

Abstract
Backround
Cigarette smoke exposure including biologically active lipopolysaccharide (LPS) in the particulate phase of cigarette smoke induces activation of alveolar macrophages (AM) and alveolar epithelial cells leading to production of inflammatory mediators. This represents a crucial mechanism in the pathogenesis of chronic obstructive pulmonary disease (COPD). Respiratory pathogens are a major cause of exacerbations leading to recurrent cycles of injury and repair. The interaction between pathogen-associated molecular patterns and the host is mediated by pattern recognition receptors (PRR's). In the present study we characterized the expression of Toll-like receptor (TLR)- 2, TLR4 and CD14 on human AM compared to autologous monocytes obtained from patients with COPD, healthy smokers and non-smokers.
Methods
The study population consisted of 14 COPD patients without evidence for acute exacerbation, 10 healthy smokers and 17 healthy non-smokers stratified according to age. The expression of TLR2, TLR4 and CD14 surface molecules on human AM compared to autologous monocytes was assessed ex vivo using FACS analysis. In situ hybridization was performed on bronchoalveolar lavage (BAL) cells by application of the new developed HOPE-fixative.
Results
The expression of TLR2, TLR4 and CD14 on AM from COPD patients, smokers and non-smokers was reduced as compared to autologous monocytes. Comparing AM we detected a reduced expression of TLR2 in COPD patients and smokers. In addition TLR2 mRNA and protein expression was increased after LPS stimulation on non-smokers AM in contrast to smokers and COPD patients.
Conclusion
Our data suggest a smoke related change in the phenotype of AM's and the cellular response to microbial stimulation which may be associated with impairment of host defenses in the lower respiratory tract.
Backround
COPD patients appear to have underlying pathologic abnormalities which facilitate bacterial colonisation and result in an increased rate of respiratory infections. Bacteria are detected in 40–60 % of exacerbations [1], and the significance of exacerbations for clinical course and decline of lung function is increasingly acknowledged [2]. The mechanisms of the increased susceptibility to bacterial infections are poorly understood. In addition to the impaired mucociliary clearance, deficient functions of the innate immune system seem to be of importance [3]. Incomplete elimination of bacterial pathogens contributes to continuing activation of immune effector mechanisms [4], possibly resulting in damage of the mucosa and parenchyma.
Tobacco smoking is known to induce inflammatory processes. Recent data demonstrated high concentrations of lipopolysaccharide (LPS) in cigarette tobacco as well as biologically active LPS in the particulate phase of cigarette smoke, suggesting a clinical relevance 5. AM play an orchestrating role in the pulmonary immune response. Pathogen recognition receptors (PRR) which are expressed on the macrophage's surface mediate the interaction between conserved patterns on microorganisms, pathogen associated molecular patterns (PAMP's), and host cells [6]. TLR- 4 together with CD14 and the MD2 adapter molecule serves as receptor for components from Gram-negative bacteria such as LPS. TLR2 predominantly recognizes components from Gram-positive bacteria such as lipoteichoic acid (LTA) and peptidoglycan (PGN) [6].
A disturbed regulation of PRR on monocytes and AM may affect the recognition of bacterial pathogens and the intracellular signaling as well as resulting effector mechanisms. A change in the PRR expression on AM from COPD patients may therefore be involved in the process of continuing inflammation and bacterial colonization. In our study we asked whether chronic cigarette smoke exposure alters the expression of PRR's in human monocytes/AM ex vivo. The surface expression of TLR 2, TLR 4 and CD14 was phenotypically characterized on circulating monocytes and AM obtained by BAL in COPD patients, healthy smokers and non-smokers. In addition, the response to LPS stimulation was evaluated at mRNA and protein level.
Methods
Study design
The study population consisted of three groups: 14 COPD patients (11 male, 3 female, FEV1 % predicted: mean 58, range 35–78), 10 healthy smokers (5 male, 5 female, FEV1 % predicted: mean 103, range 92–120), 10 young (6 male, 4 female, FEV1 % predicted: mean 108, range 98–118) and 7 elderly healthy non-smokers (6 male 1 female, FEV1 % predicted: mean 120, range 113–142). In the non-smoker group two age groups were recruited to exclude an age specific effect of PRR expression (young [A], elderly [B]).
Bronchopulmonary infection was excluded by clinical examination, systemic inflammatory markers and chest x-ray. The demographic data of the study population are summarized in table 1. This study was approved by the ethical committee of the University of Lübeck.
Table 1.
Demographic data of the study population. Data are given as mean ± SD. AM = alveolar macrophages, Ly = lymphocytes, PMN = polymorphonuclear neutrophils.
| COPD (n = 14) | Smoker (n = 10) | Non-smoker | ||
| old (n = 7) | Young (n = 10) | |||
| Age | 64.4 ± 9.2 | 30 ± 4.5 | 57.8 ± 6 | 26.8 ± 2.1 |
| GOLD stage | II (n = 9) III (n = 5) |
|||
| Pack years | 43.6 ± 13.8# | 16.2 ± 5.2 | 0 | 0 |
| Cell concentration BAL × 106/100 ml | 22.5 ± 10.8* | 29.4 ± 19.0 | 13.0 ± 3.4 | 10.7 ± 5.1 |
| Diff. Count BAL × 106/100 ml AM | 20.1 ± 9.7* | 27.7 ± 18.2 | 11.1 ± 3.3 | 9.4 ± 4.8 |
| Ly | 1.2 ± 0.41 | 0.92 ± 0.31 | 1.6 ± 2.1 | 0.9 ± 0.29 |
| PMN | 0.95 ± 0.5+ | 0.66 ± 0.2 | 0.21 ± 0.23 | 0.21 ± 0.1 |
| Gram stain smear | negative | negative | negative | Negative |
#= p < 0.01 vs. smokers, * = p < 0.01 vs. non-smokers, + = p < 0.01 vs. smokers and non-smokers.
Bronchoscopy and isolation of BAL cells
After sedation with midazolam (3–10 mg) and local anesthesia with 2% lidocain bronchoscopically guided lavage was performed according to standard conditions in the middle lobe with instillation of 300 ml 0,9% NaCl. 20 ml aliquots were instilled and immediately reaspirated, recovery was 70–90 % for smokers and non-smokers and 35–68% for COPD patients. The lavage fluid was diluted to a final volume of 50 ml and filtered through four layers of gauze to eliminate remaining mucus [7]. Cells were differentiated counting a minimum of 600 cells on a cytocentrifuge smear (Cytospin II, Shandon, Frankfurt) stained with May-Grünwald/Giemsa solution. Gram-stains were performed on a cytocentrifuge smear, and culture for bacteria and yeast was routinely performed which did not show significant growth of pathogenic microorganisms. Viability was determined by trypan blue dye exclusion and the sample was diluted to a concentration of 106 viable cells/ml.
Studies of AM were always carried out in parallel with studies of peripheral blood monocytes from the same subject.
Isolation of peripheral blood mononuclear cells (PBMC)
Peripheral venous blood was drawn 20–40 min. before bronchoscopy and PBMC were isolated from heparinized whole blood samples by Percoll density gradient centrifugation.
Cell culture and in vitro stimulation
BAL cells and PBMCs were cultured in 6-well tissue plates (Nunc, Wiesbaden, Germany) using endotoxin-free RPMI 1640 medium (Biochrome, Berlin, Germany) supplemented with 2 mM L-glutamine (Gibco, Eggenstein, Germany) and 100 mg/ml streptomycin (Gibco, Eggenstein, Germany) at a density of 1 × 106 cells/ ml at 37°C in a 5% CO2 humidified atmosphere for a period of 4 h. Nonadherent cells were then carefully removed and the pellet was again cultured in medium which was supplemented with 1 μg/ml highly purified lipopolysaccharide (LPS, Salmonella friedenau, kindly provided by Prof. Brade, Research Center Borstel) for stimulation experiments. Preliminary experiments testing increasing LPS concentrations demonstrated a dose-dependent effect (TLR expression on AM in response to 0.1 μg/ml LPS: 12.8 rMFI vs. 16.1 rMFI after 1 μg LPS/ml, mean of n = 3 experiments).
Flow cytometry
To facilitate flowcytometric analysis of the AM, we used a previously described quenching technique which reduces intracellular fluorescence and permits analysis of fluorochrome-labeled antibodies by flow cytometry [8]. After 4 h of in vitro cultivation cells were analyzed for surface antigen expression. The expression of TLR2, TLR4 and CD14 on AM and autologous monocytes was determined using a fluorescence activated cell sorter (FACS) Calibur (Becton Dickinson, Heidelberg, Germany). Data acquisition and analysis were performed with CellQuest software (Becton Dickinson, Heidelberg, Germany). Each measurement contained ≥ 20,000 cells in the AM and monocyte population determined by characteristic forward/orthogonal light scattering in a density plot and positive HLA-DR expression. For compensation of the autofluorescence of AM, cell preparations were performed using crystal violet like recommended previously [8]. For permeabilization Intraprep reagent was used (Beckman Coulter, Krefeld, Germany). Antibodies against the following epitopes were used. PE-labeled: TLR2, TLR4, isotype controls (eBioscience, San Diego, USA), CD14, isotype control; PE-CY-5-labeled: HLA-DR, isotype control (the latter all purchased from BD Pharmingen, Hamburg, Germany). The expression of surface markers was calculated as relative mean fluorescence intensity (rMFI = monoclonal antibody/ corresponding isotype control) since no bimodal distribution was found.
In situ hybridization (ISH)
In a subgroup of 13 non-smokers and six COPD patients BAL cells were attached on SuperFrost Plus microscope slides (Menzel-Gläser, Braunschweig, Germany) by centrifugation for 5 minutes at 450 rpm at high acceleration in a Cytospin 2 centrifuge (Shandon, Frankfurt, Germany) and dried for 10 min at room temperature. After overnight fixation at 4°C in Hepes-Glutamic acid buffer mediated Organic solvent Protection Effect (HOPE) solution, cells were incubated with acetone/glyoxal for 1 hr. at 4°C, 6 times dehydrated with acetone for 30 min. at 4°C, followed by two incubations in isopropanol (10 min at 60°C, 2 min. at 60°C) and air dryed. Rehydration was achieved by incubation in 70% (vol/vol) acetone for 10 min. at 4°C and DEPC treated water for 10 min. at 4°C [9-11]. Slides were air dried. A TLR2 probe for ISH was prepared as previously described [12], and ISH was carried out overnight in moist chambers at 46°C. Post hybridization washes and the detection of hybrids have been described previously [9,12]. The generation of signals was achieved in approximately 10 minutes. Slides were mounted and digitally photographed.
Statistics
Nonparametric statistics were used throughout the study. Data are given as mean ± SD, if not otherwise indicated. Surface antigen expression on AM and monocytes from COPD patients, smokers and non-smokers were tested by the analysis of variance followed by Kruskal Wallis test and the Wilcoxon signed rank test was used for comparison of paired samples (pulmonary vs. systemic cells from the same persons and stimulation experiments). A p value < 0.05 was considered as significant.
Results
Differential PRR surface pattern of monocytes and AM
The expression of CD14, TLR4 and TLR2 was higher on monocytes compared to AM in non-smokers (A and B), smokers and COPD patients (CD14: 42.92 ± 12.15 vs. 5 ± 1.56; 36.1 ± 15.4 vs. 4.7 ± 1.6 [nonsmoker groups], 32.4 ± 16.2 vs. 4.09 ± 0.69 and 40.8 ± 10.7 vs. 4.3 ± 1.53 relative mean fluorescence intensity (rMFI), p < 0.01; TLR4: 10.81 ± 2.88 vs. 5.19 ± 1.58; 8.7 ± 5.2 vs. 5.3 ± 2.1; 12.3 ± 4.6 vs. 4.28 ± 0.7 and 9.2 ± 3.9 vs. 4.62 ± 1.38 rMFI, p < 0.01; TLR2: 29.71 ± 9.01 vs. 13.98 ± 2.54; 24.8 ± 10.6 vs. 12.07 ± 3.5; 32.3 ± 8.4 vs. 6.59 ± 1.42 and 27.3 ± 10.8 vs. 6.08 ± 1.5 rMFI, p < 0.01). There was no significant difference in the PRR expression on monocytes from non-smokers (A and B), smokers, COPD patients. Figure 1a shows data from non-smokers (A), a representative histogram demonstrates the ratio of isotype control to surface molecule fluorescence (figure 1b). In addition there was also a difference in the percentage of positive cells (monocytes vs. AM, CD14: 96 vs. 9.5; TLR4: 69 vs. 11, TLR2: 87.5 vs. 22.5 % positive).
Figure 1.

(A) Flow cytometry expression of CD14, TLR4 and TLR2 on monocytes and autologous alveolar macrophages (AM). rMFI (± SD) is shown from non-smokers (n = 10). White bars = monocytes, black bars = AM. * = p < 0.01 vs. AM. rMFI = relative mean fluorescence intensity. (B) Representative histogram from experiments with monocytes.
PRR expression of AM from non-smokers, smokers, COPD patients
Comparing AM from non-smokers (A and B), smokers and COPD patients we detected a markedly lower expression of TLR2 on AM from smokers and COPD patients (13.98 ± 2.54 and 12.07 ± 3.5 [nonsmoker groups] vs. 6.59 ± 1.42 and 6.08 ± 1.5 rMFI, respectively, p < 0.01) (figure 2a). There was no difference in the expression of CD14 and TLR4 between the groups (CD14: 5 ± 1.56 and 4,7 ± 1,6 vs. 4.09 ± 0.69 and 4.3 ± 1.53 rMFI; TLR4: 5.19 ± 1.58 and 5,3 ± 2,1 vs. 4.28 ± 0.7 and 4.62 ± 1.38 rMFI, respectively). Percentage of positive cells showed analogous data (nonsmokers (A and B) vs. smokers, COPD patients, TLR2: 22.5 and 19.2 vs. 12.9 and 12.2; CD14: 9.5 and 8.5 vs. 8.1 and 8.7; TLR4: 11 and 12.5 vs. 8.9 and 10.3 % positive). A representative histogram demonstrates the ratio of isotype control to surface molecule fluorescence (figure 2b).
Figure 2.
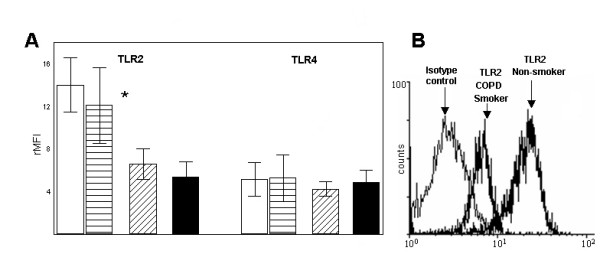
(A) Flow cytometry expression of TLR2 and TLR4 on alveolar macrophages (AM). rMFI (± SD) is shown from non-smokers (white bars [young], n = 10, horizontal hatched bars [elderly], n = 7), smokers (diagonal hatched bars, n = 10) and COPD patients (black bars, n = 14). * = p < 0.01 vs. smokers and COPD patients AM. rMFI = relative mean fluorescence intensity. (B) Representative histogram from experiments with AM.
Regulation of PRR expression on AM
To study the regulation of PRR expression after ligand stimulation, cells were exposed to LPS (1 μg/ml) which led to an increased expression of TLR2 on AM from non-smokers (18.35 ± 4.24 vs. 13.98 ± 2.54 rMFI [A] p < 0.04; 21.1 ± 6.23 vs. 12.07 ± 3.5 [B] p < 0.04). In contrast, cells of smokers and COPD patients did not respond to LPS stimulation with increased TLR2 expression (7.09 ± 1.42 vs. 6.59 ± 1.42 rMFI [smokers], p = n.s.; 6.63 ± 2.40 vs. 6.24 ± 1.71 rMFI, [COPD patients], p = n.s.) (figure 3a). Percentage of positive cells showed analogous data (nonsmokers: 28 vs. 22.5 [A], 30.1 vs. 19.2 [B], smokers: 13.8 vs. 12.9, COPD patients: 13.0 vs. 12.2 % positive). There was no effect of LPS stimulation on the surface expression of CD14 and TLR4 in all groups. Representative results of ISH targeting human TLR2-mRNA before and after LPS-stimulation in non-smokers (A and B) and COPD patients are photographically displayed in figure 3b–e. HOPE-fixed specimens showed a good preservation of morphology after the ISH procedure. The generation of signals was achieved in approximately 10 minutes. Strong signals were found in AM of non-smokers (A and B) after LPS stimulation. Nonspecific signals were not detected in control preparations, in which specific DNA probes were substituted by hybridization buffer alone or an irrelevant probe.
Figure 3.
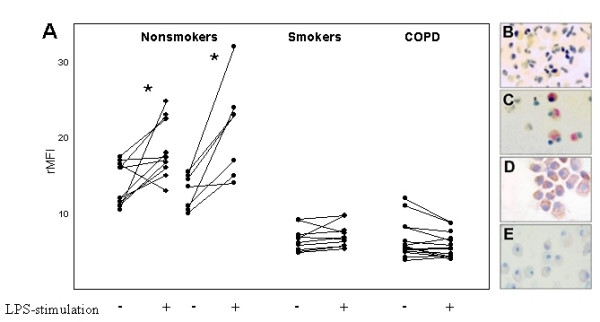
LPS-stimulation of TLR2 protein and mRNA on alveolar macrophages (AM). (A) Flow cytometry expression. rMFI (± SD) is shown from non-smokers (young n = 10, elderly n = 7), smokers (n = 10) and COPD patients (n = 14). rMFI = relative mean fluorescence intensity. * = p < 0,04. In situ hybridization targeting TLR2 mRNA of HOPE-fixated BAL cells. Cells from non-smokers before (B) and after (C [young], D [elderly]) LPS-stimulation. Cells from COPD patients (E) after LPS-stimulation (Anti-DIG-AP-New-fuchsine; 600x).
Discussion
In this study we comparatively evaluated the influence of chronic smoke exposure on the pattern of TLR2, TLR4 and CD14 expression in human AM and monocytes in COPD patients, smokers and non-smokers. We observed a significantly decreased expression of PRR's on AM compared to monocytes. The main finding was that AM from COPD patients and smokers show an equally decreased surface expression of TLR2 compared to non-smokers of two age groups. In addition, an upregulation of TLR2 after LPS stimulation was only observed on non-smokers AM.
An increased TLR2 surface expression on human monocytes in response to LPS-stimulation has been described previously [13], whereas on the transcriptional level divergent data have been reported showing either upregulation [14,15], or downregulation [16,17] of TLR2-mRNA depending on timing and dose of stimulation. In addition, differential regulation of TLR2 by IL-1, IL-10 and GM-CSF (upregulation) and IFN-gamma, TNF-α and IL-4 (downregulation) has been observed [13]. Therefore, the net result of stimulation in vivo will depend on the local balance of inflammatory mediators. What are the possible consequences of LPS-stimulated TLR2 expression? Whole gram-negative bacteria are recognized not only through TLR4 (by LPS) but also TLR 2 (by bacterial lipopeptides). In this setting upregulation of TLR2 which occurs mainly with high LPS doses, may provide an additional mechanism to sensitize cells against large microbial challenges. Moreover, TLR2 is the main PRR recognizing gram-positive bacteria, and recent studies have shown that TLR2 -deficient animals are at high risk for succumbing to invasive pneumococcal infections [18]. Thus, the blunted TLR2 expression of AM from smokers and COPD patients after LPS stimulation may impair antimicrobial defenses in the lower respiratory tract. Recently a reduced TLR expression in aging mice was demonstrated [19]. However this factor seems to be without influence on our results since comparable levels of PRR expression on monocytes and AM from healthy young and elderly non-smokers were observed.
The mechanisms of the smoking induced alteration of pulmonary immune functions are poorly understood. On the one hand smoking is known to induce inflammatory processes by activating AM and epithelial cells leading to production of TNF-α, IL-8 and LTB4 and subsequent neutrophil recruitment [20]. Accordingly, in BALF and sputum of patients with COPD neutrophil activation and elevated levels of proinflammatory cytokines and chemokines have been found [21]. On the other hand a depressed capacity for LPS-induced cytokine release of TNF-α and IL-6 from AM of smokers was described [22]. Skold and colleagues found a higher expression of CD11a, CD54 and CD71 in non-smoker's AM compared with smokers [23]. CD11a (LFA-1) and its ligand play an important role in the interaction between antigen presenting cells and T-lymphocytes. In addition the metabolic response after in vitro stimulation with phorbol myristate acetate (PMA) was higher in non-smokers than in smokers AM. Dandrea et al. observed a reduced inflammatory cytokine release in cultured AM from smokers in response to LPS by simultaneous exposure to NO2 compared to non-smokers [24]. Cultured human bronchial epithelial cells from COPD patients release lower levels of inflammatory mediators such as TNF-α and IL-8 than similar preparations from non-smokers or smokers without COPD, suggesting that downregulation of inflammatory mediator release may also occur in bronchial epithelial cells of individuals with COPD [25].
Our data demonstrating an altered AM phenotype with reduced expression of TLR2 in smokers and COPD patients suggest that a continuous exposure to microbial products in this disease provided by bacterial colonization and LPS present in tobacco smoke [5] may downmodulate the pulmonary immune response. Whether this is due to the selection of a heterogenous macrophage subpopulation in the pulmonary compartment or to a general AM phenotype change under the environmental conditions described cannot be firmly differentiated from our data. However, with regard to the continous distribution of TLR expression intensities found by flow cytometry the latter possibility seems more likely. In vitro it was shown that the TLR response is downregulated after repetitive stimulation [26]. Interestingly a hyporesponsiveness of cells to the TLR-4 ligand LPS was shown as well after preincubation with ligands for TLR2 and vice versa, which indicates the existence of common signaling pathways of the TLR system [27]. It is tempting to speculate that this phenomenon also plays a role under conditions of chronic stimulation with bacterial components in vivo as suggested by the missing effect of LPS-stimulation on TLR2 expression on cells from smokers and COPD patients. This finding was confirmed using in situ hybridization targeting TLR2, which was recently demonstrated by our group on AM and alveolar epithelial cells type II in the human lung [12]. Although there was only a small overlap between non-smokers on the one hand and smokers and COPD patients on the other we observed a large variability in the extent of response to LPS stimulation in the non-smoking group which is a well-known phenomenon with regard to the release of biologic mediators as proinflammatory cytokines [28]. In contrast, no difference in TLR4 expression between smokers, non-smokers, COPD patients was observed in our study which may be due to the generally weaker expression of this molecule on monocytes and AM.
Functionally relevant polymorphisms of TLR's have been found in persons with endotoxin hyporesponsiveness (TLR-4) and staphylococcal sepsis (TLR-2) [29,30]. Data regarding their relevance in COPD are not available. The generally lower expression of PRR's on AM's compared to monocytes is comparable to data reported for dendritic cells and may accompany the differentiation process of monocytes to macrophage populations [15]. Regarding the effect of chronic smoke exposure on blood cells we did not find any difference in the PRR expression on monocytes between smokers and non-smokers (data not shown) suggesting a compartmentalized effect of tobacco smoking. In contrast Lauener et al. demonstrated a significantly increased expression of CD14 and TLR2 on blood cells of farmers' children compared to non-farmers' children [31].
In conclusion, the altered phenotype of smokers AM could play a role in decreased cellular responses to microbial stimulation facilitating persistent infection. Further studies regarding to the functional relevance of these findings and their contribution to the pathogenesis of COPD could lead to more effective treatment regimens of this disease.
Abbreviations
AM = alveolar macrophage; BAL = bronchoalveolar lavage; COPD = chronic obstructive pulmonary disease; FACS = Fluorescence activated cell sorter; GM-CSF = granulocyte-macrophage colony-stimulating factor; HOPE = Hepes-Glutamic acid buffer mediated Organic solvent Protection Effect; IL = interleukin; IFN-γ = interferon-γ; LTA = lipoteichoic acid; LPS = lipopolysaccharide; PBMC = peripheral blood mononuclear cell; PE = phycoerythrin; PGN = peptidoglycan; PMA = phorbol myristate acetate; PMN = polymorphonuclear neutrophils; PRR = pattern recognition receptor; rMFI = relative mean fluorescence intensity; TLR = Toll-like receptor; TNF-α = tumor necrosis factor-α
Authors' contributions
DD carried out the flow cytometry and was involved in the design of the study and drafting the manuscript. TG performed the in situ hybridization and conceived of the study. TT carried out cell culture experiments and was involved in drafting the manuscript. PZ, KD and BS conducted the clinical part of the study and were involved in the design and coordination of the study. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
The authors thank S. Ross, J. Hofmeister, H. Kühl and W. Martens for excellent technical assistance.
Contributor Information
Daniel Droemann, Email: ddroemann@fz-borstel.de.
Torsten Goldmann, Email: tgoldmann@fz-borstel.de.
Thorsten Tiedje, Email: ttiedje@fz-borstel.de.
Peter Zabel, Email: pzabel@fz-borstel.de.
Klaus Dalhoff, Email: klaus.dalhoff@uni-luebeck.de.
Bernhard Schaaf, Email: schaaf@uni-luebeck.de.
References
- Sethi S, Muscarella K, Evans N, Klingman KL, Grant BJ, Murphy TF, et al. Airway inflammation and etiology of acute exacerbations of chronic bronchitis. Chest. 2000;118:1557–1565. doi: 10.1378/chest.118.6.1557. [DOI] [PubMed] [Google Scholar]
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–852. doi: 10.1136/thorax.57.10.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto A, Reyes E, Bernstein ED, Martinez B, Monserrat J, Izquierdo JL, Callol L, de Lucas P, Alvarez-Sala R, Alvarez-Sala JL, Villarrubia VG, Alvarez-Mon M. Defective natural killer and phagocytic activities in chronic obstructive pulmonary disease are restored by glycophosphopeptical (inmunoferon) Am J Respir Crit Care Med. 2001;163:1578–1583. doi: 10.1164/ajrccm.163.7.2002015. [DOI] [PubMed] [Google Scholar]
- White AJ, Gompertz S, Bayley DL, Hill SL, O'Brien C, Unsal I, Stockley RA. Resolution of bronchial inflammation is related to bacterial eradication following treatment of exacerbations of chronic bronchitis. Thorax. 2003;58:680–685. doi: 10.1136/thorax.58.8.680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasday JD, Bascom R, Costa JJ, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest. 1999;115:829–835. doi: 10.1378/chest.115.3.829. [DOI] [PubMed] [Google Scholar]
- Imler JL, Hoffmann JA. Toll receptors in innate immunity. Trends Cell Biol. 2001;11:304–311. doi: 10.1016/S0962-8924(01)02004-9. [DOI] [PubMed] [Google Scholar]
- Dalhoff K, Braun J, Wiessmann KJ, Hollandt H, Marre R. Bronchoscopic diagnosis of pneumonia with quantitative microbial count determination. Dtsch Med Wochenschr. 1990;115:1459–1465. doi: 10.1055/s-2008-1065178. [DOI] [PubMed] [Google Scholar]
- Hallden G, Skold CM, Eklund A, Forslid J, Hed J. Quenching of intracellular autofluorescence in alveolar macrophages permits analysis of fluorochrome labelled surface antigens by flow cytofluorometry. J Immunol Methods. 1991;142:207–214. doi: 10.1016/0022-1759(91)90108-R. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wiedorn KH, Kuhl H, Olert J, Branscheid D, Pechkovsky D, Zissel G, Galle J, Muller-Quernheim J, Vollmer E. Assessment of transcriptional gene activity in situ by application of HOPE-fixed, paraffin-embedded tissues. Pathol Res Pract. 2002;198:91–95. doi: 10.1078/0344-0338-00192. [DOI] [PubMed] [Google Scholar]
- Olert J, Wiedorn KH, Goldmann T, Kuhl H, Mehraein Y, Scherthan H, Niketeghad F, Vollmer E, Muller AM, Muller-Navia J, HOPE fixation. a novel fixing method and paraffin-embedding technique for human soft tissues. Pathol Res Pract. 2001;197:823–826. doi: 10.1078/0344-0338-00166. [DOI] [PubMed] [Google Scholar]
- Umland O, Ulmer AJ, Vollmer E, Goldmann T. HOPE fixation of cytospin preparations of human cells for in situ hybridization and immunocytochemistry. J Histochem Cytochem. 2003;51:977–980. doi: 10.1177/002215540305100714. [DOI] [PubMed] [Google Scholar]
- Droemann D, Goldmann T, Branscheid D, Clark R, Dalhoff K, Zabel P, Vollmer E. Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lung. Histochem Cell Biol. 2003;119:103–108. doi: 10.1007/s00418-003-0497-4. [DOI] [PubMed] [Google Scholar]
- Flo TH, Halaas O, Torp S, Ryan L, Lien E, Dybdahl B, Sundan A, Espevik T. Differential expression of Toll-like receptor 2 in human cells. J Leukoc Biol. 2001;69:474–481. [PubMed] [Google Scholar]
- Liu Y, Wang Y, Yamakuchi M, Isowaki S, Nagata E, Kanmura Y, Kitajima I, Maruyama I. Upregulation of toll-like receptor 2 gene expression in macrophage response to peptidoglycan and high concentration of lipopolysaccharide is involved in NF-kappa b activation. Infect Immun. 2001;69:2788–2796. doi: 10.1128/IAI.69.5.2788-2796.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin A, Mazzoni A, Spitzer JH, Wyllie DH, Dower SK, Segal DM. Regulation of Toll-like receptors in human monocytes and dendritic cells. J Immunol. 2001;166:249–255. doi: 10.4049/jimmunol.166.1.249. [DOI] [PubMed] [Google Scholar]
- Muzio M, Polentarutti N, Bosisio D, Prahladan MK, Mantovani A. Toll-like receptors: a growing family of immune receptors that are differentially expressed and regulated by different leukocytes. J Leukoc Biol. 2000;67:450–456. doi: 10.1002/jlb.67.4.450. [DOI] [PubMed] [Google Scholar]
- Haehnel V, Schwarzfischer L, Fenton MJ, Rehli M. Transcriptional regulation of the human toll-like receptor 2 gene in monocytes and macrophages. J Immunol. 2002;168:5629–5637. doi: 10.4049/jimmunol.168.11.5629. [DOI] [PubMed] [Google Scholar]
- Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R. Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis. 2002;186:798–806. doi: 10.1086/342845. [DOI] [PubMed] [Google Scholar]
- Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–4701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- MacNee W. Oxidants/antioxidants and COPD. Chest. 2000;117:303S–317S. doi: 10.1378/chest.117.5_suppl_1.303S-a. [DOI] [PubMed] [Google Scholar]
- Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153:530–534. doi: 10.1164/ajrccm.153.2.8564092. [DOI] [PubMed] [Google Scholar]
- McCrea KA, Ensor JE, Nall K, Bleecker ER, Hasday JD. Altered cytokine regulation in the lungs of cigarette smokers. Am J Respir Crit Care Med. 1994;150:696–703. doi: 10.1164/ajrccm.150.3.8087340. [DOI] [PubMed] [Google Scholar]
- Skold CM, Lundahl J, Hallden G, Hallgren M, Eklund A. Chronic smokeexposure alters the phenotype pattern and the metabolic response in human alveolar macrophages. Clin Exp Immunol. 1996;106:108–113. doi: 10.1046/j.1365-2249.1996.d01-805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandrea T, Tu B, Blomberg A, Sandstrom T, Skold M, Eklund A, Cotgreave I. Differential inhibition of inflammatory cytokine release from cultured alveolar macrophages from smokers and non-smokers by NO2. Hum Exp Toxicol. 1997;16:577–588. doi: 10.1177/096032719701601005. [DOI] [PubMed] [Google Scholar]
- Mills PR, Davies RJ, Devalia JL. Airway epithelial cells, cytokines, and pollutants. Am J Respir Crit Care Med. 1999;160:S38–S43. doi: 10.1164/ajrccm.160.supplement_1.11. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- Sato S, Nomura F, Kawai T, Takeuchi O, Muhlradt PF, Takeda K, Akira S. Synergy and cross-tolerance between toll-like receptor (TLR) 2- and TLR4-mediated signaling pathways. J Immunol. 2000;165:7096–7101. doi: 10.4049/jimmunol.165.12.7096. [DOI] [PubMed] [Google Scholar]
- Zabel P, Schonharting MM, Schade UF, Schlaak M. Effects of pentoxifylline in endotoxinemia in human volunteers. Prog Clin Biol Res. 1991;367:207–213. [PubMed] [Google Scholar]
- Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun. 2000;68:6398–6401. doi: 10.1128/IAI.68.11.6398-6401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauener RP, Birchler T, Adamski J, Braun-Fahrlander C, Bufe A, Herz U, von Mutius E, Nowak D, Riedler J, Waser M, Sennhauser FH, ALEX study group Expression of CD14 and Toll-like receptor 2 in farmers' and non- farmers' children. Lancet. 2002;360:465–466. doi: 10.1016/S0140-6736(02)09641-1. [DOI] [PubMed] [Google Scholar]