chemical recycling, on the other hand, is more complex and costly, posing challenges also in terms of scalability and sustainability (Muringayil Joseph et al. 2024). Recently, biorecycling (or enzymatic recycling) of PET approaches have been studied, and the best example is from the French company Carbios, which already started building a biorecycling plant with a 50k tons PET feedstock capacity in Longlaville (FR) (Tournier et al. 2020, Carbios 2024). Biological upcycling is an emerging approach to make the PET recycling process more economically viable as, in addition to the possibility of repolymerizing. Microorganisms can convert the PET monomers obtained from the enzymatic hydrolysis—terephthalic acid and ethylene glycol (EG)—into higher added-value products; an example of this approach is the upcycling of EG to glycolic acid (GA), a two-carbon α-hydroxy acid used in the textile, food, cosmetic, and pharmaceutical industries currently produced from fossil-based formaldehyde (Salusjärvi et al. 2019).
In the last years, a few microorganisms able to metabolize EG have been described and engineered; notably, the vast majority of the studies available in literature focus on bacteria, namely Pseudomonas putida, Gluconobacter oxydans, Acetobacterium woodii, and Escherichia coli, which are either natively able to grow on EG, or are easily engineered to relieve the bottlenecks for EG use as a carbon source (Mückschel et al. 2012, Trifunović et al. 2016, Franden et al. 2018, Panda et al. 2021, Yu et al. 2023, Yan et al. 2024, Zhang et al. 2016); GA is produced as a by-product by some of the abovementioned bacteria, sometimes in particularly high concentrations. Pseudomonas putida, G. oxydans, and the engineered strains of E. coli are able to metabolize EG by a series of oxidation reactions, to produce glyoxylate (GOX) (Fig. 1). For assimilation, two molecules of GOX are condensed to tartronate semialdehyde by glyoxylate carboligase (Gcl), with the release of one molecule of CO2; ultimately, tartronate semialdehyde is metabolized to pyruvate, which can be used for growth (Mückschel et al. 2012, Yan et al. 2024, Zhang et al. 2016). Bacteria lacking a functional Gcl, such as P. putida KT2440, can only oxidize GOX via the glyoxylate shunt, which can only generate energy (Franden et al. 2018). The key step in the pathway is the oxidation of EG to glycolaldehyde (GAH): P. putida and G. oxydans both express a series of pyrroloquinoline quinone (PQQ)-dependent alcohol dehydrogenases (ADH; PedE and PedH); G. oxydans also expresses a NAD+-dependent ADH, Gox0313, reported to be active on EG (Mückschel et al. 2012, Zhang et al. 2015); a similar ability has been reported for E. coli FucO and YqhD (Panda et al. 2021).
Figure 1.

Bacterial metabolic pathways for the metabolism of EG. Two metabolic pathways have been described in bacteria for EG metabolism. Acetobacterium woodii is able to assimilate EG by dehydration to acetaldehyde, subsequently disproportionated to ethanol and acetate (arrows in green); the first reaction is strongly oxygen-sensitive. Pseudomonas putida and G. oxydans metabolize EG via a series of oxidation reactions, producing GAH, GA, and GOX. Depending on the host metabolic machinery, EG can be assimilated in the biomass by condensation of two molecules of GOX in tartronate semialdehyde by Gcl and subsequent conversion to pyruvate; alternatively, GOX is dissimilated to CO2 via the GOX cycle, either by condensation to succinate (SUC) by isocitrate lyase, or to acetyl-CoA by malate synthase (arrows in blue). Since both cycles require the production of two molecules of CO2, all the carbon atoms from EG are ultimately respired, hence this pathway can only generate energy. See main text for further information.
Acetobacterium woodii, on the other hand, is able to assimilate EG by a first dehydration to acetaldehyde, which is then disproportionated to ethanol and acetate; the dehydration reaction is catalyzed by a very oxygen-sensitive diol dehydratase (PduCDE) (Trifunović et al. 2016). Figure 1 shows a representation of the bacterial EG metabolic pathways described above.
While many studies focusing on EG metabolism in bacteria are present in literature, only three works delve on EG metabolism by yeasts (Kataoka et al. 2001, Senatore et al. 2024, Carniel et al. 2023) and all report the ability of yeasts to oxidize EG to GA; this is most probably due to the activity of nonspecific ADHs, and a putative pathway for Saccharomyces cerevisiae has been proposed, but not yet proven (Senatore et al. 2024). Despite the scarce knowledge, yeasts are interesting cell factories for the bioconversion of EG to GA, since they lack the key enzyme for assimilation into biomass, Gcl, and the dissimilation of EG to CO2 is only partial, as the yields of GA on consumed EG are relatively high both in Yarrowia lipolytica, S. cerevisiae, Scheffersomyces stipitis, and Cutaneotrichosporon oleaginosus (Carniel et al. 2023, Senatore et al. 2024). Moreover, compared to bacteria, yeasts seem to be more tolerant to EG: Y. lipolytica can tolerate up to 2 M EG, while 0.3 M EG inhibits its conversion into GA in G. oxydans (Carniel et al. 2023, Zhang et al. 2016). Yeasts also seems to be more tolerant to GA, for instance S. cerevisiae can grow in the presence of up to 50 g l−1 GA, while in E. coli the growth is already inhibited when GA concentration is around 4 g l−1 (Koivistoinen et al. 2013, Yan et al. 2024).
Given the general lack of knowledge about EG metabolism in yeast, and the potential to valorize it into GA, we decided to explore wild type genes that should be responsible for the oxidation of EG to GA by deletion and constitutive expression, as well as to express two heterologous genes, gox0313 from G. oxydans and AOX1 from Komagataella phaffii. For ease of manipulation and availability of genomic data, we focused our study on the well-known S. cerevisiae.
Multiple reactions are depicted with two arrows on the same line; green arrows refer to the A. woodii pathway; blue arrows highlight the dissimilation pathway; only relevant cofactors are shown. PQQ, pyrroloquinoline quinone; CIT, citrate; ICIT, isocitrate; αKG, α-ketoglutarate; SUC-CoA, succinyl-CoA; SUC, succinate; FUM, fumarate; MAL, malate; and OXA, oxaloacetate. Adapted from Franden et al. (2018) and Trifunović et al. (2016).
Materials and methods
Strains
The S. cerevisiae parental strain used in this study was CEN.PK 113–7D (MATa; TRP1; MAL2-8c; SUC2–Dr. P. Kötter, Institute of Microbiology, Johann Wolfgang Goethe-University, Frankfurt, Germany) (van Dijken et al. 2000). All S. cerevisiae strains obtained in this work are described in the ‘Results and Discussion’ section and listed in Table 1.
Table 1.
Strains used in this study.
| Strain | Parental strain | Genotype |
|---|---|---|
| CEN.PK 113–7D | – | MATa; TRP1; MAL2-8c; SUC2 |
| deltaY | CEN.PK 113–7D | CEN.PK 113–7D; yll056cΔ |
| overY | deltaY | deltaY; X-4::pPGK1-YLL056C-tCYC1 |
| deltaG | CEN.PK 113–7D | CEN.PK 113–7D; gor1Δ |
| CER.GOX | CEN.PK 113–7D | CEN.PK 113–7D; X-2::pPGK1-gox0313-tCYC1 |
| CER.GA | CEN.PK 113–7D | CEN.PK 113–7D; X-2::tADH1-aldA-pTPI1-pPGK1-gox0313-tCYC1 |
| CER.AOX | CEN.PK 113–7D | CEN.PK 113–7D; X-2::tADH1-AOX1-pTPI1-pPGK1-CTT1-tCYC1 |
| CER.AOX_GFP | CEN.PK 113–7D | CEN.PK 113–7D; X-2::tADH1-GFP-AOX1-pTPI1-pPGK1-CTT1-tCYC2 |
Escherichia coli strain DH5α was used to clone, propagate, and store the plasmids.
Media composition
Saccharomyces cerevisiae strains were maintained in 20% (v/v) glycerol at −80°C after growth in YPD medium composed of (per liter): yeast extract 10 g, tryptone 20 g, and glucose 20 g.
When specified, EG was added to YPD at a final concentration of 150 mM, that is 9.3 g l−1 (YPD + EG). When needed, YPD was supplemented with the antibiotic G418 (200 mg l−1); agar plates were prepared with the addition of 20 g l−1 agar to the liquid media.
YP + EG medium was composed of (per liter): yeast extract 10 g, tryptone 20 g, and EG 150 mmol (9.3 g).
Defined synthetic medium base (Delft base) was composed of (per liter): (NH4)2SO4 5 g; KH2PO4 3 g; MgSO4·7H2O 0.5 g; trace elements 1X (EDTA 30 mg; ZnSO4·7H2O 9 mg; CoCl2·6H2O 0.6 mg; MnCl2·4H2O 2 mg; CuSO4·5H2O 0.6 mg; CaCl2·2H2O 9 mg; FeSO4·7H2O 6 mg; Na2MoO4·2H2O 0.8 mg; H3BO3 2 mg; and KI 0.2 mg); vitamins 1X (d-biotin 0.10 mg; calcium d-pantothenate 2 mg; nicotinic acid 2 mg; myo-inositol 50 mg; thiamine hydrochloride 2 mg; pyridoxal hydrochloride 2 mg; and para-aminobenzoic acid 0.4 mg) (Verduyn et al. 1992).
For S. cerevisiae strains cultivations, Delft base was supplemented with glucose 20 g l−1 as carbon source (Delft Glc) and GAH, as described below.
Yeast extract was purchased from Biolife Italia S.r.l., Milan, Italy. All other reagents were purchased from Merck Life Science S.r.l., Milan, Italy.
Growth conditions in shake flasks
Growth conditions in 100 ml shake flasks
The ability to oxidize EG to GA of the S. cerevisiae strains was assayed in 100 ml shake flasks, in YP + EG medium; the wild type strain was assayed as a reference condition.
Seed cultures from YPD plates were grown in 50 ml glass tubes filled with 10 ml YP for 24 h; cells were then harvested, washed with dH2O, and inoculated in shake flasks (final OD = 0.25) filled with 20 ml of YP + EG. All growths were performed in a rotary shaker at 160 rpm and 30°C. Samples were collected at regular time intervals for OD measurement and HPLC analysis.
Growth conditions in 250 ml shake flasks
EG metabolism in the presence of glucose was studied with S. cerevisiae strains in 250 ml shake flasks, in YPD and YPD + EG media.
Seed cultures from YPD plates were grown in glass tubes filled with 2 ml YPD for 8 h; cells were then inoculated for the intermediate inoculum (starting OD = 0.01) in 50 ml glass tubes containing 10 ml of YPD, and grown for 16 h. Cells were then harvested, washed with dH2O and inoculated in shake flasks (final OD = 0.5) filled with 50 ml of medium under investigation. All growths were performed in a rotary shaker at 160 rpm and 30°C. Samples were collected at regular time intervals for OD and HPLC analysis.
Growth conditions in Growth Profiler (96-deepwell microplates)
Assessment of GAH toxicity
To assess GAH toxicity, S. cerevisiae strains were grown in Delft Glc with the addition of GAH at a final concentration of 0, 5, 10, or 20 mM, from a 1 M stock solution. The 1 M GAH solution was obtained by dissolving 0.301 g of GAH dimer in 5 ml of demineralized water, obtaining a 0.5 M solution of GAH dimer (Jayakody et al. 2018); prior to addition to the medium, the GAH dimer solution was heated (10 min at 65°C) to produce the GAH monomer, as described by Anderson et al. (2007).
Seed cultures from YPD plates were grown in a 24-deepwell microplate (CR1424, manufactured by Enzyscreen, Heemstede, The Netherlands) filled with 2 ml YPD for 24 h, until saturation. Cells were then harvested, washed with dH2O and inoculated in 96-deepwell microplate (CR1496dg, manufactured by Enzyscreen) at a final OD of 0.1, with each well filled with 250 µl of medium. Growth was performed at 30°C under constant agitation (250 rpm) in the Growth Profiler 960 (Enzyscreen). HPLC samples were collected at the end of the experiment (72 h).
Maximum specific growth rate and lag phase duration were calculated with a custom R script.
Design of plasmids and constructs
The Easy-MISE toolkit (Addgene Kit number 1000000230) (Maestroni et al. 2023a) was utilized for the preparation of all the necessary plasmids and constructs. Briefly, pEM plasmids (level 0) carrying the codon-optimized version of the genes of interest (pEM.YLL056C, pEM.gox, pEM.aldA, pEM.AOX, and pEM.CTT) and the homology regions for the locus X-2 (pEM.H07L and pEM.H07R) (Table S1) were built by a Golden Gate Assembly reaction as recommended in the Easy-MISE protocol; subsequently, a second GGA reaction with the newly built pEM plasmids, pGA-red-Maxi and the pEM plasmids from the ready-to-use library (homology regions, promoters, terminators, adaptors, and fluorescent proteins genes) was performed to build (double) transcription units in the level 1 plasmids, obtaining plasmids pOverY, pGA, pAOX, and pAOX_GFP (Table S2). The specific genotypes of each strain are reported in Table 1.
Sequences for open reading frames of gox0313 from G. oxydans, aldA from E. coli, and AOX1 from K. phaffii were translated into DNA sequences, codon-optimized for S. cerevisiae and synthesized by Twist Bioscience; CTT1 from S. cerevisiae was domesticated for Esp3I, BsaI, and NheI sites and synthesized by Twist Bioscience; the DNA sequence of YLL056C was amplified directly from genomic DNA of CEN.PK 113–7D by PCR (Table S1). All synthetic sequences used in this study are listed in Table S3.
Yeast transformation
Yeast transformants were obtained by exploiting the pCEC-red plasmid (Addgene kit number 196040) (Maestroni et al. 2023b) and the constructs obtained with the Easy-MISE toolkit. The 20mer sequence for the integration locus X-2 was obtained from the EasyClone-MarkerFree vector set, a gift from Irina Borodina (Addgene kit number 1000000098); the 20mer sequences targeting the YLL056C and GOR1 loci were designed with Benchling (https://www.benchling.com/crispr); the gRNA sequences used are listed in Table S4. The pCEC-gRNA plasmids (pCEC-X2, pCEC-YLL056C, and pCEC-GOR1) were constructed as described in the published protocol and are listed in Table S5. The repair fragments for the deletion of YLL056C (RF_YLL056C) and GOR1 (RF_GOR1) were constructed by amplifying homology regions about 500 bp upstream and downstream each gene with BsaI recognition sites and the correct protruding ends to form the final repair fragment into pGA-red-Maxi acceptor vector obtaining plasmids pRF_YLL056C and pRF_GOR1 (Tables S4 and S5), as described in the pCEC-red protocol.
All yeast transformations were performed following the pCEC-red protocol. Correct integration of the vectors into the genome or gene deletion was verified by colony PCR.
Colony PCRs
To perform colony PCRs, at least five different E. coli colonies were picked for each transformation plate and dissolved (i) in 20 µl of growth media with the proper antibiotic as a colony backup and (ii) into the PCR tube with the appropriate PCR mix. To boost cell disruption, the initial denaturation step must last at least 5 min. The positive E. coli clones were then inoculated starting from the 20 µl liquid cultures prepared at the beginning.
To perform colony PCRs on S. cerevisiae colonies, genomic DNA was extracted following the LiOAc-SDS optimized procedure of Lõoke et al. (2011). After obtaining the genomic DNA, 1 µl of the supernatant was used as the PCR template. The positive clones were then inoculated in the correct growth media.
OneTaq® 2X Master Mix (New England Biolabs) was used on a ProFlex PCR System (Life Technologies) to perform colony PCR reactions.
RNA preparation and quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) assays were performed to measure the expression levels of GOR1, YLL056C, GRE2, ADH1, ADH2, and ADH6 in the presence of EG, during the glucose consumption phase (Glc), after the diauxic shift (EtOH) and after the depletion of ethanol and acetate, when EG is consumed (EG), in the wild-type (wt) strain. To avoid problems with RNA extraction from old cells, a short fermentation was simulated, by growing the cells in Delft base supplemented with glucose 1 g l−1 and EG 150 mM (9.3 g l−1). This experimental set up allowed to monitor the three growth phases during the working day (< 8 h) and ensured efficient RNA extraction.
Seed cultures from YPD plates were grown in 2000 ml shake flasks filled with 400 ml YPD for 16 h; cells were then harvested, washed twice with dH2O and inoculated in 10 ml of Delft base with glucose 1 g l−1 in glass tubes, at a starting OD of 5. All growths were performed in a rotary shaker at 160 rpm and 30 °C. Samples for RNA extraction were collected after 20 min of growth and then every 1 h until 7 h of fermentation. The growth phase at each time point was characterized by HPLC analysis of the supernatants, monitoring the presence of glucose, ethanol, acetate, GA and the consumption of EG. Samples taken after 20 min (growth on glucose), 3 h (growth on ethanol and acetate), and 6 h (EG consumption) were processed for RNA extraction. Briefly, 4 ml of culture were centrifuged (4000 × g, 2 min, 4 °C) and the pellet was flash-frozen in dry ice; samples were stored at −80 °C until RNA extraction the following day.
RNA extraction was performed with GeneJET RNA Purification Kit (Thermo Scientific) following the yeast total RNA purification protocol. For genomic DNA removal from RNA preparations, 1000 ng of RNA samples were treated with DNAse (Thermo Scientific). cDNA was retrotranscribed with the ProtoScript® II First Strand cDNA Synthesis Kit, according to manufacturer indications; 750 ng of total RNA were used as template. As negative controls, the same reactions were performed without adding the ProtoScript® II Reverse Transcriptase enzyme. qRT-PCRs were performed by combining 100 ng of cDNA template and Luna® Universal qPCR Master Mix (New England Biolabs) with specific oligonucleotides (Table S6). To monitor double-stranded DNA synthesis, the CFX Real-Time PCR (BioRad) with SYBR scan mode was used. The qRT-PCR conditions were set to 95 °C for 60 s, followed by 40 cycles of 95 °C for 15 s, 55 °C for 15 s, and 60 °C for 30 s. At the end, a melt curve step was done following the real-time instrument recommendations. As reference gene, a fragment of ACT1 was amplified. Values were normalized by the internal reference gene according to the equation
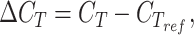 |
where 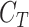 is the threshold cycle of the gene of interest and
is the threshold cycle of the gene of interest and 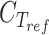 is the threshold cycle of the reference gene (ACT1) in the same condition. Relative expression was calculated according to the equation
is the threshold cycle of the reference gene (ACT1) in the same condition. Relative expression was calculated according to the equation
 |
where 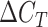 refers to the condition of interest (i.e. expression after the diauxic shift and during EG consumption) and
refers to the condition of interest (i.e. expression after the diauxic shift and during EG consumption) and 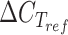 refers to the reference condition (i.e. expression during growth on glucose) (Livak and Schmittgen 2001). Values are the mean of three independent biological replicates and two technical replicates.
refers to the reference condition (i.e. expression during growth on glucose) (Livak and Schmittgen 2001). Values are the mean of three independent biological replicates and two technical replicates.
In vitro assay for the activity of Gox0313
The activity of Gox0313 was assayed in vitro by comparing the ability of cell raw extracts of the strains CER.GOX and wt to reduce NAD+ in the presence of EG.
Seed cultures from YPD plates were grown in glass tubes filled with 5 ml YPD for 8 h; cells were then inoculated in 250 ml glass tubes containing 50 ml of YPD starting OD = 0.0005, and grown for 16 h, when the cultures reached OD 1. All growths were performed in a rotary shaker at 160 rpm and 30 °C. For protein extraction, 10 OD of cells were harvested by centrifugation (4000 × g, 2 min) and washed with dH2O. The pellet was resuspended in 500 µl of lysis buffer [Tris–HCl pH8 50 mM, PMSF 1 mM (Thermo Scientific) and cOmplete™ Protease Inhibitor 1X (Merck)] and transferred to a screwcap vial containing 100 µl of glass beads. Mechanical lysis was performed with FastPrep®-24 (Thermo) with the following cycles: 20 s homogenization, 30 s on ice, for five times. After clarification (20 000 × g, 20 min, 4°C), total protein concentration in the supernatant (raw extract) was measured with the Bradford method. The reaction mixture consisted of Tris–HCl pH8, 50 mM; EG, 5 mM; NAD+, 5 mM; raw extract, 5 mg ml−1. The reaction was run in 1 ml cuvettes incubated at 30°C to monitor the change in absorbance at 340 nm due to NAD+ reduction. Five independent replicates were performed.
Fluorescence microscopy analyses
Yeast cells were grown in Delft Glc medium and harvested in the exponential phase. The pellet of 1 ml of culture was washed with water and then resuspended in phosphate-buffered saline solution (PBS; NaH2PO4 53 mM, Na2HPO4 613 mM, and NaCl 75 mM). Cells were then observed with a THUNDER Imager 3D Live Cell –LEICA equipped with a 100× objective. Images were acquired with a Leica DFC9000 camera. GFP-tagged Aox1 was observed using the DFT51010 Cube (EX 479/33 DM 500 EM 519/25) filter. Digital images were acquired with the LAS X-software.
Metabolites quantification by HPLC
HPLC analysis was performed to quantify the amount of glucose, EG, GAH, GA, glycerol, ethanol, and acetate. Prior to analysis, all samples were centrifuged (21 000 × g, 5 min) and diluted when necessary. The HPLC was equipped with a Rezex ROA-Organic Acid H+ (8%) Ion Exclusion column 300 × 7.8 mm, 8 µm (Phenomenex); 10 µl of samples were injected in the column. The mobile phase was H2SO4 0.005 N, at a flow of 0.8 ml min−1; column temperature was set to 80°C. Separated components were detected after 20 min by a refractive index detector, and by a variable wavelength detector set at 210 nm. Peaks were identified by comparison with reference standards dissolved in ultrapure H2O (18 MΩ). Calibration curves for peak quantification were prepared in a range between 0.625 and 40.0 g l−1.
Statistical analysis
All the experiments were performed with three independent replicates, unless differently stated. GraphPad PRISM 10.1.0 was used for the statistical analyses. For the toxicity test, two-way ANOVA (Analysis of variance) analyses were performed to analyze the effect of “strain” and “GAH concentration” on maximum specific growth rate, lag phase duration, and production of EG and/or GA, followed by a post hoc Tukey–Kramer test for multiple comparisons. The remaining statistical analyses were performed using a two-tailed, unpaired, heteroscedastic Student’s t-test.
Results and discussion
Deletion and constitutive expression of yeast endogenous genes for EG metabolism
In our previous study (Senatore et al. 2024), we described the physiology of EG metabolism by S. cerevisiae and nine other yeasts, all capable of oxidizing EG to GA; in the same work, a putative pathway was also proposed for S. cerevisiae, suggesting that Yll056c (coded by YLL056C) might be responsible for the oxidation of EG to GAH. Moreover, we also speculated that glyoxylate reductase (encoded by GOR1) might be able to catalyze the oxidation of GA to GOX (Rintala et al. 2007, Salusjärvi et al. 2019), as consumption of GA without accumulation of intermediates was observed in the same study (Fig. 2A). To validate our hypothesis, in this work three different S. cerevisiae strains were constructed (see the section “Materials and methods” for more details and Table 1): strain deltaY harboring a marker-free deletion of YLL056C, strain overY harboring YLL056C under a medium strength constitutive promoter, and strain deltaG harboring a marker-free deletion of GOR1.
Figure 2.

Metabolic engineering strategies to improve EG metabolism. (A) Deletion and constitutive expression of wt genes for EG metabolism. Deletion of the genes YLL056C and GOR1 resulted in the strains deltaY and deltaG, respectively; constitutive expression of YLL056C under a medium strength promoter resulted in strain overY. Yll056c is putatively involved in the oxidation of EG to GAH, while Gor1 is responsible for the reduction of GOX to GA. (B) Engineered strains CER.GA and CER.AOX. Strain CER.GA expresses the NAD+-dependent ADH gox0313 from G. oxydans and aldA from E. coli under medium strength constitutive promoters; strain CER.AOX expresses the alcohol oxidase AOX1 from K. phaffii and the endogenous catalase CTT1 under medium strength constitutive promoters for detoxification of hydrogen peroxide. (C) GA production by the engineered S. cerevisiae strain after 144 h of cultivation in YP + EG; no significant difference can be observed between the engineered strains and the wt control.
Since we previously described that glucose, ethanol, and acetate inhibit EG consumption by S. cerevisiae (Senatore et al. 2024), we opted to screen the performances of the new strains by evaluating their ability to produce GA when grown in YP + EG. As it can be observed from Fig. 2(C), no significant differences could be observed between the engineered strains and also in comparison to the parental strain in terms of GA production after 144 h of cultivation; the same was true for the growth profiles (Fig. S1). To further investigate the behavior of the strains, shake flasks cultivations in the presence of glucose were performed, to assess if the constitutive expression of YLL056C allowed for EG consumption during the exponential growth phase, relieving glucose inhibition. As shown in Fig. 3, no EG consumption and GA production could be observed in the presence of glucose, and strain overY did not show any difference when compared to the wild type. Both strains started consuming EG and producing GA at a late fermentation stage, after ethanol and acetate exhaustion (data not shown), as reported previously (Senatore et al. 2024).
Figure 3.

Exponential growth profiles of engineered strains in the presence of glucose. Strains wt, overY, CER.GA, and CER.AOX were grown in YPD + EG in the presence of glucose to assess whether the constitutive expression of YLL056C, gox0313, or AOX1 could relieve the glucose inhibition of EG consumption. In each panel, the left y-axis shows OD (blue, circles), and glucose (red, squares), ethanol (green, triangles) and EG (dark blue, hollow circles) concentration in g l−1; the right y-axis shows glycerol (pink, diamonds), acetate (orange, triangles), and GA (yellow, hollow squares) concentration in g l−1.
These results suggest that, at least in the tested conditions, YLL056C and GOR1 are not good targets to improve EG metabolism in S. cerevisiae. While the oxidation of EG to GAH is reported as a reversible reaction, the equilibrium in vitro is shifted toward the reduction of the aldehyde (Wang et al. 2017). Considering the reaction equation, we initially speculated that Yll056c might be able to work in the desired direction in vivo, given the very low concentration of GAH: however our experimental data suggest that the equilibrium might be shifted toward the reduction of the aldehyde in vivo, too, in accordance with classification of this protein as an aldehyde reductase (Wang et al. 2017). This strongly suggests that other ADHs may be responsible for EG oxidation to GA in S. cerevisiae.
A similar situation occurs for the oxidation of GA to GOX, catalyzed by Gor1, as the equilibrium of the reaction is shifted toward the reduction of GOX to GA (Rintala et al. 2007). Nevertheless, S. cerevisiae is able to consume GA in specific conditions (Senatore et al. 2024), most probably thanks to the activity of Gor1. Lack of differences in GA production between the strain deltaG and the wt (Fig. 2C) might suggest an upstream bottleneck, or lack of expression of Gor1 in the control condition in the first place: indeed, it is reported that GOR1 expression is repressed by glucose (Koivistoinen et al. 2013). To clarify the role of this protein in GA oxidation, the expression of GOR1 was evaluated during growth on glucose (Glc), during the ethanol consumption phase (EtOH), and after the depletion of EtOH and acetate, when EG consumption finally starts (EG). The expression of GOR1 was assayed in the wt strain, in the presence of EG. Figure S2 shows the fermentation profiles and the sampling times. As it can be observed in Fig. 4, no significant differences in the expression of GOR1 were observed in any of the growth phases. Moreover, assay on GOR1 showed positive ΔCT values (>2) compared to ACT1 whose promoter is constitutive but weak (Zhang et al. 2020), suggesting a general low expression of this gene in the assayed conditions. Therefore, the lack of differences observed between the wt and deltaG strains is most likely due to the low expression of GOR1. Additionally, the expression of other genes might contribute to the oxidation of GA to GOX observed by Senatore et al. (2024), such as the not annotated YPLL13C and YGL185C, which were shown to have activity toward GOX by Rintala et al. (2007). Taken together, these findings suggest that GOR1 is not involved in a significant way in EG metabolism in S. cerevisiae.
Figure 4.

Expression profile of GOR1 under different growth phases in wt S. cerevisiae. The expression of GOR1 was assayed by qRT-PCR in the presence of EG, during the glucose consumption phase (Glc), after the diauxic shift (EtOH) and after the depletion of ethanol and acetate, when EG is consumed (EG). The y-axis shows the fold change in the expression of GOR1 with respect to its expression on glucose. No significant differences were observed. Values are the mean ± standard deviation of five independent experiments.
Constitutive expression of gox0313 and AOX1
Since we were not able to identify an endogenous gene that could improve either EG catabolism or GA accumulation, we decided to further focus our engineering efforts on the first reaction of the pathway, that is the oxidation of EG to GAH. Once again, the available literature about enzymes active on EG is scarce, and some of the enzymes have only been tested in vitro. Two classes of enzymes have been reported, ADHs and alcohol oxidases. Regarding ADHs, the most studied ones are PedE and PedH from P. putida, and mADH and Gox0313 from G. oxydans. PedE and PedH are periplasmic dehydrogenases, while mADH is a membrane-bound ADH, all requiring the specific cofactor PQQ (Mückschel et al. 2012, Zhang et al. 2016) that S. cerevisiae is not able to natively synthetize; Gox0313, on the other hand, is cytosolic and requires NAD+ (Zhang et al. 2015). Among the enzymes involved with EG, it also worth mentioning the NAD+-dependent FucO (variant I7L/L8V) and YqhD from E. coli (Lu et al. 1998, Alkim et al. 2015), which have been reported to be aldehyde reductases, despite the reaction being reversible. Alcohol oxidases belong to the second class of enzymes reported to be able to catalyze the oxidation of EG; to the best of our knowledge, three alcohol oxidases with this property have been described—only in in vitro studies—namely Aox1 from K. phaffii, an alcohol oxidase from Candida sp. (Isobe and Nishise 1994), and a glycerol oxidase from Aspergillus japonicus (Isobe 1995). These alcohol oxidases are also able to oxidize GAH to glyoxal, however in a less efficient way (Isobe and Nishise 1994); the glycerol oxidase from A. japonicus is reported to be able to oxidize GA to GOX, as well (Isobe 1995). It is interesting to mention that contrary to dehydrogenases, alcohol oxidases require oxygen as cofactor, producing hydrogen peroxide as a by-product and making the reaction nonreversible (Goswami et al. 2013).
With this information in hand, we decided to express in S. cerevisiae the NAD+-dependent ADH Gox0313 from G. oxydans and the alcohol oxidase Aox1 from K. phaffii. Gox0313 was selected for being cytosolic and for its cofactor requirement; moreover, expression of Gox0313 in E. coli proved to be the only suitable strategy to increase EG oxidation in a very recent publication (Yan et al. 2024); FucO and YqhD were discarded as they most likely catalyze the reaction in the unwanted direction. Among the alcohol oxidases, we selected Aox1 because it is the most characterized one among the three and because of its specificity for EG and GAH. Thus, we created two strains (Table 1, Fig. 2B): CER.GA expressing gox0313 under a constitutive medium strength promoter, coupled with expression of aldA from E. coli; CER.AOX expressing AOX1 from K. phaffii, coupled with constitutive expression of the endogenous catalase CTT1 to reduce the oxidative stress from the production of hydrogen peroxide. See the section “Material and methods” and Table S2 for an accurate description of the expression cassettes.
To benchmark the newly obtained strains, their ability to produce GA from EG was assessed in YP + EG, as described for the previous experiment (Fig. 2C). No significant differences were observed when compared to the wt strain in terms of GA production after 144 h of cultivation; the same was true for the growth profiles (Fig. S1). To further investigate the behavior of the engineered strains, shake-flask cultivations in the presence of glucose were performed, to assess if the constitutive expression of gox0313 and AOX1 allowed for EG consumption during the exponential growth phase, relieving glucose inhibition (Fig. 3): no EG consumption in the presence of glucose was observed, and the strains showed the same phenotype as the wt control; for these strains, EG consumption and GA production started at a late fermentation stage as well, after ethanol and acetate exhaustion and showed no differences with the wt strain (data not shown).
These data are in contrast with the results previously obtained with bacteria and in vitro. Indeed, while heterologous expression of gox0313 in E. coli allowed for the total consumption of 10 g l−1 of EG in 96 h (Yan et al. 2024), no improvement was observed in the strain CER.GA; it might be possible that Gox0313 catalyzes the opposite reaction, similarly to what FucO and YqhD do in E. coli. To better investigate this aspect, an in vitro assay was set up to understand if gox0313 is expressed and functional in yeast; as the assay relies on the change in absorbance due to the reduction of NAD+, a strain expressing only gox0313 (CER.GOX) was constructed, to exclude the contribution of aldA to the change in absorbance. Cells were harvested during the early exponential phase and the activities of the raw cell extracts were compared by providing EG and NAD+ as substrates and monitoring the change in absorbance at 340 nm over time (ΔA); CER.GOX was compared to the wt strain. As it can be observed in Fig. 5, CER.GOX shows a significantly higher ΔA compared to the wt strain (P-value < .001), being more than twice as high. This result indicates a higher reduction rate of NAD+, and thus a higher oxidation rate of EG in the strains CER.GOX, suggesting that gox0313 is correctly expressed and functional even when the cells are grown in the presence of glucose. However, it remains unclear why EG oxidation is not observed in vivo under the assayed conditions. Indeed, while EG uptake might be considered a limiting aspect, Scherrer et al. (1974) demonstrated that S. cerevisiae membranes are permeable to this compound, and thus transport should not be the issue under the examined conditions. Another possible reason for this behavior could be the redox power in the cytosol, as NAD+ is required by Gox0313 for the oxidation of EG. Finally, it is possible that the in vivo oxidation of EG by Gox0313 is inefficient due to competition with the native proteome for the cofactor NAD+.
Figure 5.

In vitro assay for the activity of Gox0313. The activity of Gox0313 was assayed in vitro by measuring the reduction of NAD+ in the presence of EG by raw cell extracts of the strains CER.GOX (blue. squares) and wt (orange, circles). The y-axis shows the absorbance at 340 nm in arbitrary units (AU); the change in absorbance (ΔA) was calculated by fitting a regression line, and it is expressed as mAU min−1; dotted lines represent the 95% confidence interval. Values are the mean ± standard deviation of five independent experiments.
A similar behavior was observed for AOX1. While in vitro studies showed the ability of alcohol oxidases to oxidize EG to GAH (and glyoxal) (Isobe 1995), such activity was not observed in vivo with the strain CER.AOX. Functional expression of AOX1 in its native host K. phaffii requires localization to the peroxisomes for the oligomerization, and targeting of AOX1 to the cytosol results into inactive aggregates (Waterham et al. 1997). Therefore, we decided to check the localization of Aox1 in S. cerevisiae, by constructing a strain expressing a GFP-tagged version of the enzyme (CER.AOX_GFP). Fluorescence microscopy revealed that Aox1 is cytosolic (Fig. S3), in accordance with previous studies in which AOX1 was expressed in S. cerevisiae (Dai et al. 2017, Zhan et al. 2023). It is worth noting that the deletion of K. phaffii’s peroxisome localization sequence at the C terminus (LARF) is not necessary for cytosolic expression of AOX1 in S. cerevisiae, as confirmed also by our fluorescence microscopy results. Moreover, cytosolic Aox1 is not active in K. phaffii, but this is not the case for S. cerevisiae: indeed, both studies showed that engineered strains of S. cerevisiae are able to grow on methanol thanks to the expression of cytosolic KpAox1 (Dai et al. 2017, Zhan et al. 2023), proving that targeting to the peroxisomes is not necessary in S. cerevisiae. From these observations and the fluorescence microscopy data, we concluded that Aox1 is correctly expressed in the strain CER.AOX, but it remains unclear why no EG oxidation was observed. Further studies in its natural host could more directly elucidate whether AOX1 can catalyze this reaction in vivo, as proposed in a very recent paper by Carneiro et al. (2024).
Further investigations on the metabolic roles of Yll056c, Gox0313, and Aox1
From the growth phenotypes, we speculate that Yll056c functions as an aldehyde reductase in S. cerevisiae. To test if this was the case, we decided to evaluate the ability of the strains to tolerate GAH. Indeed, GAH is toxic as it reacts with amine and thiol groups in proteins causing cross-linking, it induces DNA damage by glycation of the amine groups of nucleic acids and it causes lipid peroxidation; this results in the inactivation of key enzymes—such as Pgk1–and in the G2-M cell cycle arrest (Jayakody and Jin 2021). Since GAH can be detoxified both via EG and via GA, the engineered strains should exhibit differences in terms of tolerance and accumulation of either EG or GA. To test our hypothesis, the strains wt, overY, deltaY, CER.GA, and CER.AOX were grown in the presence of different concentrations of GAH (Fig. 6, Fig. S4), since S. cerevisiae shows different sensitivity to GAH depending on the growth conditions (Jayakody et al. 2013, 2018). CER.GA was included in the experiment as well, to evaluate the contribution of gox0313 and aldA in GAH detoxification; CER.AOX was included in the experiment, too, since GAH is reported to be a substrate of Aox1 as well, producing glyoxal which can cause DNA damage by forming adducts with (deoxy)ribonucleosides (Jayakody and Jin 2021).
Figure 6.

Effects of GAH toxicity in the engineered strains. (A) Maximum specific growth rate in the presence of GAH. Light-blue columns represent the data obtained with 10 mM of GAH; blue columns represent the data obtained with 20 mM of GAH; and the y-axis show the maximum specific growth rate in h−1. The level of statistical significance is indicated only for differences between the samples. (B) Production of EG and GA in the presence of GAH. After 72 h of growth, the supernatants were analyzed by HPLC to quantify EG (blue), GA (yellow), and GAH (orange); concentrations of each compound are reported in mM; columns are stacked.
Maximum specific growth rate (µmax) was calculated to evaluate the toxic effect of GAH. Since 5 mM did not result in a toxic effect, we included in Fig. 6 only the results obtained with 10 and 20 mM GAH for a clearer visualization; Fig. S4 shows the complete set of results. When exposed to 10 mM GAH, all the strains showed a reduced growth rate (0.24–0.27 h−1) compared to the control condition (0.31–0.33 h−1), except for CER.GA, which showed a growth rate of 0.314 ± 0.004 h−1, comparable to the growth rate without exposure to GAH (0.313 ± 0.003 h−1). No significant difference was observed between the strains overY and deltaY (Fig. 6A). More differences in terms of growth rates were observed when the strains were exposed to 20 mM GAH; the wt growth rate was reduced to 0.192 ± 0.006 h−1. Notably, no significant differences were observed between the wt strain and CER.GA: however, the engineered strain (CER.GA) showed a much shorter lag phase compared to the wt (12 ± 1 h versus 32 ± 1 h). Overexpression of YLL056C did not result in an improved growth rate or a shorter duration of the lag phase; strain deltaY, on the other hand, showed a further reduction in growth rate when increasing GAH concentration to 20 mM (0.077 ± 0.011 h−1), suggesting that Yll056c is involved in GAH detoxification in S. cerevisiae. Finally, also CER.AOX showed a reduced growth rate (0.095 ± 0.003 h−1), which might be explained by the combined toxic effect of GAH and glyoxal, produced by Aox1 itself.
To further analyze our hypothesis, we examined the production of EG and GA at the end of the experiment, which was stopped after 72 h of cultivation (Fig. 6B). It is interesting to notice that all the strains detoxified GAH mostly by reducing it to EG; EG is a diol, and alcohols are in general less toxic than organic acids; nevertheless, small quantities of GA were produced by all strains and in all conditions. When exposed to 10 mM GAH, all the engineered strains produced slightly less EG than the control (around 7–10 mM); overY produced more EG than deltaY, however the difference was not statistically significant. This might be explained by the fact that the multiple aldehyde reductases present in S. cerevisiae might be able to compensate for the lack of Yll056c. Among all strains, CER.GA was the only one showing a different phenotype: the strain produced a much lower amount of both EG (1.97 ± 0.76 mM), and GA, detectable in only one of the three replicates. Indeed, while for most strains the sum of EG and GA concentrations was close to the initial concentration of GAH, this was not the case for CER.GA: we speculate that AldA is responsible for the detoxification of GAH to GA, which is then consumed by oxidation to GOX (and respiration to CO2), as suggested in Senatore et al. (2024).
When exposed to 20 mM GAH, a similar behavior was observed between the strains. With a higher concentration of GAH, it was possible to appreciate a small, yet significant, difference in the production of EG by the strains overY and deltaY, in line with the previous observations suggesting that Yll056c is an aldehyde reductase. Interestingly, the difference is due to the higher production of EG by the overY strain, rather than to an impeded GAH detoxification by the deltaY strain. Indeed, the latter showed the same EG production as the wt strain, as observed with 10 mM GAH; the mutant strain, however, showed a significantly lower growth rate. These observations might be explained by the expression of GRE2. A few studies described Gre2 to be involved in GAH detoxification as a NAD(P)H-dependent reductase (Jayakody et al. 2013, 2018), contrary to Yll056c which requires NADH as a cofactor: the lower growth rate (yet same EG production) of the strain deltaY might be explained by the compensation of Gre2 in the detoxification of GAH toward EG, consuming a fraction of the NADPH destined for growth. The reason why this is not observed at a lower concentration of GAH could be explained by the fact that several aldehyde-reducing enzymes switch the cofactor preference from NADH to NADPH at high aldehyde concentrations, Gre2 being one of the examples (Jayakody and Jin 2021).
These observations uncover the bottleneck of EG utilization by S. cerevisiae, which is the first oxidation step toward GAH: S. cerevisiae has probably evolved redundant enzymatic activities to protect against aldehydes (therefore including GAH) toxicity. Most likely, both Gox0313 and Aox1 are capable of oxidizing EG in vivo, but the endogenous aldehyde reductases efficiently detoxify the intermediate back to EG, creating a dynamic equilibrium, which masks the low activity of Gox0313 and Aox1.
Role of ADHs in EG catabolism
Given the inability of Yll056c to oxidize EG in the assayed conditions, in an effort to elucidate the native EG catabolic pathway we decided to direct our focus on ADHs as well. We initially did not focus our attention to this class of enzymes as they are mostly involved in maintaining the redox balance between the different compartments of the cell during growth (Bakker et al. 2001). Additionally, (over)expression of (native) ADHs in S. cerevisiae resulted in increased titers of EG from GAH (Uranukul et al. 2019), suggesting that ADHs are involved in the reduction of GAH to EG, rather than the oxidation of EG to GAH. In S. cerevisiae there are seven genes encoding ADHs. ADH1 is repressed in the absence of glucose during the growth on ethanol, and its overexpression was reported to increase resistance to GAH stress, with increased EG production (Jayakody et al. 2013). On the other hand, ADH2 is strongly induced during growth on ethanol (yeastgenome.org), but no EG consumption during this phase was observed in our previous study. These observations suggest that at least ADH1 and ADH2 are not involved in the oxidation of EG. S. cerevisiae’s genome also encodes for ADH6 and ADH7, members of the cinnamyl family of ADHs and putatively involved in the synthesis of fusel alcohols, aldehyde tolerance, and NADPH homeostasis (Larroy et al. 2002a, b). Both ADH6 and ADH7 have a broad substrate specificity, however showing a preference for aromatic aldehydes (Generoso et al. 2015). Finally, S. cerevisiae’s genome encodes for ADH3, ADH4, and ADH5, as well. ADH3 is mitochondrial and it plays a role in maintaining the redox cofactor balance during anaerobic growth (Young and Pilgrim 1985). The role of ADH4 is less clear, however it is only expressed at low levels in laboratory strains (Drewke and Ciriacy 1988, Larroy et al. 2002a, Dickinson et al. 2003). Finally, ADH5 is a paralog of ADH1 and its activity is only evident in adh1Δ adh3Δ double null mutants (Smith et al. 2004). Considering the metabolic role of these alcohol oxidases, we decided to investigate the expression profiles of ADH1, ADH2, and ADH6 in the presence of EG. Additionally, we also measured the expression of YLL056C and GRE2, to better clarify their role in GAH detoxification during EG catabolism. The expression level of the abovementioned genes was evaluated during growth on glucose (Glc), during the ethanol consumption phase (EtOH), and after the depletion of EtOH and acetate, when EG consumption finally starts (EG). None of the genes showed strong expression exclusively during the EG consumption phase, which would suggest a potential candidate for EG oxidation. (Fig. 7). The expression profiles of ADH1 and ADH2 are coherent with literature data. ADH1 is repressed after the diauxic shift, while ADH2 is strongly induced after the depletion of glucose, and it remains strongly expressed even after the depletion of EtOH. Despite Adh2 being the key enzyme for the oxidation of EtOH to acetaldehyde after the diauxic shift, these results indicate that Adh2 is not involved in the oxidation of EG: if this was the case, consumption of EG should start with the consumption of EtOH. ADH6 expression is strongly repressed after the diauxic shift. Taken together, these results suggest that the native ADHs of S. cerevisiae are not involved in the oxidation of EG in the assayed conditions.
Figure 7.

Expression profile of ADHs, YLL056C, and GRE2 under different growth phases in wt S. cerevisiae. The expression of ADHs (ADH1, ADH2, and ADH6), YLL056C, and GRE2 was assayed in the presence of EG by qRT-PCR, in the presence of glucose (Glc), ethanol (EtOH), and after the depletion of ethanol and acetate, when EG is consumed (EG). The y-axis shows the fold change in the expression with respect to expression on glucose. The level of statistical significance is indicated only for differences between the samples. Values are the mean ± standard deviation of three independent experiments.
Finally, it is worth mentioning that the expression of YLL056C is significantly higher only during the EtOH phase (P-value = .0259); GRE2, on the other hand, is repressed after the depletion of EtOH and acetate (P-value = .0276). The expression pattern suggests that neither of these two genes are involved in EG catabolism, coherently with the phenotype of the overY and deltaY strains in the presence of EG, and are most likely only involved in response to high GAH concentrations.
Conclusions
The scope of this work was to investigate the pathway that leads to the oxidation of EG to GA in S. cerevisiae, first by assessing the potential contribution to this metabolism of two endogenous genes, YLL056C (a putative ADH) and GOR1 (glyoxylate reductase). Additionally, two heterologous genes (gox0313 from G. oxydans and AOX1 from K. phaffii) were expressed in an effort to increase EG oxidation toward GA.
Constitutive expression of YLL056C did not improve EG conversion to GA, and deletion did not cause a loss in the ability of S. cerevisiae to produce GA. Similarly, deletion of GOR1 did not increase GA accumulation. qRT-PCR data showed that GOR1 is not expressed in high levels in the assayed conditions, which explains why the deletion resulted in an unchanged phenotype with respect to the wt strain. Hence, GOR1 is not significantly involved in GA catabolism. To improve EG oxidation to GA, two engineered strains were created: CER.GA expressing a previously reported bacterial EG dehydrogenase (gox0313) and a GAH dehydrogenase (aldA), and CER.AOX, expressing alcohol oxidase AOX1 from the methylotrophic yeast K. phaffii and the endogenous catalase 1 (CTT1) for H2O2 detoxification. The newly obtained strains did not show an improved GA production in the assayed conditions, despite the functionality of Gox0313 (as demonstrated by the in vitro assay) and Aox1 (as reported in literature). It remains unclear why EG is not oxidized in the presence of other carbon sources, even when EG oxidizing enzymes are constitutively expressed.
To further investigate the roles of Yll056c, Gox0313, and Aox1, the strains were grown in the presence of GAH, product (and/or substrate) of these enzymes. We concluded that in vivo Yll056c acts most probably as an aldehyde reductase, which is not involved in the EG catabolism pathway in S. cerevisiae. Moreover, CER.GA showed the most different phenotype, with a very low production of EG from GAH: most likely, Gox0313 catalyzes the reoxidation of EG to GAH, counteracting the detoxification machinery at high concentrations of GAH; a similar conclusion can also be drawn for Aox1.
Finally, the potential contribution of ADHs in the EG catabolic pathway was considered, too. qRT-PCR data shows that ADH1, ADH2, and ADH6 are not involved in the pathway: ADH1 and ADH2 are expressed either during growth on glucose or after diauxic shift, while ADH6 expression is repressed after the depletion of glucose. None of the tested genes was exclusively induced during the EG consumption phase. If these genes were involved in the pathway, their expression would enable EG consumption during the previous phases. Thus, we conclude that S. cerevisiae native ADHs are most likely not involved in EG catabolism.
The (probably nonspecific) endogenous EG oxidation pathway toward GA production remains unknown in S. cerevisiae. We uncovered that the bottleneck is the first oxidation step of EG to GAH, possibly due to the endogenous aldehyde detoxification system. Further efforts could be directed toward combining the expression of gox0313 (or AOX1) with the deletion of (some) aldehyde reductases, such as Yll056c and Gre2, finding a good compromise with the robustness of the strain. Alternatively, increasing the expression levels of gox0313 (or AOX1) (stronger promoter, increase of copy number in the genome, expression on a multicopy plasmid) might outcompete the detoxification system, unlocking the bottleneck and S. cerevisiae potential toward the upcycling of EG to GA.
As EG is not generally found in its natural niches, S. cerevisiae most likely did not evolve specific enzymes and a related signaling pathway for this molecule. Therefore, EG metabolism possibly exploits the promiscuous activity of some native proteins, and its regulation may happen at a different level than transcriptional and translational processes. A multiomics-based approach is necessary to definitively elucidate this pathway, clarifying why EG consumption is only observed in the late stationary phase. More specifically, metabolomics and fluxomics (possibly including ethylene-d4 glycol) will be necessary to understand and model the first steps of the EG catabolic pathway.
Supplementary Material
Acknowledgment
We thank Ilaria Spreafico and Monica Squicciarini for their help in the creation of the engineered strains.
Contributor Information
Vittorio Giorgio Senatore, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy.
Fiorella Masotti, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy.
Riccardo Milanesi, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy.
Sofia Ceccarossi, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy; Department of Earth and Marine Sciences, University of Palermo, Via Archirafi 22, 90123 Palermo, Italy.
Letizia Maestroni, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy.
Immacolata Serra, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy.
Paola Branduardi, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy.
Conflict of interest
None declared.
Funding
This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under grant agreement number 101036838, by the Ministero dell'Istruzione e del Merito (MIUR) PRIN number 2020SBNHLH, and by National Center 5 “National Biodiversity Future Center” (Award Number: Project code CN_00000033, Concession Decree No. 1034 of 17 June 2022 adopted by the Italian Ministry of University and Research, CUP H43C22000530001, Project title “National Biodiversity Future Center – NBFC. Funder: Project funded under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.4 - Call for tender No. 3138 of 16 December 2021, rectified by Decree n.3175 of 18 December 2021 of Italian Ministry of University and Research funded by the European Union – NextGenerationEU).
References
- Alkim C, Cam Y, Trichez D et al. Optimization of ethylene glycol production from (d)-xylose via a synthetic pathway implemented in Escherichia coli. Microb Cell Fact. 2015;14:127. 10.1186/s12934-015-0312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SE, Wells J, Fedorowicz A et al. Evaluation of the contact and respiratory sensitization potential of volatile organic compounds generated by simulated indoor air chemistry. Toxicol Sci. 2007;97:355–63. 10.1093/toxsci/kfm043. [DOI] [PubMed] [Google Scholar]
- Bakker BM, Overkamp KM, van Maris AJA et al. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol Rev. 2001;25:15–37. 10.1111/j.1574-6976.2001.tb00570.x. [DOI] [PubMed] [Google Scholar]
- Carbios . CARBIOS corporate presentation—January 2024. Clermont-Ferrand, 2024, accessed July 31st, 2024. https://www.carbios.com/wp-content/uploads/2022/01/carbios_presentation-january-2022.pdf. [Google Scholar]
- Carneiro CVGC, Trichez D, Bergmann JC et al. Engineering Komagataella phaffii for ethylene glycol production from xylose. AMB Expr. 2024;14:131. 10.1186/s13568-024-01795-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carniel A, Santos AG, Chinelatto LS et al. Biotransformation of ethylene glycol to glycolic acid by Yarrowia lipolytica: a route for poly(ethylene terephthalate) (PET) upcycling. Biotechnol J. 2023;18:2200521. 10.1002/biot.202200521. [DOI] [PubMed] [Google Scholar]
- Conroy S, Zhang X. Theoretical insights into chemical recycling of polyethylene terephthalate (PET). Polym Degrad Stab. 2024;223:110729. 10.1016/j.polymdegradstab.2024.110729. [DOI] [Google Scholar]
- Dai Z, Gu H, Zhang S et al. Metabolic construction strategies for direct methanol utilization in Saccharomyces cerevisiae. Bioresour Technol. 2017;245:1407–12. 10.1016/j.biortech.2017.05.100. [DOI] [PubMed] [Google Scholar]
- Dickinson JR, Salgado LEJ, Hewlins MJE. The catabolism of amino acids to long chain and complex alcohols in Saccharomyces cerevisiae *. J Biol Chem. 2003;278:8028–34. 10.1074/jbc.M211914200. [DOI] [PubMed] [Google Scholar]
- Drewke C, Ciriacy M. Overexpression, purification and properties of alcohol dehydrogenase IV from Saccharomyces cerevisiae. Biochim Biophys Acta Gene Struct Express. 1988;950:54–60. 10.1016/0167-4781(88)90072-3. [DOI] [PubMed] [Google Scholar]
- Franden MA, Jayakody LN, Li W-J et al. Engineering Pseudomonas putida KT2440 for efficient ethylene glycol utilization. Metab Eng. 2018;48:197–207. 10.1016/j.ymben.2018.06.003. [DOI] [PubMed] [Google Scholar]
- Generoso WC, Schadeweg V, Oreb M et al. Metabolic engineering of Saccharomyces cerevisiae for production of butanol isomers. Curr Opin Biotechnol. 2015;33:1–7. 10.1016/j.copbio.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Goswami P, Chinnadayyala SSR, Chakraborty M et al. An overview on alcohol oxidases and their potential applications. Appl Microbiol Biotechnol. 2013;97:4259–75. 10.1007/s00253-013-4842-9. [DOI] [PubMed] [Google Scholar]
- Isobe K, Nishise H. Enzymatic production of glyoxal from ethylene glycol using alcohol oxidase from methanol yeast. Biosci Biotechnol Biochem. 1994;58:170–3. 10.1271/bbb.58.170. [DOI] [PubMed] [Google Scholar]
- Isobe K. Oxidation of ethylene glycol and glycolic acid by glycerol oxidase. Biosci Biotechnol Biochem. 1995;59:576–81. 10.1271/bbb.59.576. [DOI] [PubMed] [Google Scholar]
- Jayakody LN, Horie K, Hayashi N et al. Engineering redox cofactor utilization for detoxification of glycolaldehyde, a key inhibitor of bioethanol production, in yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2013;97:6589–600. 10.1007/s00253-013-4997-4. [DOI] [PubMed] [Google Scholar]
- Jayakody LN, Jin Y-S. In-depth understanding of molecular mechanisms of aldehyde toxicity to engineer robust Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2021;105:2675–92. 10.1007/s00253-021-11213-1. [DOI] [PubMed] [Google Scholar]
- Jayakody LN, Turner TL, Yun EJ et al. Expression of Gre2p improves tolerance of engineered xylose-fermenting Saccharomyces cerevisiae to glycolaldehyde under xylose metabolism. Appl Microbiol Biotechnol. 2018;102:8121–33. 10.1007/s00253-018-9216-x. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Sasaki M, Hidalgo A-RGD et al. Glycolic acid production using ethylene glycol-oxidizing microorganisms. Biosci Biotechnol Biochem. 2001;65:2265–70. 10.1271/bbb.65.2265. [DOI] [PubMed] [Google Scholar]
- Koivistoinen OM, Kuivanen J, Barth D et al. Glycolic acid production in the engineered yeasts Saccharomyces cerevisiae and Kluyveromyces lactis. Microb Cell Fact. 2013;12:82. 10.1186/1475-2859-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larroy C, Fernández MR, González E et al. Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: relevance in aldehyde reduction. Biochem J. 2002a;361:163–72. 10.1042/bj3610163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larroy C, Parés X, Biosca JA. Characterization of a Saccharomyces cerevisiae NADP(H)-dependent alcohol dehydrogenase (ADHVII), a member of the cinnamyl alcohol dehydrogenase family. Eur J Biochem. 2002b;269:5738–45. 10.1046/j.1432-1033.2002.03296.x. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lõoke M, Kristjuhan K, Kristjuhan A. Extraction of genomic DNA from yeasts for PCR-based applications. BioTechniques. 2011;50:325–8. 10.2144/000113672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Cabiscol E, Obradors N et al. Evolution of an Escherichia coli protein with increased resistance to oxidative stress. J Biol Chem. 1998;273:8308–16. 10.1074/jbc.273.14.8308. [DOI] [PubMed] [Google Scholar]
- Maestroni L, Butti P, Milanesi R et al. Easy Modular Integrative fuSion-ready Expression (Easy-MISE) toolkit for fast engineering of heterologous productions in Saccharomyces cerevisiae. ACS Synth Biol. 2023a;12:1508–19. 10.1021/acssynbio.3c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestroni L, Butti P, Senatore VG et al. pCEC-red: a new vector for easier and faster CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. FEMS Yeast Res. 2023b;23:foad002. 10.1093/femsyr/foad002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mückschel B, Simon O, Klebensberger J et al. Ethylene glycol metabolism by Pseudomonas putida. Appl Environ Microbiol. 2012;78:8531–9. 10.1128/AEM.02062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muringayil Joseph T, Azat S, Ahmadi Z et al. Polyethylene terephthalate (PET) recycling: a review. Case Stud Chem Environ Eng. 2024;9:100673. 10.1016/j.cscee.2024.100673. [DOI] [Google Scholar]
- Nisticò R. Polyethylene terephthalate (PET) in the packaging industry. Polym Test. 2020;90:106707. 10.1016/j.polymertesting.2020.106707. [DOI] [Google Scholar]
- Panda S, Fung VYK, Zhou JFJ et al. Improving ethylene glycol utilization in Escherichia coli fermentation. Biochem Eng J. 2021;168:107957. 10.1016/j.bej.2021.107957. [DOI] [Google Scholar]
- Rintala E, Pitkänen J, Vehkomäki M et al. The ORF YNL274c (GOR1) codes for glyoxylate reductase in Saccharomyces cerevisiae. Yeast. 2007;24:129–36. 10.1002/yea.1434. [DOI] [PubMed] [Google Scholar]
- Salusjärvi L, Havukainen S, Koivistoinen O et al. Biotechnological production of glycolic acid and ethylene glycol: current state and perspectives. Appl Microbiol Biotechnol. 2019;103:2525–35. 10.1007/s00253-019-09640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer R, Louden L, Gerhardt P. Porosity of the yeast cell wall and membrane. J Bacteriol. 1974;118:534–40. 10.1128/jb.118.2.534-540.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatore VG, Milanesi R, Masotti F et al. Exploring yeast biodiversity and process conditions for optimizing ethylene glycol conversion into glycolic acid. FEMS Yeast Res. 2024;24:foae024. 10.1093/femsyr/foae024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MG, Des Etages SG, Snyder M. Microbial synergy via an ethanol-triggered pathway. Mol Cell Biol. 2004;24:3874–84. 10.1128/MCB.24.9.3874-3884.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier V, Topham CM, Gilles A et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature. 2020;580:216–9. 10.1038/s41586-020-2149-4. [DOI] [PubMed] [Google Scholar]
- Trifunović D, Schuchmann K, Müller V. Ethylene glycol metabolism in the acetogen Acetobacterium woodii. J Bacteriol. 2016;198:1058–65. 10.1128/JB.00942-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranukul B, Woolston BM, Fink GR et al. Biosynthesis of monoethylene glycol in Saccharomyces cerevisiae utilizing native glycolytic enzymes. Metab Eng. 2019;51:20–31. 10.1016/j.ymben.2018.09.012. [DOI] [PubMed] [Google Scholar]
- van Dijken JP, Bauer J, Brambilla L et al. An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strains. Enzyme Microb Technol. 2000;26:706–14. 10.1016/S0141-0229(00)00162-9. [DOI] [PubMed] [Google Scholar]
- Verduyn C, Postma E, Scheffers WA et al. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–17. 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- Wang H-Y, Xiao D-F, Zhou C et al. YLL056C from Saccharomyces cerevisiae encodes a novel protein with aldehyde reductase activity. Appl Microbiol Biotechnol. 2017;101:4507–20. 10.1007/s00253-017-8209-5. [DOI] [PubMed] [Google Scholar]
- Waterham HR, Russell KA, Vries Yd et al. Peroxisomal targeting, import, and assembly of alcohol oxidase in Pichia pastoris. J Cell Biol. 1997;139:1419–31. 10.1083/jcb.139.6.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Qi X, Cao Z et al. Biotransformation of ethylene glycol by engineered Escherichia coli. Synth Syst Biotechnol. 2024;9:531–9. 10.1016/j.synbio.2024.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ET, Pilgrim D. Isolation and DNA sequence of ADH3, a nuclear gene encoding the mitochondrial isozyme of alcohol dehydrogenase in Saccharomyces cerevisiae. Mol Cell Biol. 1985;5:3024–34. 10.1128/mcb.5.11.3024-3034.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Liu X, Lv Y et al. Tandem production of high-purity sodium glycolate via the dual purification technology of crystallization and active carbon adsorption. Chem Eng J. 2023;452:138994. 10.1016/j.cej.2022.138994. [DOI] [Google Scholar]
- Zhan C, Li X, Lan G et al. Reprogramming methanol utilization pathways to convert Saccharomyces cerevisiae to a synthetic methylotroph. Nat Catal. 2023;6:435–50. 10.1038/s41929-023-00957-w. [DOI] [Google Scholar]
- Zhang H, Shi L, Mao X et al. Enhancement of cell growth and glycolic acid production by overexpression of membrane-bound alcohol dehydrogenase in Gluconobacter oxydans DSM 2003. J Biotechnol. 2016;237:18–24. 10.1016/j.jbiotec.2016.09.003. [DOI] [PubMed] [Google Scholar]
- Zhang J, Petersen SD, Radivojevic T et al. Combining mechanistic and machine learning models for predictive engineering and optimization of tryptophan metabolism. Nat Commun. 2020;11:4880. 10.1038/s41467-020-17910-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang B, Lin J et al. Oxidation of ethylene glycol to glycolaldehyde using a highly selective alcohol dehydrogenase from Gluconobacter oxydans. J Mol Catal B Enzym. 2015;112:69–75. 10.1016/j.molcatb.2014.12.006. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.