

Abstract
We examine the role of electronic polarizability in water on short (tens of femtoseconds), intermediate (hundreds of femtoseconds), and long (≈1 ps) time scales by comparing molecular dynamics results to experimental data for vibrational spectroscopy of HOD in liquid D2O. Because the OH absorption frequency is sensitive to the details of the atomic forces experienced in the liquid, our results provide important quantitative comparisons for several popular empirical water potentials. When compared with their fixed-charge counterparts, the polarizable models give similar slower long time constants for the decay of vibrational correlations and reorientational motion that is in better agreement with experiments. Polarizable potentials yield qualitatively dissimilar predictions for frequency fluctuations and transition dipole moment fluctuations at equilibrium. Models that confine the polarizability to the plane of the molecule (i.e., TIP4P–FQ) overestimate the width of the distribution describing frequency fluctuations by more than a factor of two. These models also underestimate the amplitude of the hydrogen-bond stretch at 170 cm–1. A potential that has both an out-of-plane polarization and fluctuating charges, POL5–TZ, compares best with experiments. We interpret our findings in terms of microscopic dynamics and make suggestions that may improve the quality of emerging polarizable force fields for water.
Keywords: hydrogen-bond dynamics, molecular dynamics, polarizable force fields
Molecular dynamics simulations of empirical models have guided our understanding of how water molecules play their essential roles in chemistry, biology, and physics. Using classical molecular dynamics (MD) simulations, Rahman and Stillinger (1) made predictions of bulk thermodynamic, atomic pair correlation functions and transport properties of liquid water that were impressive for their time. Stillinger (2) described the peculiar chemical and physical properties of water in terms of the microscopic details of the hydrogen-bond network and provided a consistent molecular level description of hydrogen bonding in water. The potential energy models developed in these early studies provided guidelines for the design of molecular potentials for water that researchers have used for decades. Most empirical water potentials replace electron densities with point charges situated either at the atomic positions or at “virtual” sites near the molecule, rigid intramolecular bonds take the place of intramolecular vibrations, and a Lennard–Jones potential represents the excluded molecular volume.
In real molecular systems, particularly those with large or polarizable atoms, the electronic degrees of freedom are not stationary. The electric fields in the liquid distort the molecular electron densities and polarize molecules. Even though water has a modest molecular polarizability (≈1 Å3), molecular electric field strengths are high enough (≈1 V/Å) to induce dipole moment fluctuations (≈1 debye) that approach the average value of the molecular dipole in the liquid (≈2–3 debye) (3). The fixed-charge models inspired by Rahman and Stillinger's (1) early work ignore electronic fluctuations. These models average away the fluctuations and replace them with averaged “mean field” values assigned to atomic or virtual sites. Such models are evidently unable to model energetic or dynamic effects arising from distorted and fluctuating electron densities. To incorporate these effects, Rick et al. (4) have developed fluctuating charge (FQ) models and methods for MD (5–7). These protocols and potentials allow the electronic degrees of freedom to deform during the course of the simulation but only introduce a modest additional computational burden (4) over fixed-charge models, providing an attractive alternative to ab initio MD when electronic polarizability is important.
Experimental data used to parameterize and validate both fixed-charge and FQ water potentials are either thermodynamic (i.e., enthalpies of vaporization), transport (i.e., diffusion coefficient), or structural (radial distribution functions) quantities. Such quantities are spatial and temporal averages over a large number of molecules and are not related to the microscopic details of a water potential in a simple way.
Computer simulations of atomistic models examine chemistry in molecular detail and are often a surrogate when experiments are unavailable. MD studies are seldom concerned with mesoscopic or macroscopic behaviors; instead, they often focus on microscopic motions that facilitate electron and proton transfer or fold proteins or participate in biological catalysis between an enzyme and its substrate (8–10). On molecular length scales (Å), the microscopic details of a molecular potential should be important.
The characteristic time scales of intermolecular motions in water are faster than those of any other liquid at standard temperature and pressure. The fastest motion, the librations, have a periodicity of ≈60 fs (11). Femtosecond Raman (12) and electronic solvation dynamics experiments (13) that do have enough time resolution to observe fast intermolecular motions in water probe collective motions of the liquid or view them through a non-native probe molecule. Inelastic neutron and x-ray scattering probes trade high spatial resolution for poor temporal resolution.
Vibrational Spectroscopy from Computer Simulations
The OH stretch frequency in water is sensitive to both the local hydrogen bonding environment (14, 15) and the polarization field generated by the liquid. By isotopically labeling a small number of water molecules, femtosecond IR spectroscopy on the OH stretch of HOD in liquid D2O views water's structural evolution on molecular time scales with an ostensibly native and vibrationally distinct probe. Several reports have appeared recently that use a mixture of nonlinear IR femtosecond spectroscopies to probe microscopic structural evolution in water (16, 17). Similarly, MD studies on the vibrational spectroscopy of water have identified computationally efficient methods for computing the frequency of the OH stretch of HOD in D2O based on an adiabatic separation between the fast vibrational motion and the translations and rotations of the D2O molecules (15, 18, 19). Simulations of fixed-charge models have revealed that the frequency of the OH stretch is sensitive to the electric field strength at the proton in the direction of the OH bond (15, 20). On short time scales (<200 fs), spectroscopy of the OH stretch is sensitive to distance fluctuations between the HOD molecule and its hydrogen-bonding partner (bonded to the H). On time scales >200 fs, the OH stretch probes the configurational fluctuations on length scales larger than a molecular diameter.
Two functions characterize equilibrium fluctuations and provide a connection between experiments and computer simulation. The frequency–frequency time correlation function, Cωω(t), measures the equilibrium fluctuations of the OH vibrational frequency (13), Cωω(t) =〈δωOH(t)δωOH(0)〉, where 〈... 〉 is an equilibrium ensemble average, ωOH is the transition frequency for the |1〉 ← |0〉 transition of the OH stretch, and δωOH(t) = ωOH(t) –〈ωOH〉. Within several reasonable approximations, one can retrieve this correlation function from a three-pulse photon echo peak-shift measurement (13). The second function describes the reorientational motion of the transition dipole moment (TDM), Cor(t) = 〈P2(û(t)·û(0))〉, where û(t) is the unit direction vector of the TDM. Within the Condon approximation, the square of the TDM is proportional to the oscillator strength of the vibrational transition. The TDM is u(t) = ∂μ(t)/∂Q|Q=0. Here, μ(t) is the dipole moment of the HOD molecule at time t, and Q is the OH stretch vibrational coordinate that minimizes the potential energy of the molecule at Q = 0.¶ P2(x) is the second Legendre polynomial, P2(x) = 3(x2 – 1)/2. One obtains Cor(t) by measuring the anisotropy in polarization-selective femtosecond vibrational pump-probe spectroscopy.
Although simple point charge-type models show qualitatively similar features in Cωω(t) and Cor(t) to those extracted from experiments, the results from simulation are not in quantitative agreement with the experiments. Fecko et al. (15, 17) have recently performed nonlinear vibrational spectroscopies with sufficiently fast time resolution to measure water's fastest intermolecular motions. The results from MD simulations with the fixed-charge SPC–E (21) water model underestimated the width of the frequency distribution and predicted a time constant for the long time decay in Cωω(t) that is approximately half of the experimentally measured value.
Because ωOH is sensitive to the electric field strengths, femtosecond vibrational spectroscopies provide a point of comparison on microscopic length scales where the effects of electronic polarizability should be most pronounced. For example, Xu et al. (22) showed that large discrepancies between polarizable and fixed-charge models appear in time correlation functions on picosecond time scales. Xu et al. (22) showed that electronic polarizability strengthens hydrogen bonds between molecules, lengthening the time that pairs of molecules spend hydrogen-bonded to one another.
In this report, we examined the role of electronic polarizability in vibrational spectroscopy with computer simulations of fixed-charge and polarizable potentials of water. We have chosen some of the most popular fixed-charge models for water and, where appropriate, made comparisons to predictions from their polarizable counterparts. In Methods, we describe the protocol developed to compute vibrational spectroscopies. In Results and Discussion, we compare recent experimental results for Cωω(t), Cor(t), and the IR absorption spectrum to predictions from simulations of several fixed-charge and polarizable water models. We conclude by discussing the results and interpreting our findings in terms of predicted and measured microscopic dynamics.
Methods
Polarizable Models of Water. The traditional way of introducing polarizability into a molecular simulation is to allow the dipoles to polarize but to keep the values of the molecule's charges fixed (23, 24). Instead, the FQ method allows the charges to fluctuate so that the electronic degrees of freedom stay near the minimum of the electrostatic potential energy. “Hybrid” models allow both dipoles and charges to fluctuate. Let us consider a parameterization for a water model that places point charges and dipoles at atomic or virtual sites close to the atomic positions. The multipole expansion of the total potential energy of the assembly of charges and dipoles (6, 7) gives
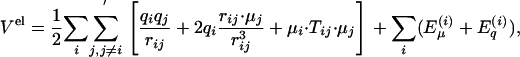 |
[1] |
where {μ} is the set of site dipoles, {q} is the set of site charges, rij = ri – rj, and rij = |rij|. The indices i and j denote the sites in the model, and the prime on the sum over j indicates that this sum skips over sites on the same molecule as i. The first term in the first summand is the familiar Coulomb energy between monopoles, the second is the monopole–dipole energy, and the third is the dipole–dipole energy. Tij is the dipole interaction tensor, 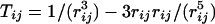 . Polarizable dipoles have both a permanent and induced part. The remaining terms are the self-energies of the sites that define the parameter set of the model. The energies may be approximated by
. Polarizable dipoles have both a permanent and induced part. The remaining terms are the self-energies of the sites that define the parameter set of the model. The energies may be approximated by  . The parameter γi generates a permanent dipole isolated moment on the site i. The polarizability tensor of the dipole site, αi, the Mulliken electronegativity, χi, and twice the hardness of the electronegativity,
. The parameter γi generates a permanent dipole isolated moment on the site i. The polarizability tensor of the dipole site, αi, the Mulliken electronegativity, χi, and twice the hardness of the electronegativity, 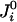 , are also parameters (4, 6). If there is no dipole at site i,
, are also parameters (4, 6). If there is no dipole at site i, 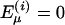 . In the models studied, intramolecular atomic distances are fixed, and the electrostatic interaction between sites on the same molecule are additional parameters (4, 6).
. In the models studied, intramolecular atomic distances are fixed, and the electrostatic interaction between sites on the same molecule are additional parameters (4, 6).
For a given atomic configuration, the values for the charges and dipoles, and thus the forces in the simulation, can be determined by minimizing Eq. 1 with respect to the electronic degrees of freedom under the constraint that the molecules remain electroneutral.
Simulation Details. We have studied several polarizable models of water that are popular in computer simulations and have qualitatively different parameterizations. Specifically, we have examined the three-site fixed-charge models, SPC–E (21), along with the fluctuating charge SPC–FQ (4) model; the four-site fixed-charge models, TIP4P (25), with the fluctuating charge TIP4P–FQ (4) model and the inducible dipole Dang–Chang (DC) (24) model; and the five-site fixed-charge model, TIP5P–E (26), with the combined fluctuating charge and inducible dipole POL5–TZ (6) model. Both the three-site and four-site FQ models confine the charges to the molecular plane and do not allow for an out-of-plane polarization. The DC and POL5–TZ models, however, do allow the molecule to polarize out of the plane. All of the potential models used in this study ignore intramolecular vibrations. Intramolecular vibrations can distort the molecular polarizability, but we ignore such effects and focus only on the electronic contribution. The electronic part of the polarizability should dominate over the vibrational part because the vibrational frequencies are so much larger than kBT that intermolecular interactions do not significantly alter the intramolecular nuclear positions.
We performed simulations at the thermodynamic state point of liquid D2O at ambient conditions, where T = 298.15 K and the density is 1.104 g/cm3. During the initial equilibration period of 10 ps, Nose–Hoover chain thermostats (27) kept a system of 1 HOD molecule dissolved in 107 D2O molecules at constant temperature (298.15 K). The equilibrated trajectories provided initial conditions to generate 17 independent realizations of the system. We ran each trajectory at constant volume and energy for a duration of 500 ps. In the constant energy simulation, a time step of 1 fs kept the fluctuations in the total energy to O(10–4) of the average.
For the polarizable potentials, we minimized Eq. 1 with respect to the electronic degrees of freedom by iteration at the beginning of the simulation and then used the extended Lagrangian method to ensure that the electronic coordinates remained near the minimum of the electrostatic potential throughout the simulation. In the extended Lagrangian method, one assigns a fictitious mass to the fluctuating charges and inducible dipoles to dynamically propagate the electronic degrees of freedom and the nuclei during the course of the simulation. The fictitious electronic mass is chosen small enough so that there is an adiabatic separation between the electronic and nuclear degrees of freedom (7) but also large enough (here, 1 fs) to efficiently sample classical phase space. To compute the long-ranged electrostatics, we used the particle-mesh approximation, P3ME, to include dipolar terms in the Ewald sum (7).
Calculating Vibrational Frequencies. A previous study introduced a computationally efficient strategy for calculating the vibrational frequency of an HOD molecule in liquid D2O (15) based on the adiabatic separation of the OH stretch from the translational and rotational degrees of freedom. We follow a similar approach here but modify the protocol to handle electronic degrees of freedom in the polarizable models. Other researchers have pursued a different strategy for computing vibrational frequencies and transition dipoles that relies on a mixture of MD simulation and quantum chemical calculations (28, 29). But this approach does not provide a direct test of the molecular polarizability in the water models, because the frequencies and TDMs are extracted from an electronic structure calculation.
We partition the total Hamiltonian (ℋ) into the quantal system Hamiltonian Hs (OH oscillator), the classical bath Hamiltonian Hb, and the system–bath coupling Hsb.  . Hs is the Hamiltonian of the OH stretch for HOD in the gas phase, which we take from a spectroscopic gas phase Hamiltonian (30). The bath Hamiltonian, Hb describes the translations, rotations, and electronic coordinates of all molecules. Hsb is the system–bath Hamiltonian that couples the OH oscillator with the classical degrees of freedom, both nuclear and electronic. The vibrational eigenstates and energies are solutions to the adiabatic Schrödinger equation for a given frozen configuration of the nuclei at time t, R(t), (Hs + Hsb)|Ψ[R(t)]〉 = ε[R(t)]|Ψ[R(t)]〉.
. Hs is the Hamiltonian of the OH stretch for HOD in the gas phase, which we take from a spectroscopic gas phase Hamiltonian (30). The bath Hamiltonian, Hb describes the translations, rotations, and electronic coordinates of all molecules. Hsb is the system–bath Hamiltonian that couples the OH oscillator with the classical degrees of freedom, both nuclear and electronic. The vibrational eigenstates and energies are solutions to the adiabatic Schrödinger equation for a given frozen configuration of the nuclei at time t, R(t), (Hs + Hsb)|Ψ[R(t)]〉 = ε[R(t)]|Ψ[R(t)]〉.
In the polarizable models, the electronic coordinates are fast relative to the vibrational coordinate. For both the fixed-charge and polarizable models, we solve the total potential energy (Eq. 1) as a function of the vibrational coordinate by translating the H and O atoms that define Q along the OH bond with all other atomic positions fixed. We displaced Q in 0.1-Å intervals while leaving the center of mass between H and O constant. The system–bath coupling at each value of Q is Hsb = V(Q) – V(Q = 0), where Q = 0 corresponds to the OH bond length of a given water model. At each position Qα, the values of {q} and {μ} are solved by minimizing Eq. 1 with respect to the electronic degrees of freedom subject to the constraint of molecular electroneutrality, ensuring that the electronic degrees of freedom respond adiabatically to displacements in Q.
Finally, we fit a third-order polynomial to the discretely sampled Hsb, Hsb = FQ + GQ2 + HQ3, and solve the fitted Hsb to second order in time-independent perturbation theory to obtain the vibrational frequencies for the |1〉 ← |0〉 transition at 12-fs intervals in the simulation. The quantities F, G, and H are functions of the bath degrees of freedom. While acquiring Hsb, we simultaneously obtain the TDM from the slope of a linear fit of the dipole moment of HOD as a function of Q.
Results and Discussion
In this section, we compare our results to available experimental data for Cωω(t), Cor(t), and the OH absorption spectrum of HOD in liquid D2O. A previous report (15) argued that for fixed-charge models, the OH frequency essentially measures the strength of the electric field in the direction of OH. We can make a similar argument for polarizable models. Likewise, the TDM will be a function of the electric field in the direction of the OH bond. A stronger applied field in the direction of the OH bond will induce a larger TDM.
Absorption Line Shape. Perhaps the simplest infrared experiment with which to compare our data is the IR absorption spectrum. In the low-temperature limit, the dipole or E1 approximation to the absorption spectrum (31) is
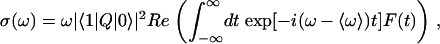 |
[2] |
where, in the adiabatic scheme, F(t) =〈u(t)·u(0) exp[i∫t0δω(t′)dt′]〉. Because F(t) decays on an ≈100-fs time scale (roughly the inverse of the linewidth), including the lifetime (≈500 fs) does not change the spectrum qualitatively. Fig. 1 is a plot of the calculated absorption spectra for the four- and five-site models along with the experimental results.
Fig. 1.

Absorption spectra of HOD in D2O and comparison to experiment. The absorption spectrum is slightly asymmetric, with a steeper tail on the blue side than on the red side. The width (FWHM) of the experimental spectrum is 245 cm–1. (A) Computed absorption spectra of TIP4P and TIP4P–FQ models of water. The TIP4P–FQ model predicts a spectral width in stark contrast with experiment (FWHM ≈ 806 cm–1). (B) The spectrum of the TIP5P–E model slightly underestimates the width (FWHM ≈ 190 cm–1). Molecular polarizability increases the width of the absorption line shapes dramatically, but the notable disagreement of TIP4P–FQ with experiment implies that it is a poor model of molecular polarizability.
To compare spectra, we computed the full width at half maximum (FWHM) and the average frequency red-shift (Δωred = ω0 – 〈ω〉, and ω0 is the experimental gas phase transition frequency). The results for all of the models studied appear in Table 1. The FWHM of the spectra are considerably different for the fixed-charge and polarizable models. The fixed-charge models underestimate the width of the line by ≈30–50 cm–1, and the polarizable models overestimate the width by between 50 cm–1 (POL5–TZ) and 600 cm–1 (TIP4P–FQ).
Table 1. Comparison of experiment and various water models for the frequency fluctuation time scales, caging times, linewidth, distribution width, and solvent frequency red-shift (Δωred).
| Models | τfast, fs | τslow, fs | τor, ps | τcage, ps | Γ (I(ω)), cm-1 | Γ (P(ωOH)), cm-1 | Δωred, cm-1 |
|---|---|---|---|---|---|---|---|
| Experiment | 35 | 1,450 | 3.0 | N/A | 245 | 297 | 294 |
| SPC-E | 39 | 700 | 2.4 | 3.0 | 207 | 295 | 241 |
| SPC-FQ | 36 | 1,380 | 3.2 | 3.4 | 877 | 640 | 628 |
| TIP4P | 35 | 500 | 1.8 | 2.3 | 214 | 300 | 277 |
| TIP4P-FQ | 34 | 1,200 | 3.0 | 3.3 | 806 | 770 | 696 |
| DC | 34 | 770 | 2.7 | 3.1 | 334 | 390 | 368 |
| TIP5P-E | 32 | 690 | 2.0 | 2.6 | 190 | 210 | 140 |
| POL5-TZ | 43 | 1,150 | 3.6 | 3.7 | 307 | 365 | 333 |
The symbol Γ denotes the full width at half the height of the distributions. The experimental width of P(ωOH) comes from the equal time value of Cωω (t) extracted from ref. 17. The time scales come from the fits to a biexponential. This is a different functional form than was used in ref. 17 for Cωω (t), and, thus, the time scales reported here are slightly different. N/A, no data.
If the frequency fluctuations are static on the time scale of the decay for F(t), the absorption spectrum is the equilibrium distribution of absorption frequencies. In Cωω(t), both calculated and experimental, there are both fast (50 fs) and slow (≈1 ps) time scales, but F(t) decays on an ≈100-fs time scale. Therefore, it is natural that a spectrum with a large FWHM should correspond to a broad distribution of frequencies. Fig. 1 and Table 1 confirm this notion. The FWHM of the frequency distribution (see Table 1) in the FQ models (≈700–800 cm–1) significantly overestimates the experimental value of 297 cm–1, as estimated by assuming a Gaussian distribution of ωOH and taking the t = 0 value of Cωω(t). The extra degrees of freedom in polarizable models produce a broader distribution of forces on the proton and hence a broader distribution of ωOH. These forces also change the molecular dipole moment (Fig. 2 Inset) for polarizable water models. The width of the distribution, P(|μ|), is similar for the TIP4P–FQ, DC, and POL5–TZ models. But in Fig. 2, which shows the distribution of the dipole moment along the OH bond, μQ = r̂HO·μ, P(μQ) indicates that the fluctuations of the dipole moment have different anisotropies for different models. This distribution is considerably wider for the TIP4P–FQ model than for the DC and POL5–TZ models and thus should arise from a qualitative difference in the model.
Fig. 2.

Molecular dipole moment and transition dipole moment distribution. (Upper) The distribution of the projection of the dipole moment along the direction of the OH stretch (μQ) along with the distribution of the molecular dipole moment (Inset). The variance in the molecular dipole moment is similar for all of the polarizable models, but the projection into the molecular plane reveals the anisotropy of the polarization response of charges that are confined to lie in the molecular plane. (Lower) The equilibrium TDM distribution. The distribution from the TIP4P–FQ model is much wider than that of either the DC or POL5–TZ models.
In the gas phase, the polarizability tensor of water is almost isotropic (32). In TIP4P–FQ, the charges are confined to lie in the molecular plane so the isotropy is manifestly broken. An electric field applied out of the plane of the molecule does not induce a molecular dipole moment. If we take the gas phase molecular dipole moment to lie along the z axis, then αzz = 0.82 Å3 is the constant of proportionality between the z component of the induced dipole and the electric field directed along z, and αyy = 2.55 Å3 is the proportionality constant between the applied field and the magnitude of the induced dipole in the orthogonal y direction of the molecular plane (4). In TIP4P–FQ, αyy is roughly twice the size of the gas phase value (≈1.44 Å3) and three times the size of the value of αzz. The dipole along the OH bond, which is roughly 45° to both z and y, has a comparatively large susceptibility because αyy is so large. Natural electric field fluctuations at equilibrium thus produce a much larger distribution of μQ in TIP4P–FQ than in the other models.
If the fluctuating charge paradigm for the TIP4P–FQ (and, similarly, the SPC–FQ) model is to be followed, it would be best to allow charges to move out of the molecular plane. We also suggest that screening the bare Coulomb intermolecular interactions at short distances, as in the POL5–TZ model for the out-of-plane virtual sites, may improve the results. Preliminary results indicate that using this screening function to calculate ωOH for the TIP4P–FQ model narrows the spectrum by ≈10, slightly improving the agreement between simulation of the model and the experiment.
In contrast to TIP4P–FQ, the POL5–TZ and DC models do allow the molecule to polarize out of plane. Interestingly, the components of the molecular polarizability tensor correspond closely with the experimental gas phase value. Although an improvement over the TIP4P–FQ model, the absorption spectra for the POL5–TZ and DC models predict a spectrum that is still too wide. The fact that the absorption spectra are consistently wider in polarizable models than they are in fixed-charge models suggests that current polarizable models overestimate the degree of polarizability in the liquid phase. Electronic structure calculations further support this notion. These studies have found that Pauli repulsion distorts the electron cloud on neighboring molecules and reduces the polarizability in the condensed phase (33).
IR experiments do not measure the molecular dipole moment directly; instead, they interact with the molecule through the TDM. The distribution of the TDM as a function of ωOH appears in Fig. 2 Lower. The trends among the polarizable models for the TDM are similar to those for μQ and the distribution of ωOH. The fluctuations of the TDM in TIP4P–FQ make the line more asymmetric than what one would expect if the TDM were time-independent. The peak of the absorption spectrum is ≈200 cm–1 to the red of 〈ωOH〉. The difference between the other polarizable models POL5–TZ (19 cm–1) and DC (6 cm–1) is much more modest.
The red-shift (Δωred) is closely correlated to the mean strength of the electric force on the proton of HOD. The average red-shifts for SPC–E and TIP4P are in close agreement with the experimental value, but the TIP5P–E model underestimates Δωred by ≈150 cm–1, indicating that this model does not provide an accurate representation of the local electric force. In TIP5P–E, the negative point charges are relatively distant (0.7 Å) from the oxygen atom. This charge geometry prevents the model from accurately reproducing both the dipole and quadrupole moments of water. As a result, the quadrupole moment (26) is too small to accurately determine the electric force between the “lone pairs” and hydrogen atom distances typical of hydrogen-bonding geometries. The five-site POL5–TZ model incorporates an additional dipole at the oxygen atom. This parameterization gives both a more accurate quadrupole moment and solvent red-shift.
Frequency Fluctuations at Short and Intermediate Times. We present the normalized frequency time correlation function, Cωω(t)/Cωω(0) from simulations in Fig. 3. Table 1 shows the time constants from a biexponential fit. The experimental time correlation function (17) derived from the vibrational peak-shift experiment initially decays with a time scale of ≈60 fs and shows an underdamped oscillation with a peak at 170 fs. The short time dephasing of the hydroxyl stretch frequency is related to the critically damped internuclear vibration along the hydrogen-bonding coordinate (15, 18, 34) or hydrogen-bond vibration. With the exception of SPC–FQ and TIP4P–FQ, all models examined, both polarizable and fixed charge, exhibit an oscillation in Cωω(t) from the hydrogen-bond vibration.
Fig. 3.

Frequency–frequency correlation functions for the OH stretch of HOD in liquid D2O for various polarizable and fixed-charge models compared with experiment (logarithmic vertical scale). (A) Normalized Cωω(t) for TIP4P (red), TIP4P–FQ (orange), and DC (blue) models. The beat in Cωω(t) from TIP4P–FQ is barely noticeable. (B) Normalized Cωω(t) for TIP5P–E (brown) and POL5–TZ (green) models. The beat frequency in these models is slightly closer to that measured in experiment. The long time decay for both polarizable models is similar, with a time constant of ≈1 ps; for both fixed-charge models, the value is ≈600 fs. The short time exponential decay time for all models is ≈30 fs. The time constants for all of the models studied are presented in Table 1.
But in the TIP4P–FQ model [results for SPC–FQ are not reported here but are analogous (20)], the beat in Cωω(t) is overdamped. Because the TIP4P–FQ model is polarizable, the overdamping might come from fluctuations of molecular charges that do not significantly affect the dynamics of the center of mass. Fig. 4 is a plot of the velocity–velocity autocorrelation functions for the center of mass, Cvv(t), for various polarizable and nonpolarizable water models. This figure indicates that like Cωω(t), Cvv(t) does not show an oscillation. The oscillation appearing in the density fluctuations of water at this frequency has been confirmed experimentally (35) and implicated in the transmission of high-frequency acoustic waves in water (36). More importantly, the disappearance of the beat in Cωω(t) and Cvv(t) indicates that the TIP4P–FQ model incorrectly models these high-frequency, short-ranged internuclear dynamics.
Fig. 4.

Center of mass velocity autocorrelation function for the fixed-charge and FQ models examined in the text. (A) TIP4P (red), TIP4P–FQ (orange), and DC (blue) models of water. The hydrogen-bond vibrations that generate the beat in Cωω(t) for these models also appear in Cvv(t). The black arrow illustrates that the beat absent in Cωω(t) from TIP4P–FQ is nearly absent in Cvv(t). (B) Cvv(t) for TIP5P–E (brown) and POL5–TZ (green). In both of these models, the beat frequency is slightly higher than in TIP4P, mirroring the same behavior for the beat in Cωω(t).
The attractive electrostatic forces between the oxygen and the hydrogen of a bonding pair must balance against the repulsive steric forces that keep molecules from overlapping. The balance of these forces implies that the most probable oxygen–oxygen distance in the liquid is less than the minimum of the potential that describes the steric hindrance of molecular contacts (e.g., Lennard–Jones) (36). The hydrogen-bond vibration is an underdamped motion along the direction of these competing forces. In the TIP4P–FQ model, the Lennard–Jones potential is stiffer than in the models that do display a beat in both Cωω(t) and Cvv(t). This stiff repulsion is to compensate the increased attractive electrostatic force imposed by the large value of the polarizability directed along the OH···O bond. In TIP4P–FQ, the coefficient of the repulsive term for the Lennard–Jones potential is nearly twice that of its fixed-charge counterpart, and 30% larger than the DC model (24). The imbalance of these attractive and repulsive forces weakens the beat. Instead of using a Lennard–Jones function to parameterize the steric interactions, POL5–TZ uses a softer exponential to mimic the repulsion from overlapping electron clouds. This model comes the closest to quantitatively predicting the experimental Cωω(t) in the region of the beat.
Long Time Dynamics. Cωω(t) exhibits relaxations on both subpicosecond and picosecond time scales. Although the short and intermediate time dynamics may be assigned to specific molecular processes, fluctuation time scales of ≈1 ps correspond to more collective density and polarization fluctuations (15). The onset of cooperative behavior in Cωω(t) occurs at times as short as 200 fs (15).
In addition to Cωω(t), we have calculated Cor(t) for the fixed-charge and polarizable models. To quantify the long time relaxation of the various models studied, we fit to biexponential functions for Cωω(t) and Cor(t), resulting in the fitting functions Cωω(t) = Ae–t/τfast + Be–t/τslow and Cor(t) = Ae–t/τor-fast + Be–t/τor. The results appear in Table 1. The long time decay of Cωω(t) and Cor(t) is generally longer for polarizable models than for fixed-charge models. The fixed-charge models predict a τslow of ≈600 fs, but the experiment has extracted a value of 1,400 fs. Polarizable models that incorporate fluctuating charges relax more slowly, with a time τslow closer to 1 ps, as might have been anticipated from earlier work (4, 22). The DC polarizable model, which does not allow for fluctuating charges, has a τslow that is only marginally slower than for the fixed-charge models.
The results for the rotational anisotropy are qualitatively similar to those for Cωω(t). In the fixed-charge models, τor is smaller than the experimental value, whereas the polarizable models show a slower τor. The polarizable models predict a τor that is slower than that measured by experiment, but these values are still closer to the experimental result than their fixed-charge counterparts. Even though different polarizable models can give vastly different results for statics [e.g., P(μQ)] and for intermediate time dynamics, the decay time for the long time relaxations is similar (with the exception of the DC polarizable model). This observation is consistent with the notion that, in water, time scales of ≈1 ps are sensitive to collective phenomena and are not sensitive to the details of the microscopic potential energy.
To characterize the configurational relaxation in the picosecond regime of Cωω(t) and Cor(t), we computed the rate of change of the molecular surroundings for a tagged water molecule. Rabani et al. (37) introduced the following function to describe how a solvent “cage” around a molecule breaks apart, Ccage(t) = 1/N∑i〈Θ(1 – ni(0, t))〉. Here, N is the number of molecules, Θ is the Heaviside step function, ni(0, t) = |li(0)|2 – li(0)·li(t), and li is a vector that enumerates the hydrogen-bonding neighbors for molecule i.∥ We plot the cage correlation function for the four- and five-site models in Fig. 5, and the long time constants of biexponential fits appear in Table 1. The molecular environment, including the first solvation shell, of the fixed-charge water models changes more rapidly than in the polarizable models. This observation is consistent with the trends for τor and τslow, as well as our interpretation that the long time part of Cωω(t) corresponds to collective phenomena, not to the details of the local configuration.
Fig. 5.

The cage correlation function for the models discussed here (plotted on a logarithmic vertical scale). Similar to the long time relaxation of Cωω(t) and Cor, the polarizable models exhibit a slower decay of the local molecular environment relative to the nonpolarizable models.
Conclusions
We have developed a method of calculating vibrational frequencies “on the fly” in a MD simulation for polarizable potentials. Our simple protocol is based on the adiabatic separation of fast electronic motions, the slower vibrational motion of the OH stretch, and the still slower translations and rotations of molecules. It is simple to implement and can easily be extended to other molecular systems with variable electronic degrees of freedom.
Including molecular polarization and fluctuating charges in models of water increases the decay time of the picosecond dynamics in reorientational motion and in the vibrational dephasing of the OH stretch. For relaxation on picosecond time scales, the role of molecular polarizability is clear. On this time scale, models that parameterize the electronic response of a water molecule in qualitatively different ways give results that are in better agreement with experiment than those from fixed-charge models. At first glance, these results would seem to indicate that molecular polarizability must be included so that models of water can depict realistic time scales for electric field fluctuations at equilibrium. The results for statics and short time and intermediate time dynamics paint a very different picture. For such quantities, there is considerable variation among the results for the polarizable water models. Indeed, it would be difficult to have anticipated these variations given the similarity of the thermodynamic and bulk properties calculated from the polarizable models.
When compared with vibrational spectroscopy experiments, the most quantitatively accurate model is POL5–TZ. The salient features of this model that set it apart from the others are the softened steric repulsion between molecular centers and the out-of-plane polarization response that results from a combination of both fluctuating charges and inducible dipoles. In contrast, the FQ models derived from point charge potentials, such as TIP4P–FQ and SPC–FQ, that do not allow charges to move out of the molecular plane give absorption line shapes that are in stark disagreement with experiments because the predicted equilibrium distribution of frequencies is too broad. One may be able to improve results from the polarizable models by screening the intermolecular electrostatic interactions at short distances by adding an exponential softening function to the steric interaction between molecules and renormalizing the polarizability for the condensed phase. Fixed-charge models are surprisingly accurate for statics and short time and intermediate time dynamics. Our observations argue that electronic fluctuations decrease the rates of change for the molecular environments and increase the distribution of frequencies in vibrational spectroscopies.
The results from TIP4P–FQ underscore an important point. Most potentials are fine-tuned to reproduce particular features in water. It is unlikely that any one potential will provide a robust computer model for arbitrary properties of the liquid. Despite the good agreement between experiments and predictions from TIP4P–FQ for thermodynamic properties (i.e., location of the density maximum), low-frequency dielectric response, and solvation enthalpies, TIP4P–FQ does not accurately model the vibrational spectroscopy of water. For some applications that are insensitive to the molecular details of the parameterization, an oversimplified but computationally efficient representation of molecular polarizability would certainly be preferred.
Acknowledgments
We thank Jim Skinner for interesting conversations. E.H. thanks Noelle Ibrahim, Byungchan Kim, and Tom Young for helpful discussions. This work was supported by National Science Foundation Grant CHE-03-16896 (to B.J.B.) and U.S. Department of Energy Grant DE-FG02-99ER14988 (to A.T.).
Author contributions: E.H. and J.D.E. performed research; E.H., J.D.E., and B.J.B. wrote the paper; and A.T. initiated collaboration.
Abbreviations: MD, molecular dynamics; TDM, transition dipole moment; DC, Dang–Chang; FWHM, full width at half maximum.
Footnotes
There are small differences (≈0.05 Å) in the value of Q that minimizes the potential energy of the molecule in the gas phase and the value that minimizes the total potential energy of the system and bath in the liquid phase. These differences in Q are small, as judged by the quantum mechanical uncertainty for Q (0.07 Å) or by the variation in Hsb as a function of Q. Evaluating the TDM at either place does not change the results reported here.
The vector li has elements [f(ri1)... f(riN)]. The elements of li are functions of the interatomic oxygen distance (rij) defined again by the Heaviside function, f(rij) =Θ(rnlist – rij). We choose a value of rnlist = 3.5 Å to encompass the first solvation shell of the water molecules.
References
- 1.Rahman, A. & Stillinger, F. H. (1974) Phys. Rev. A 10, 368–378. [Google Scholar]
- 2.Stillinger, F. H. (1980) Science 209, 451–457. [DOI] [PubMed] [Google Scholar]
- 3.Silvestrelli, P. L & Parrinello, M. (1999) J. Chem. Phys. 111, 3572–3580. [Google Scholar]
- 4.Rick, S. W., Stuart, S. J. & Berne, B. J. (1994) J. Chem. Phys. 101, 6141–6156. [Google Scholar]
- 5.Rick, S. W. & Berne, B. J. (1996) J. Am. Chem. Soc. 118, 672–679. [Google Scholar]
- 6.Stern, H. A., Berne, B. J. & Friesner, R. A. (2001) J. Chem. Phys. 115, 2237–2251. [Google Scholar]
- 7.Harder, E., Kim, B., Friesner, R. A. & Berne, B. J. (2005) J. Chem. Theory Comput. 1, 169–180. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt, U. W. & Voth, G. A. (1999) J. Chem. Phys. 111, 9361–9381. [Google Scholar]
- 9.Margulis, C. J. & Berne, B. J. (2002) J. Phys. Chem. B 106, 10748–10752. [Google Scholar]
- 10.Warshel, A. (2003) Annu. Rev. Biophys. Biomol. Struct. 32, 425–443. [DOI] [PubMed] [Google Scholar]
- 11.Stillinger, F. H. (1975) Adv. Chem. Phys. 31, 1–101. [Google Scholar]
- 12.Fecko, C. J., Eaves, J. D. & Tokmakoff, A. (2002) J. Chem. Phys. 117, 1139–1154. [Google Scholar]
- 13.Fleming, G. & Cho, M. (1996) Annu. Rev. Phys. Chem. 47, 109–134. [Google Scholar]
- 14.Novak, A. (1974) Structure and Bonding, eds. Dunitz, J., Hemmerich, P., Holm, R., Ibers, J., Jorgensen, C., Neilands, J., Reinen, D. & Williams, R. (Springer, New York), Vol. 18, pp. 177–216. [Google Scholar]
- 15.Fecko, C. J., Eaves, J. D., Loparo, J. J., Tokmakoff, A. & Geissler, P. L. (2003) Science 301, 1698–1702. [DOI] [PubMed] [Google Scholar]
- 16.Asbury, J. B., Steinel, T., Stromberg, C., Corcelli, S. A., Lawrence, C. P., Skinner, J. L. & Fayer, M. D. (2004) J. Phys. Chem. A 108, 1107–1119. [Google Scholar]
- 17.Fecko, C. J., Loparo, J. J., Roberts, S. T. & Tokmakoff, A. (2005) J. Chem. Phys. 122, 54506. [DOI] [PubMed] [Google Scholar]
- 18.Lawrence, C. P. & Skinner, J. L. (2003) J. Chem. Phys. 118, 264–272. [Google Scholar]
- 19.Moller, K. B., Rey, R. & Hynes, J. T. (2004) J. Phys. Chem. A 108, 1275–1289. [Google Scholar]
- 20.Harder, E. (2004) Ph.D. thesis (Columbia University, New York).
- 21.Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. (1987) J. Phys. Chem. 91, 6269–6271. [Google Scholar]
- 22.Xu, H., Stern, H. A. & Berne, B. J. (2002) J. Phys. Chem. B 106, 2054–2060. [Google Scholar]
- 23.van Belle, D., Froeyen, M., Lippens, G. & Wodak, S. J. (1992) Mol. Phys. 77, 239–255. [Google Scholar]
- 24.Dang, L. X. & Chang, T. (1997) J. Chem. Phys. 106, 8149–8159. [Google Scholar]
- 25.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1983) J. Chem. Phys. 79, 926–935. [Google Scholar]
- 26.Rick, S. W. (2004) J. Chem. Phys. 120, 6085–6093. [DOI] [PubMed] [Google Scholar]
- 27.Tuckerman, M. E., Martyna, G. J. & Klein, M. L. (1992) J. Chem. Phys. 97, 2635–2643. [Google Scholar]
- 28.Corcelli, S. A., Lawrence, C. P., Asbury, J. B., Steinel, T., Fayer, M. D. & Skinner, J. L. (2004) J. Chem. Phys. 121, 8897–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corcelli, S. A., Lawrence, C. P. & Skinner, J. L. (2004) J. Chem. Phys. 120, 8107–8117. [DOI] [PubMed] [Google Scholar]
- 30.Reimers, J. R. & Watts, R. O. (1984) Mol. Phys. 52, 357–381. [Google Scholar]
- 31.Gordon, R. G. (1968) Adv. Magn. Reson. 3, 1–42. [Google Scholar]
- 32.Murphy, W. F. (1977) J. Chem. Phys. 67, 5877–5882. [Google Scholar]
- 33.Morita, A. & Kato, S. (1999) J. Chem. Phys. 110, 11987–11998. [Google Scholar]
- 34.Klaus, R., Moller, K. & Hynes, J. (2002) J. Phys. Chem. A 106, 11993–11996. [Google Scholar]
- 35.Sette, F., Ruocco, G., Krisch, M., Bergmann, U., Masciovecchio, C., Mazzacurati, V., Signorelli, G. & Verbeni, R. (1995) Phys. Rev. Lett. 75, 850–853. [DOI] [PubMed] [Google Scholar]
- 36.Balucani, U., Ruocco, G., Torcini, A. & Vallauri, R. (1993) Phys. Rev. E 47, 1677–1684. [DOI] [PubMed] [Google Scholar]
- 37.Rabani, E., Gezelter, D. & Berne, B. J. (1997) J. Chem. Phys. 107, 6867–6876. [Google Scholar]