

Abstract
Meibomian gland dysfunction (MGD) is a leading cause of dry eye disease and one of the most common ophthalmic conditions encountered in eye clinics worldwide. These holocrine glands are situated in the eyelid, where they produce specialized lipids, or meibum, needed to lubricate the eye surface and slow tear film evaporation – functions which are critical to preserving high-resolution vision. MGD results in tear instability, rapid tear evaporation, changes in local microflora, and dry eye disease, amongst other pathological entities. While studies identifying the mechanisms of MGD have generally focused on gland obstruction, we now know that age is a major risk factor for MGD that is associated with abnormal cell differentiation and renewal. It is also now appreciated that immune-inflammatory disorders, such as certain autoimmune diseases and atopy, may trigger MGD, as demonstrated through a T cell-driven neutrophil response. Here, we independently discuss the underlying roles of gland and immune related factors in MGD, as well as the integration of these two distinct mechanisms into a unified perspective that may aid future studies. From this unique standpoint, we propose a revised model in which glandular dysfunction and immunopathogenic pathways are not primary versus secondary contributors in MGD, but are more fluid, interactive and dynamic, which we likened to the Yin and Yang of MGD.
1. Introduction
Meibomian gland dysfunction (MGD) is characterized by insufficient production and/or altered secretion of meibum, predominantly lipids, to the tear film, producing instability, rapid tear evaporation, and dry eye disease (DED) symptoms, including blurry vision, redness, ocular pain, and foreign body sensation [1]. Population and clinical-based studies with varying design, patient characteristics, and definitions report an MGD prevalence of 38.9% in the US, but as high as 69.3% in subjects aged 60 years or older [2]. Despite being one of the most frequent ophthalmic conditions encountered in eye clinics worldwide, treatment is mostly palliative in nature, and currently there are no FDA approved pharmacotherapies indicated for MGD [3].
This unmet medical need can be partially explained by our incomplete understanding around the early triggers which initiate the disease process. The predominating hypothesis is that MGD begins with hyperkeratinization of the meibomian gland duct epithelium. This event, which has been extensively reviewed by other authors [3, 4], is thought to lead to obstruction, increased intraglandular pressure, cystic dilation of the duct, acinar cell dysfunction, disuse atrophy, and gland dropout, with subsequent abnormal meibum composition and decreased secretion [5–9]. However, in counter distinction to the obstructive hyperkeratinization hypothesis, ductal hyperplasia, characterized by ductal thickening and epithelial cell desquamation without orifice hyperkeratinization, has also been proposed in some forms of MGD [2]. Moreover, how any of these early changes and downstream consequences relate to inflammation that is co-incident with most forms of MGD is poorly understood. In fact, whether immunopathogenesis is necessary and/or sufficient in the disease process is a topic of current debate [10].
The original purpose of this perspective piece was to delineate the distinct perspectives for the role of non-immune versus immune processes in MGD pathobiology. Subsequently, this discussion expanded to encompass a unified perspective that integrates these two seemingly disparate views into a novel, unified framework focused on the interplay between glandular-driven and immune-driven dysfunction. In contrast to the existing ‘vicious cycle’ perspective [1, 11, 12], which argues a more sequential process in MGD pathogenesis, we postulate a highly fluid model to represent the inter-relationship between certain pathological immune responses and aberrant glandular cell functions – we have likened this interaction to the Yin and Yang of MGD.
2. Anatomy and physiology
The meibomian glands are branched, tubuloacinar holocrine glands embedded in the tarsal plates of the upper and lower lids. In humans, each gland consists of a long, blind-ended, central duct that connects to individual glandular acini by means of short ductules [3]. In mice, meibomian gland structure is similar to humans – histology and gross anatomy of glands from a young, healthy C57Bl/6 mouse can be seen in Figure 1. The central duct terminates at the eyelid margin, anterior to the mucocutaneous junction, where dry keratinized skin transitions to wet, non-keratinized conjunctiva. The distal portion of the meibomian gland duct, at the orifice to the gland, is lined by keratinized stratified squamous epithelium expressing keratins (Krt) 1 and 10, whereas the proximal ductal epithelium is non-keratinized and expressing Krt6. The acini are comprised of meibocytes that synthesize and secrete meibum through a holocrine mechanism involving cell proliferation, differentiation, and cell death – a process requiring continual cell renewal. Meibum comprises a mixture of polar and nonpolar lipids, along with cellular proteins, that are secreted onto the ocular surface to form the lipid layer of the tear film. Specifically, the lipid layer renders the tear film a smooth optical surface and decreases surface tension during blinking, thus stabilizing the aqueous layer and delaying evaporation. Inadequate meibomian gland function leads to a decreased quality and/or quantity of meibum, resulting in tear film instability, increased tear evaporation, and DED [3].
Figure 1. The healthy meibomian gland.
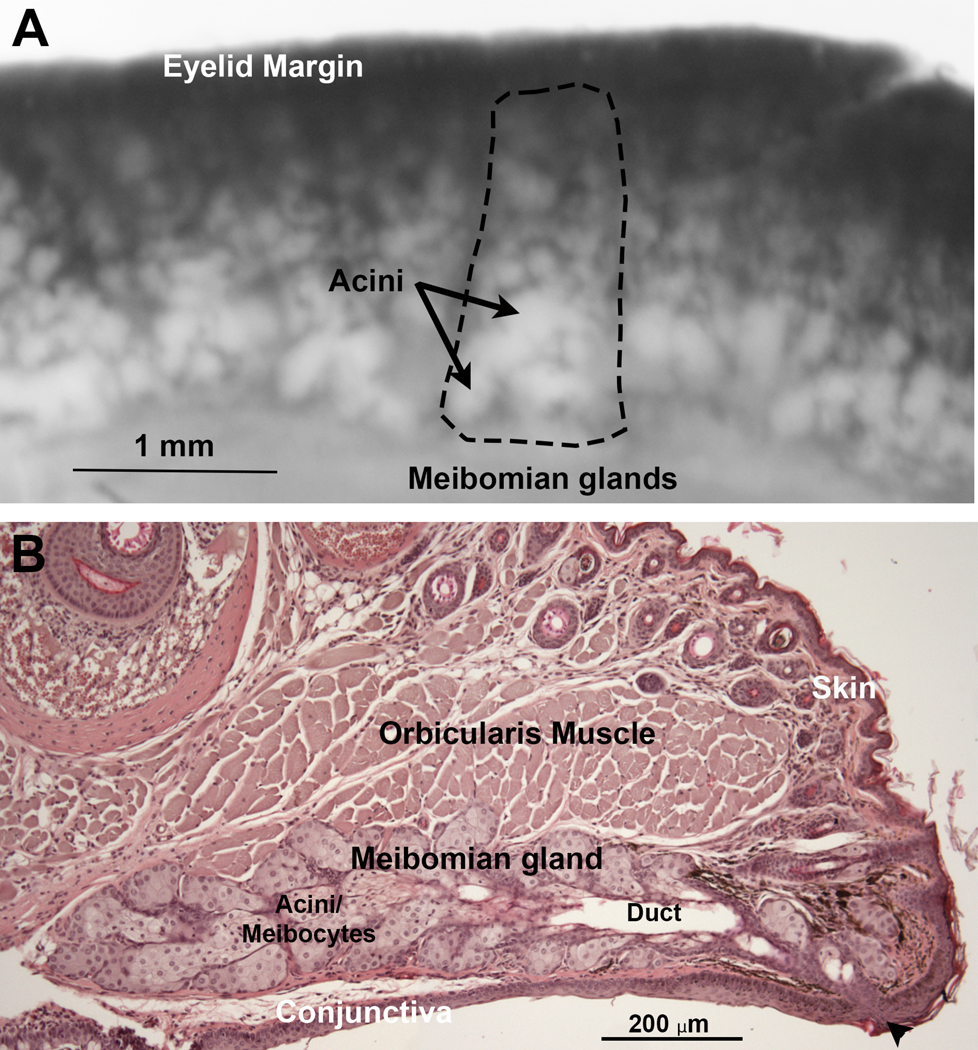
A) The meibomian glands (dashed lines) can be seen as a row of white structures embedded in the distal eyelid that extend from the eyelid margin and are comprised of clusters of small round acini. B) H&E-stained tissue section of eyelid from a normal wild-type mouse showing the meibomian gland underlying the orbicularis muscle. The gland has a blind ended central duct (Duct) connecting acini to the gland orifice (Arrowhead) at the junction between the skin and conjunctiva.
3. Historical perspective of MGD pathology
Historically, the anatomical distinction between anterior and posterior blepharitis and MGD was not always made, thus these terms were used indistinctively to name lid margin disorders, including the meibomian glands [3]. Anterior blepharitis involves the eyelid skin and eyelash base and follicles, and posterior blepharitis affects the meibomian glands. Meibomian gland disorders were initially described as an inflammatory, hypersecretory condition of adults, with or without bacterial induced seborrheic blepharitis, mainly by Staphylococcus aureus [13]. In 1977, McCulley and Sciallis first described the clinical consequences of meibum stagnation in a group of patients with chronic blepharitis caused by meibomian gland obstruction in the absence of anterior blepharitis, including superficial punctate keratopathy (SPK) and decreased tear-film breakup time. They named this condition “meibomian keratoconjunctivitis” [5]. The authors suggested that SPK was caused by tear film instability rather than by S. aureus toxins, which is more common in the context of anterior blepharitis. However, these findings, based on clinical observations, did not explain the role of inflammation in MGD.
Three years later, Korb and Henriquez were the first to provide evidence of MGD in a cluster of patients with apparent contact lens intolerance, but without signs of inflammation or infection in the eyelid margin. Cytological analysis revealed that MGD was associated with obstruction of the meibomian gland orifices by desquamated epithelial cells [14]. Later development of meibomian gland imaging (meibography), initially using eyelid transillumination and infrared light photography, began to identify blepharitis patients that showed atrophy and distortion of meibomian glands along with changes in meibum quality [15, 16]. Later studies by Mathers in 1993 established a link between meibomian gland atrophy in chronic blepharitis patients and increased tear evaporation, confirming the importance of meibomian gland function in maintaining tear film homeostasis [17]. Based on further meibographic and clinical evidence of altered meibum quality in blepharitis patients, it is generally thought that MGD is caused by obstruction of the meibomian gland duct in the absence of inflammation, thus leading to blockage of meibum secretion, ductal dilation, and later atrophy of the meibomian gland. As dysfunction progresses in severity, altered meibum secretion then leads to disruption of the ocular surface tear film homeostasis, increased tear evaporation, increased tear osmolarity and other signs and symptoms of dry eye [1].
Generally, obstruction of the meibomian gland is thought to involve hyperkeratinization of the meibomian gland duct. This model is based, in part, on ultrastructural and histologic study of the meibomian glands of human, rabbit, primate, and steer specimens showing evidence of partial keratinization in portions of the meibomian gland duct [6]. Later studies of animal models of MGD following epinephrine and polychlorinated biphenyl exposure, as well as the rhino mouse, all reported ductal dilation of the meibomian gland with thickened, keratinized epithelium and accumulation of cellular, desquamated epithelial debris, supporting a possible mechanism of hyperkeratinization without signs of inflammation [6, 7, 18]. In humans, Gutgesell et al. performed a case-control histopathological analysis of patients with severe MGD undergoing eyelid repair [8]. They reported obstruction and dilation of the meibomian gland ducts, foreign-body reaction with granuloma formation, and enlargement of the acini with squamous metaplasia, keratinization, and absence of secretory content. These changes were hypothesized to be caused by pressure atrophy due to prolonged MGD obstruction. The authors further reported minimal inflammatory cells, despite observing granuloma formation, suggesting that inflammation is mildly associated with MGD [8]. In 2002, Obata reported basement membrane thickening and atrophy of the acini, cystic dilation of acini and/or central duct, and minimal granulation tissue and lipogranulomatous inflammation after performing meibomian gland histopathological analysis of human cadavers [9]. At this point, the pathogenesis of MGD was best explained by hyperkeratinization of the meibomian gland duct, leading to meibomian gland obstruction and acinar cell dysfunction, resulting in decreased meibum secretion and/or altered composition. In contrast, it should be noted that Reneker et al. using immunohistopathology targeting selective biomarkers in a small series of 4 cases [19] observed that one individual with severe obstruction showed evidence of ductal epithelial hyperproliferation with no evidence of hyperkeratinization. A similar finding has been noted in aging mouse meibomian glands using immuno tomography [20], together suggesting multiple pathways for leading to obstructive MGD, perhaps involving hyperplasia and not hyperkeratinization.
4. Glandular-Driven MGD
While the role of inflammation in the development of DED has received increased attention, this has been based on animal models that induce a primary acute and chronic inflammatory response that leads to classical signs of dry eye, including corneal fluorescein staining and decreased tear break-up time [10, 21, 22]. However, the link between ocular surface inflammation and dysfunction of the meibomian glands remains unclear as the cellular and molecular mechanisms controlling these processes have yet to be elucidated. Regarding the relative importance of inflammation to DED, there are several lines of evidence supporting a primary role for meibomian glands in the development of chronic blepharitis that include three that will be discussed below, including: 1) effects of age on meibomian gland renewal and differentiation, 2) knockout mouse models and meibum secretion, and 3) isotretinoin/Accutane and MGD.
4A. Effects of age on meibomian gland renewal and differentiation
Subject age is an important, if not the most significant, risk factor for the development of DED [23]. While the effects of age on the meibomian gland are incompletely understood, studies of aging mice have shown that meibomian gland acinar cells, or meibocyte progenitor cells, exhibit a significant decrease in cell proliferation and renewal with age, decreasing over 50% by 9 to 12 months of age [24]. Aging mice also show atrophy of the meibomian gland similar to that observed in humans, suggesting that loss of meibocyte renewal may lead to meibomian gland atrophy [20, 25]. A similar decrease in proliferative potential has also been identified in the human meibomian glands based on tissue obtained from blepharoplasty patients and correlated to meibomian gland atrophy and altered meibum quality for which patient age was the most significant correlating factor [26]. Cell renewal is critical to meibomian gland function, given that meibomian glands are holocrine glands producing lipid through a process of cell differentiation, accumulation of intracellular meibum, and release through an undefined cell death program, or ‘meiboptosis’. Given that progenitor meibocytes in young mice are highly proliferative with over 25% of acinar basal cells showing evidence of cell cycling [24], combined with a cell cycle rate of approximately 4 days and a differentiation half-life of 9 days as measured in the rat meibomian gland by Olami et al. [27], a loss of proliferation rate with aging most likely severely impacts the delivery of meibum to the eyelid margin and ocular surface.
In addition to the decrease in proliferative potential, both mice and human meibomian glands show a distinct change in the overall expression and post-translational modification of the nuclear receptor, peroxisome proliferator activated receptor gamma (PPARγ) [28]. Specifically, in the mouse, aging leads to a complete loss of sumoylated, cytoplasmic PPARγ and a 75% decrease of nuclear PPARγ [28]. PPARγ is a lipid sensitive, nuclear receptor that regulates the expression of enzymes involved in lipid synthesis and is absolutely required for the differentiation of adipocytes and sebocytes [29, 30]. PPARγ once transported to the nucleus forms a transcriptional complex with the retinoid X receptor (RXR) and is involved in gene repression and activation that governs cell differentiation depending on ligand binding. Cell culture studies using both mouse and human immortalized meibomian gland epithelial cells have also established that activation of PPARγ by lipid ligands or synthetic agonists induce cell cycle exit, expression of lipid synthesizing genes involved in meibum synthesis, and the accumulation of lipid droplets stored in the endoplasmic reticulum, similar to that detected in intact mouse and human meibomian glands (Figure 1) [28, 31, 32]. Taken together, the findings that PPARγ is required for meibocyte differentiation and the loss of PPARγ signaling with aging further suggests that aging impacts the ability of the meibomian gland to deliver meibum to the ocular surface.
While meibomian gland stem cell renewal has recently been the subject of a review article [33], it should be noted that lineage tracing studies suggest the meibomian gland acini are self-renewing and apparently derived from a single stem/progenitor cell located at the interface between the acini and the short ducts that lead to the central meibomian gland duct [34]. Noting that meibomian glands undergo hyper-proliferation in response to desiccating stress [35], it is likely that meibomian stem/progenitor cells may also be subject to exhaustion with age, although this possibility requires further study. Together, the loss of meibocyte progenitor cell proliferative potential, changes in PPARγ expression, and the limited number of stem/progenitor cells available for renewal of meibocytes would suggest these age-related changes might play an important role in the development of age-related meibomian gland dysfunction leading to ocular surface disease, a process not directly involving an immune-driven mechanism.
4B. Knockout mouse models with inhibited meibum secretion
While various knockout and transgenic mouse models have been developed as previously reviewed [36], two knockout mice targeting enzymes critical to meibum synthesis are of particular interest and include acyl-CoA wax alcohol acyltransferase 2 (AWAT2) and the fatty acyl-CoA reductase (FAR2) [37, 38]. Both enzymes are critical for the synthesis of wax esters, a major lipid constituent of meibum for which mice lacking these enzymes are deficient in the synthesis of this lipid species. Importantly, the major phenotype is thickening and retention of meibum within the gland duct leading to marked ductal dilation and plugging of the gland. Meibum is known to have a wide melting range starting at 10oC and extending to 40°C with three phase transitions at 12°C, 21°C and 32°C [39]. The thickening of meibum in the Awat2 and Far2 knockout mouse models suggest a change in the melting point of meibum that likely affects meibum viscosity and secretion onto the eyelid margin. This conclusion is supported by reported differences between the melting temperature of wax esters and cholesterol esters (the other major lipid species in meibum) [40, 41] as well as by differences in the ratio of wax/cholesterol esters in meibum from patients with blepharitis [42]. Since thickening of meibum is one of the major clinical signs of altered meibum quality, it may be noteworthy that small increases have been noted in the melting point of meibum from blepharitis patients [39, 43], although it is not clear what controls the melting point of meibum. Together these finding suggest that changes in meibum lipid synthesis affect meibum melting point and, hence, meibum viscosity may also be a possible mechanism underlying meibomian gland dysfunction.
Interestingly, rheology studies of normal human meibum by Rosenfeld et al. have established that meibum is a non-Newtonian fluid whose secretion is dependent on the minimum yield stress and plastic viscosity that is affected by the physical state of the meibum from fluid/liquid to viscose/solid [44]. Finite element modeling of meibum flow rates suggests that more viscous meibum shows an exponential increase in the minimum yield stress and plastic viscosity which leads to an exponential decrease in meibum secretion through the terminal duct of the meibomian gland [45]. These findings suggest that shifts in the meibum melting curve may be traced to changes in lipid synthesis and ratio of wax esters to cholesterol esters, which may explain the finding of increased meibum viscosity and decreased expressibility in patients with meibomian gland dysfunction and the development of duct dilation and gland obstruction. Such a hypothesis may help explain the known association of dyslipidemia and hypercholesterolemia with meibomian gland dysfunction [46, 47].
Modeling of meibum flow rates also identified that eyelid pressure exerted on the meibomian gland during blinking also affects meibum secretion. Studies measuring eyelid pressure exerted on the eye have identified an age-related decrease in static eyelid pressure of 2–3 mmHg per decade of life [48]. This age-related decrease in static eyelid pressure, if similarly affecting eyelid blink pressure, would translate into a 50% to 70% reduction of meibum secretion in subjects aged 60 to 90, respectively, based on finite element modeling studies [42]. Of course, ductal diameter also greatly affects meibum flow rate and is linearly related to the fourth power of the excretory duct radius. This finding suggests that small changes in the ductal epithelial thickness measuring 5 microns or only an increase of less than 15% in the ductal epithelial thickness could decrease meibum flow by as much as 35% to over 90% depending on the initial duct diameter and eyelid blink pressure. It is interesting to note that recent work by Reneker et al. [19] found that in a young subject with severely obstructed meibomian glands, there was an increase in hyperproliferation biomarkers without signs of keratinization in MGD. These findings support a mechanism for obstructive MGD that focuses on ductal epithelial thickening leading to restricted meibum secretion, in counter distinction to the conventional obstructive hyperkeratinization, that does not require an inflammatory response for pathogenesis.
4C. Isotretinoin/Accutane and MGD
Isotretinoin is a widely used, and highly effective therapy for acne vulgaris that is composed of the prodrug, 13-cis retinoic acid, which is converted by isomerization to all-trans retinoic acid (ATRA) that interacts with the retinoic acid and retinoid X receptors (RAR/RXR) [49]. This receptor complex can then act to selectively repress/activate genes leading to the suppression of sebaceous gland function and differentiation [50]. While isotretinoin reduces the size of the sebaceous gland, restores normal hair follicle keratinization, and inhibits inflammation, a common complaint of patients receiving therapy is ocular irritation with signs/symptoms of dry eye [49]. Studies of patients treated with Accutane have shown that in addition to the reduced size of the sebaceous glands, there is also an apparent atrophy of the meibomian gland during treatment, as detected by meibography. Furthermore, subjects also showed decreased and thickened meibum secretions, as well as increased tear osmolarity without any change in lacrimal gland function [51]. Earlier studies using rabbit and hamster models also document meibomian gland acinar atrophy with ductal epithelial hyperplasia after 13-cis retinoic acid exposure [52, 53], and more recent rat studies have shown isotretinoin to significantly decrease the expression of PPARγ [54]. These findings, along with other more recent confirmatory studies of isotretinoin, strongly suggest that meibomian gland function can be pharmacologically targeted and directly inhibited by other therapeutic drugs, inducing DED in the absence of apparent inflammation.
While the mechanisms underlying the effect of 13-cis retinoic acid on sebaceous and meibomian gland function is incompletely understood, current hypotheses have centered on the role of ATRA and RAR/RXR interactions targeting the expression of FoxO3 and secondary upregulation of FoxO1, leading to the initiation of a gene expression cascade that causes down regulation of PPARγ (Figure 2). Specifically, FoxO1 appears to directly bind to PPARγ promoter regions to repress expression and function of PPARγ regulated genes. Additionally, FoxO1 can directly block binding of the PPARγ/RXR transcription factor complex to the DNA binding domain and consequently inhibit PPARγ function and regulation of cell differentiation. While this pathway has not been studied in the context of meibomian gland function, the possible interaction between FoxO1 and PPARγ gene regulation suggests an intriguing pathway for the control of meibomian gland function. In particular, the noted effects of growth hormone and other growth factors leading to FoxO1 phosphorylation and nuclear export would lead to the promotion of meibocyte differentiation and meibum synthesis by the opening of PPARγ/RXR DNA bind sites. By contrast, loss of growth factor signaling during aging and disease would have opposite effects, leading to persistent DNA binding by FoxO1 and thus blocking downstream meibocyte differentiation. Certainly, studies focusing on this pathway may help explain not only DED with isotretinoin therapy but also potentially the effects of aging, as well as suggesting novel targets for treating meibomian gland atrophy.
Figure 2. Potential mechanism controlling meibocyte differentiation and acinar atrophy.
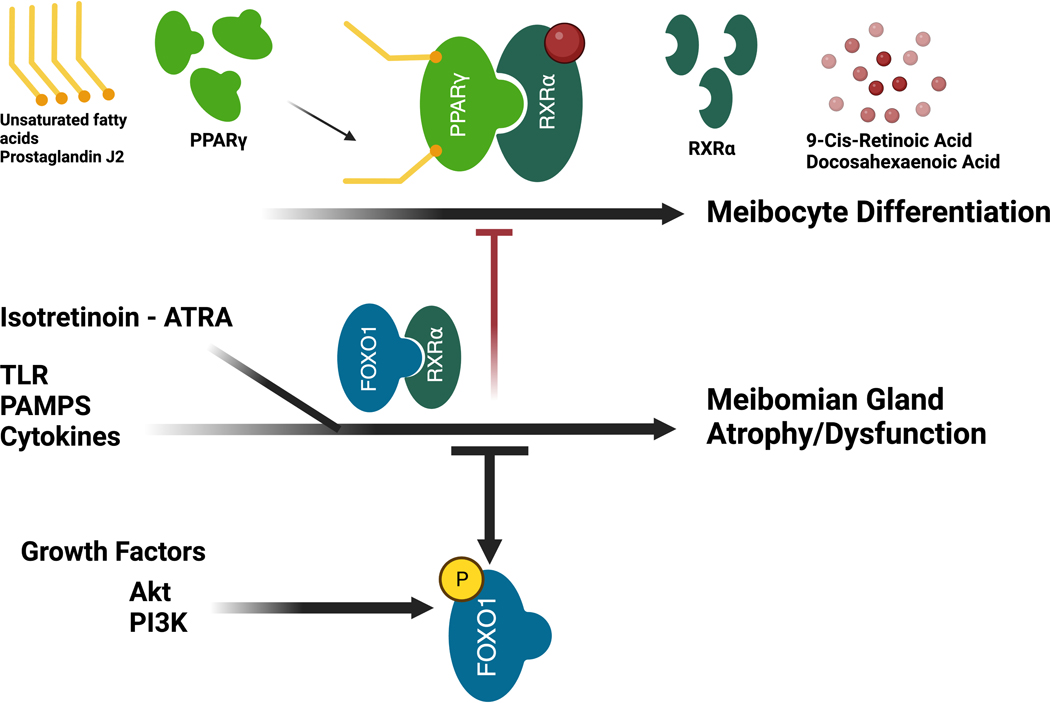
Meibocyte differentiation requires binding of lipid sensitive nuclear receptor, PPARγ, and RXRα to the PPARγ DNA binding domain. Activation of FOXO1 by isotretinoin (Accutane), inflammatory cytokines, TLRs, or PAMPs leads to replacement of PPARγ from the DNA binding site and the inhibition of meibocyte differentiation. Growth factor signaling leading to phosphorylation of FoxO1 leads to the nuclear exit of FoxO1 and facilitates downstream PPARγ signaling.
Abbreviations: PPARγ, peroxisome proliferator activated receptor gamma; RXRα, retinoid; ATRA, all-trans retinoic acid; TLR, toll-like receptor; PAMPs, pathogen-associated molecular patterns; FOXO1, forkhead box protein O1; Akt, protein kinase B; PI3K, phosphoinositide 3-kinases.
4D. Summary for glandular-driven MGD
The three mechanisms discussed above clearly show that inflammation is not necessary to explain meibomian gland atrophy, altered meibum quality or obstructive MGD. Furthermore, it is not clear that some of the putative models supporting inflammation as the primary mechanism, do not directly affect the meibomian gland, and hence lead to a secondary inflammatory response. For instance, the very well-studied desiccating stress and pharmacological lacrimal gland inhibition model show a marked increase in meibomian gland acinar proliferation detected as early as the increased inflammation [35]. Furthermore, the effects of scopolamine, a muscarinic receptor antagonist, on the meibomian gland have never been studied even though the meibomian gland expresses all five muscarinic receptors in addition to other neural receptors and most likely is controlled by yet to be discovered neural regulatory mechanisms [55]. More recently, other models of inflammatory dry eye have used vitamin A deficiency and the Pinkie mutant RXR mouse model [56]. However, as noted above, meibomian gland function critically involves retinoic acid signaling through both RAR and RXR transcriptional complexes with PPARγ, therefore these models most likely have upstream effects on the meibomian gland that need to be considered in the analysis of secondary inflammation.
5. Immune-Driven MGD
A direct role for inflammation as an early trigger in MGD pathogenesis remains an area of debate [1]. However, we posit that specific, pathologic immune responses can act as an early driver of MGD given the co-occurrence of MGD secondary to immune diseases with ocular involvement [e.g., ocular graft-versus-host disease (oGVHD), Stevens-Johnson Syndrome (SjS), Sjogren’s Syndrome and chronic/severe allergic eye disease (AED)]. One argument against the conclusion that inflammation is an important driver of MGD originates from the identification of obstructive MGD in patients without clinically apparent inflammation, referred to as non-obvious or non-inflamed obstructive MGD [57]. However, while the characterization of this patient setting has indeed advanced the field, continued research is still needed to fully describe the exact prevalence of this form of MGD. Also, germane will be whether subclinical inflammation is present in these patients [58]. Addressing these knowledge gaps is critical if we are to extrapolate this into a generalizable understanding in MGD pathogenesis. Meanwhile, for in-depth mechanistic and causative studies in complex disease settings, our most robust tool is animal models. While various MGD models have successfully recapitulated cardinal features of MGD via advanced aging, orifice cauterization, genetic disruption of meibum synthesis and composition, induction of sterile inflammation, and other methods [4], in this section we primarily focus on the AED mouse model [10, 59–66]. The AED model was the first model to demonstrate that an immune response can induce the cardinal features of MGD, including orifice obstruction, ductal dilation, some level of glandular atrophy, and meibum inspissation, observed as toothpaste-like meibum upon manual gland expression that hinders its ability to evenly coat the ocular surface [10]. This model has enabled in-depth mechanistic studies into the immunopathogenesis of MGD.
5A. Allergic eye disease (AED) mouse model
The AED model uses an exogenous antigen-driven systemic immune response followed by ocular instillations of antigen to model chronic-like ocular allergy, including associated neutrophil recruitment and MGD pathogenesis similar to that observed in patients with atopic keratoconjunctivitis [10]. Specifically, C57Bl/6 (B6) mice are immunized against the model allergen, ovalbumin (OVA), with adjuvants aluminum hydroxide and pertussis toxin. Allergy and MGD is then induced 2 weeks later through a 7-day course of daily, topical ocular instillation with OVA [10, 67].
5B. A role for Type 3 lymphocyte responses in MGD pathogenesis in the AED model
The inflammatory processes underpinning ocular surface manifestations in the AED model involves parallel, but distinct, lymphocyte responses that fall under the classification of type 2 and type 3 lymphocyte-mediated immune responses, referred to here as simply type 2 and type 3 immune responses (Table 1) [68]. Briefly, type 2 immunity relies on production of IL-4, IL-5, and IL-13 by GATA-3+ lymphocytes and downstream eosinophil recruitment, while type 3 immunity leverages IL-17A produced by RORγt+ lymphocytes and downstream neutrophil recruitment [68]. This dual-pronged inflammatory profile is similarly observed in tear washes from patients with atopic keratoconjunctivitis [10]. However, Reyes et al., demonstrated that MGD pathogenesis in the AED model is specifically driven by a type 3 immune response through numerous lines of evidence [10] (Figure 3). First, characteristic of type 3 immune responses, T cells in the draining lymph node of AED mice are skewed towards a T helper 17 (Th17) phenotype. Furthermore, inhibition of Th17 skewing or effector function through genetic deletion of interleukin (IL)-17A or pharmacologic blockade of IL-23 (Th17 survival/differentiation factor) led to significant reduction of the meibomian gland orifice obstruction, suggesting a causative role for these lymphocytes. Concordantly, adoptive transfer of in-vitro expanded Th17 cells into mice with a mild allergic conjunctivitis without MGD resulted in meibomian gland obstruction, thereby demonstrating that Th17 cells are sufficient to induce MGD. In contrast, meibomian gland obstruction was not induced with transfer of in-vitro expanded Th2 cells into this mild allergic conjunctivitis model, suggesting that type 2 responses are unable to drive MGD pathogenesis [10]. Independently, Lou et al. demonstrated that lymphatic endothelial deletion of Vegfr3 in AED mice inhibits development of clinical allergy signs, but not meibomian gland obstruction [66], thereby lending further credence to a role for type 3 lymphocyte responses in MGD pathogenesis.
Table 1.
Overview of type 1, 2, and 3 lymphocyte responses.
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| Master transcription regulator | T-bet | GATA3 | RORγt |
| Response-mediating lymphocytes | Th1 ILC1 Tbet+ γδ T cell |
Th2 ILC2 GATA3+ γδ T cell |
Th17 ILC3 RORγt+ γδ T cell |
| Effector cytokines | IFNγ TNF |
IL-4 IL-5 IL-13 |
IL-17A IL-17F IL-22 |
| Downstream effector leukocytes | Macrophages CTLs | Mast cells Eosinophils B cells | Neutrophils |
Abbreviations: T-bet, T-box transcription factor 21; GATA3, GATA Binding Protein 3; ROR, retinoic acid-related orphan receptor; Th, T helper; ILC, innate lymphoid cell; IFNγ, interferon gamma; TNF, tumor necrosis factor; IL, interleukin; CTLs, cytotoxic T-cells.
Figure 3. Immune-mediated MGD is driven by type 3 immune responses.
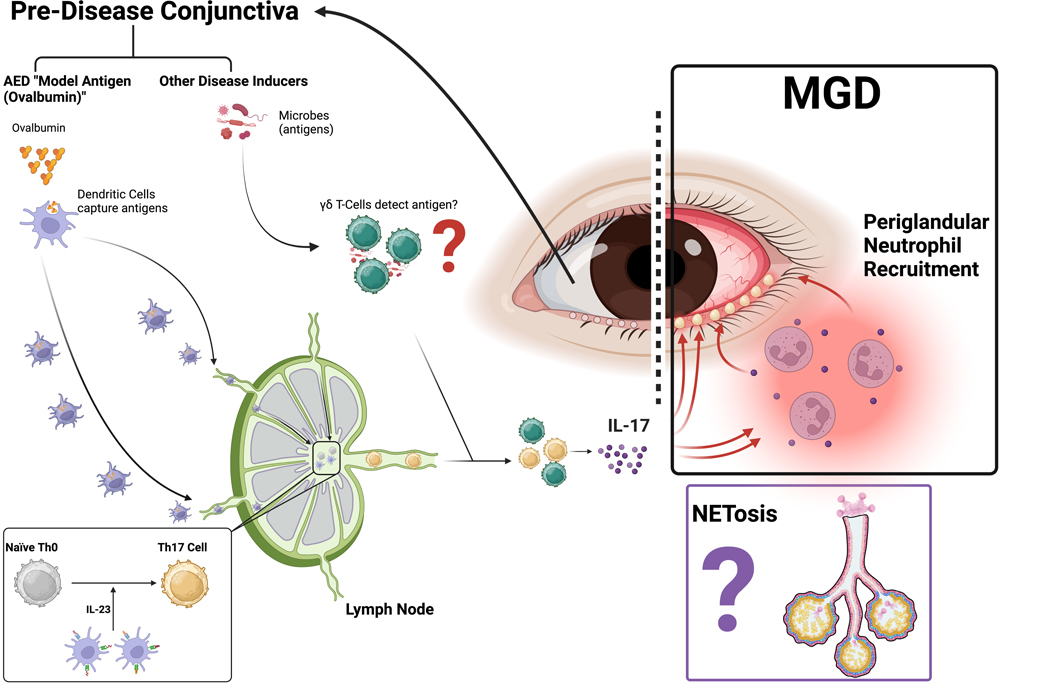
Similar to other type 3 responses, immune mediated MGD appears to rely on conjunctival IL-17A production leading to downstream neutrophil recruitment, which induces glandular dysfunction through mechanisms that are still being studied. NET formation is a leading hypothesis, but the specific mechanism requires further study.
Abbreviations: AED, allergic eye disease; IL, interleukin; Th, T helper; MGD, Meibomian gland dysfunction; NET, neutrophil extracellular traps.
Future research is needed to further resolve this type 3 response. For example, T helper cell expression of IFN-γ is classically categorized as type 1 lymphocyte response. However, as in hyperdessication-induced DED models [69] (and other autoimmune models [70, 71]), polyfunctional Th17 cells expressing both IL-17A and IFN-γ have been identified in the AED model [10], though an isolated role for IFN-γ in MGD pathogenesis in the AED model has not been examined. Interestingly, like AED, Perez et al. demonstrated that a mouse model of oGVHD associated with MGD contains CD4+ T cells in the draining lymph node that significantly express both IL-17A and IFN-γ [72]. The oGVHD setting is particularly important because patients experience aggressive MGD [72].
Though pathogenic Th17 responses are central to MGD causation in the AED model, and independently in DED models [73], the role of other lymphocytes that produce IL-17A, such as gamma-delta (γδ) T cells [74], in MGD pathobiology have not yet been closely evaluated. This may be important, as recent evidence from the Pinkie mouse model suggests that conjunctival γδ T cells expressing IL-17A can contribute to ocular surface disease [75]. Pinkie mice possess a retinoid X receptor alpha (RXRα) mutation that results in exacerbated signs of age-related DED [75] – including meibomian gland orifice obstruction [Pflugfelder, personal communication]. Moreover, young bone marrow chimeric mice that are wildtype for RXRα, but with Pinkie mouse bone marrow, exhibit a more severe DED phenotype compared to normal B6 mice when exposed to desiccating stress, thereby demonstrating that the RXRα mutation in immune cells is sufficient to exacerbate the phenotype [75].
The role of IL-17A in patients with MGD is not yet fully understood, but several studies have reported direct correlations between IL-17A levels in the tears and disease severity [76, 77]. One study comparing patients with MGD to healthy controls showed elevated levels of IL-17A in their tears, like those of ocular surface inflammatory disorders including oGVHD, SjS, and Sjögren’s syndrome. Interestingly, the anti-inflammatory effect of intense pulsed light treatment was able to decrease IL-17A tear levels, especially 1 week after treatment. Additional literature regarding tear IL-17A concentrations in MGD patients with various clinical scenarios is summarized in Table 2 [78–85].
Table 2.
Studies reporting elevated levels of IL-17 in patients with MGD associated with different clinical scenarios.
| Author (year) | Study objective | Study findings |
|---|---|---|
| Kang et al. (2011) [78] | Cytokine levels patients with MGD, OSIDs, and healthy controls |
Significantly higher concentration of IL-17 in MGD and inflammatory disorders compared to controls. |
| Lee et al. (2011) [79] | Oral minocycline and ATs vs. ATs only in moderate to severe MGD | Significant reduction in levels of IL-1b, IL-6, IL-7, IL-8, IL-12p70, IL-17a, IFNγ, TNFα, and MCP-1 in patients who received minocycline. |
| Lee et al. (2014) [80] | Topical loteprednol etabonate and lid hygiene vs. lid hygiene only for moderate-severe MGD | Loteprednol group: Significant decreases in IL-1b, IL-6, IL-8. Slight, but not significant decrease in IL-17. Lid hygiene group: Significant decrease in IL-6 and IL-8. |
| Landsend et al. (2018) [81] | Inflammatory cytokine levels in congenital aniridia with MGD vs healthy controls | Increased levels of IL-1b, IL-9, IL-17a, bFGF, and MIP-1α in aniridia patients, all had a positive correlation with MGD parameters. Decreased IL-1RA/IL-1b ratio in aniridia patients. |
| Gurumurthy et al. (2018) [82] | To compare pre- and post-MMG cytokine profiles in SjS patients | Pre-MMG: Increased GM-CSF, IL-1b, IL-2, IL-8, IL-15, IL-17a, MCP-1, and bFGF. Decreased IP-10. Post-MMG: Increased IL-15, IL-17a, and bFGF. |
| Choi et al. (2019) [83] | Anti-inflammatory effect of IPL for MGD | Decreased levels of IL-4, IL-6, IL-10, IL-17a, and TNFα after completing three sessions. |
| Gao et al. (2019) [84] | Comparison of IPL vs. tobramycin/dexamethasone plus warm compresses for MGD | Lower level of IL-1b and IL-17 at 1-week post IPL, but levels returned to baseline after 1 month. |
| Roy et al. (2023) [85] | Correlation of cytokine levels with DED symptoms | Lower corneal staining correlated with higher IL-17a, IL-10, and IFNγ
Higher IFNγ levels correlated with lower conjunctival staining. Higher IL-17a levels correlated with higher TBUT score |
Abbreviations: MGD, meibomian gland dysfunction; OSIDs, ocular surface inflammatory disorders; IL, interleukin; ATs, artificial tears; IFNγ, interferon gamma; TNFα, tumor necrosis factor alpha; MCP-1, macrophage chemoattractant protein 1; bFGF, basic fibroblast growth factor; MIP-1α, macrophage inflammatory protein 1 alpha; MMG, mucous membrane grafting; SjS, Stevens-Johnson syndrome; GM-CSF, granulocyte macrophage colony stimulating factor; IP-10, interferon gamma-induced protein 10; IPL, intense pulsed light; TBUT, tear film breakup time.
5C. Type-3 Lymphocyte – Neutrophil Axis in MGD Pathogenesis
Neutrophils can function as downstream effector cells in type 3 responses [68, 86], similarly making them a target for future study in MGD. However, neutrophils are considered unable to respond directly to IL-17A, as they do not express the IL-17RA – IL-17RC heterodimers that enable response to IL-17A [87]. Instead, non-hematopoietic cells are often recognized as the primary downstream responders to IL-17A, with subsequent neutrophil responses being shaped by IL-17A-driven gene expression changes, including release of neutrophil-influencing chemokines [88, 89]. Indeed, neutrophils were shown to infiltrate the conjunctival tissues in the AED model, and Th17 inhibition reduced this recruitment. Also, systemic depletion of neutrophils (via anti-Ly6G antibody) inhibited the formation of meibomian gland orifice obstructions in the AED model [10]. Further supporting the role of a Th17-neutrophil axis in MGD, adoptive transfer of in vitro differentiated Th17 cells in the mild allergic conjunctivitis model led to increased neutrophil recruitment and development of MGD [10]. It was therefore concluded that Th17 responses result in neutrophil recruitment, and that these neutrophils in turn cause MGD in the AED model. Supporting this conclusion, Singh et al., showed in the AED model that administration of topical lifitegrast (LFA1-ICAM1 inhibitor indicated for DED treatment) led to reduced MGD signs, but not allergy clinical scores. Concordantly, in the conjunctiva of these mice, a reduction in neutrophils, but not eosinophils, was demonstrated [90]. These findings also fit with work by Sun et al., who demonstrated that lifitegrast likewise reduces corneal neutrophil recruitment in an infectious keratitis model in mice [91].
In patients, the role of neutrophils in MGD is less well understood than in mice, but it was observed that the number of neutrophils in patient tears directly correlated with meibum viscosity, indicating that the type 3 immune skewing observed in mice may similarly be relevant in the clinic [10]. Interestingly, a recent study demonstrated greater improvement in signs of meibomian gland function with lifitegrast compared with thermal pulsation procedure in patients with inflammatory MGD [92]. Additional studies evaluating neutrophils in patients have been summarized in Table 3 [10, 65, 72, 93–95]. Hence, these links between MGD neutrophil abundance, lifitegrast neutrophil targeting, and MGD amelioration implicate a possible role for neutrophils in certain forms of human MGD.
Table 3.
Studies reporting elevated levels of neutrophils at the ocular surface of patients with MGD associated with different clinical scenarios.
| Author (year) | Study objective | Study findings |
|---|---|---|
| Sonawane et al. (2012) [93] | To determine the level of nucleases and their relation to eDNA and NET formation in ocular surface inflammation. | Increased eDNA strand length and amount in DED compared to controls, with co-localization of PMNs and elastase to the eDNA strands. Increased conjunctival TLR-9, MyD88, IFN-type 1, IL-6, and TNFα in DED compared to controls. |
| Reyes et al. (2018) [10] | To determine if inflammation has a role in MGD. | Increased quantities of neutrophils in MGD patient tear fluid samples strongly correlated with MGD clinical severity. Tear cytology of AKC and BKC patients revealed populations of leukocytes including neutrophils, eosinophils, and mononuclear cells. |
| Postnikoff et al. (2020) [94] | To describe the phenotype and activation pattern of closed-eye neutrophils in DED versus controls. | Tear neutrophils from DED patients had higher expression of CD66b and higher monocytes than controls. Elevated extracellular MMP-9 and neutrophil elastase. |
| Nair et al. (2021) [95] | To determine the proportion of immune cell subsets in the ocular surface wash samples of DED patients. | Flow cytometry revealed higher proportions of neutrophils, CD4, CD8 T-cells, and double-positive T-cells than controls. Higher neutrophil/NK ratio in evaporative and aqueous DED than in controls. Positive correlation of flow cytometry findings with clinical indices. |
| Mahajan et al. (2021) [65] | To investigate the pathogenesis of obstructive MGD in ocular surface inflammation. | Ocular discharge samples from patients with MGD show aggregated NETs and occlude meibomian gland orifices as shown via eyelid biopsy fluorescent confocal microscopy. Increased tear C5a, IL-6, IL-8, and IL-18. Levels of C5a and IL-8 correlated with tear deficiency. |
| Perez et al. (2023) [72] | To investigate the role of MGD in the pathogenesis of oGVHD. | Spectral flow cytometry on tear samples from oGVHD patients revealed a substantial cell population composed primarily of neutrophils, along with mononuclear, and CD4 cells compared with healthy controls. |
Abbreviations: eDNA, extracellular DNA; NET, neutrophil extracellular trap; DED, dry eye disease; TLR-9, toll-like receptor 9; MyD88, mutant myeloid differentiation primary response 88; IFN, interferon; IL, interleukin; TNFα, tumor necrosis factor alpha; MGD, meibomian gland dysfunction; AKC, atopic keratoconjunctivitis; BKC, blepharokeratoconjunctivitis; CD, cluster of differentiation; MMP-9, matrix metalloproteinase 9; NK, natural killer cell; oGVHD, ocular graft-versus-host disease.
The study included “tear-deficient patients” with ocular cicatricial pemphigoid, oGVHD, and neurotrophic keratitis.
Though the precise way neutrophils cause MGD is not fully understood, a role for neutrophil extracellular traps (NETs) in MGD has been proposed. Mahajan et al., demonstrated that MGD severity in the AED model is reduced through topical application of DNase-1, which breaks down NETs via degrading extracellular DNA. The authors demonstrated similar findings with pharmacological inhibition or gene deletion of peptidylarginine deiminase 4, which plays a notable role in NET generation, and concluded that NETs were mediating MGD through obstructing the gland orifice [65]. In another study, An et al. showed that neutrophil and NET levels at the ocular surface of oGVHD patients are elevated relative to healthy controls. They also found that NETs inhibit meibocyte proliferation and meibum production, providing another possible mechanism through which NETs may induce MGD [96]. Relatedly, Perez et al. demonstrated that oGVHD is strongly correlated with MGD development, reporting an incidence of 97% in oGVHD and an incidence of 93% in their oGVHD mouse model [72]. More studies are required to understand the mechanism underpinning possible neutrophil and NET interactions with meibomian gland cells that may be driving long term dysfunction.
6. Towards a unified perspective on the future of MGD pathogenesis research
The underlying motivation for this piece was to present two opposing views on the initial drivers of MGD: one view or “pathway” focused on immune-driven MGD and the other glandular-driven MGD. However, it must be acknowledged that neither pathway truly acts in a ‘vacuum,’ particularly in the chronic disease setting. Hence, our discussions further explored how these two pathways may interact in the MGD disease process. Below is the synthesis of our discussion, which we based on the following three points: (1) that either pathway can serve as the early trigger for MGD; (2) that both pathways interact in MGD; and (3) that some stimuli may trigger both pathways simultaneously.
-
(1)
Either pathway can serve as the early trigger of MGD: Examples of glandular-driven MGD induced pharmacologically (e.g. isotretinoin) or genetically (e.g. Awat2 KO) in animal models have already been reviewed herein. Likewise, we reviewed findings from the AED model, which provided direct evidence of immune-driven MGD. Hence, it can be concluded that either pathway (immune or non-immune) may serve as an early trigger of MGD.
-
(2)
Both pathways interact with one another in MGD: For the immune pathway, immune-driven MGD must trigger pathologic glandular activity to cause disease. As reviewed in the earlier sections, one possible mechanism by which type 3 immune responses can negatively affect glandular cells involves NETosis that leads to MG obstruction in the AED model [65], or via the demonstrated in vitro effect that NETs can directly alter meibocyte physiology [96]. Evidence for the reverse scenario, that glandular-driven MGD can involve secondary pathological immune responses, comes from the Awat2 KO setting [38]. In these mice it was reported that an exacerbated MGD phenotype was associated with infiltration of immune cells [38], suggesting a role for immune responses in amplifying the disease process. Though the precise mechanism by which this secondary immune response amplified MGD was not investigated, it is conceivable that there may be overlapping activities from the AED model with respect to neutrophil infiltration.
-
(3)
Some stimuli may affect both compartments simultaneously in MGD: Interaction between these two pathways do not need to be sequential. For example, meibocyte expression levels of PPARγ may impact meibocyte physiology and immune cell responses simultaneously. Meibocyte expression of PPARγ is reduced with aging, and this reduction impairs meibocyte differentiation [28, 31, 32]. Separately, it is also known that PPARγ can exert anti-inflammatory effects [97]. As such, age associated reduction in PPARγ may simultaneously impair meibocyte activity while being permissive to otherwise restricted inflammatory activity. Moreover, exposure of meibocytes to IL-1β was shown to decrease cell proliferation, lipid synthesis, and expression of PPARγ [98]. Relatedly, IL-1β is also known to promote type 3 immune response severity [68].
We illustrate the synthesis of these three points in the form of a simple Yin and Yang diagram, whereby each color represents a given pathway (Figure 4). Because either pathway can serve as the early trigger, can interact, or can be simultaneously affected by stimuli, the benefit of the Yin and Yang is that it de-emphasizes the sequence of these events. This point is critical because it is often assumed that inflammation is not an early trigger. The Yin and Yang analogy also acknowledges the differential contribution from each pathway in the disease process, depending on the underlying condition and/or disease, while also highlighting that both components generally exhibit some level of contribution to MGD. In short, the Yin and Yang captures a more fluid concept which recognizes the interplay between both immune-driven and glandular-driven factors to help contextualize and promote the study of the synergies between these two seemingly disparate compartments.
Figure 4. The Yin and Yang of MGD: Dynamics of immune and non-immune contributions to the MGD disease process.

The Yin and Yang analogy captures the 3 points we argued regarding the need for a more fluid and dynamic model to represent the disease process. These points are: (1) that the early initiators of MGD can either be immune-driven or glandular-driven pathologic pathways; (2) that both pathways can interact in the disease process; and (3) that some pathologic stimuli may trigger both pathways simultaneously. The level of contribution from each pathway can change depending on the etiology and disease state, but both compartments must be considered to holistically understand MGD pathogenesis.
Abbreviations: MGD, Meibomian gland dysfunction.
7. Conclusion
In this perspective piece we lay out the evidence and significance of both glandular-driven and immune-driven pathways in the pathobiology of MGD and discussed mechanistic insights for both. Moreover, that neither pathway truly acts in a vacuum, particularly in the chronic disease setting, we discussed how both pathways could be interactive and further amplify the disease process. The points raised clearly suggest an important relationship between pathologic immune responses and glandular cell dysfunction – regardless of which process serves an upstream role. It is hoped that through more diligent research that focuses on the nexus between these two pathways, we will come to a better understanding of the events leading to dry eye disease. Certainly, it is true that we do not see that for which we are not looking, and future research needs to combine both meibomian gland functional and immunological investigations if we are not going to continue to simply repeat the studies of the past. Such work would enhance our understanding of the disease-driving synergies observed between immune and non-immune mediated dysfunction, especially regarding how both components contribute, to varying degrees, in distinct disease settings – the Yin and Yang of MGD.
Support by:
NEI EY021798 (DRS), NEI EY021510 (JVJ), NEI EY024484 (VLP), P30 EY034070 (JVJ), P30 EY005722 (DRS/VLP), and unrestricted grants from Research to Prevent Blindness, Inc, to the Gavin Herbert Eye Institute at the University of California Irvine (JVJ) and the Duke Eye Center at Duke University (DRS/VLP).
Footnotes
Disclosure of Conflicts of Interest
DRS is a consultant for Roche, Genentech, and Tarsus Pharmaceuticals, and has previously performed investigator-initiated research for Novartis. VLP is a consultant for BrightStar, BRIM Biotechnology, Kala Pharmaceuticals, Novartis, Quidel, and Thea, as well as a consultant and advisory board member for Dompe, consultant and equity holder for Kiora and Trefoil Therapeutics, and has received research support from Alcon. For all other authors, there are no disclosures to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
9. References
- [1].Nichols KK, Foulks GN, Bron AJ, Glasgow BJ, Dogru M, Tsubota K, et al. The international workshop on meibomian gland dysfunction: executive summary. Invest Ophthalmol Vis Sci. 2011;52:1922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schaumberg DA, Nichols JJ, Papas EB, Tong L, Uchino M, Nichols KK. The international workshop on meibomian gland dysfunction: report of the subcommittee on the epidemiology of, and associated risk factors for, MGD. Invest Ophthalmol Vis Sci. 2011;52:1994–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Knop E, Knop N, Millar T, Obata H, Sullivan DA. The international workshop on meibomian gland dysfunction: report of the subcommittee on anatomy, physiology, and pathophysiology of the meibomian gland. Invest Ophthalmol Vis Sci. 2011;52:1938–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sun M, Moreno IY, Dang M, Coulson-Thomas VJ. Meibomian Gland Dysfunction: What Have Animal Models Taught Us? Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McCulley JP, Sciallis GF. Meibomian keratoconjunctivitis. Am J Ophthalmol. 1977;84:788–93. [DOI] [PubMed] [Google Scholar]
- [6].Jester JV, Nicolaides N, Smith RE. Meibomian gland studies: histologic and ultrastructural investigations. Invest Ophthalmol Vis Sci. 1981;20:537–47. [PubMed] [Google Scholar]
- [7].Jester JV, Rife L, Nii D, Luttrull JK, Wilson L, Smith RE. In vivo biomicroscopy and photography of meibomian glands in a rabbit model of meibomian gland dysfunction. Invest Ophthalmol Vis Sci. 1982;22:660–7. [PubMed] [Google Scholar]
- [8].Gutgesell VJ, Stern GA, Hood CI. Histopathology of meibomian gland dysfunction. Am J Ophthalmol. 1982;94:383–7. [DOI] [PubMed] [Google Scholar]
- [9].Anatomy Obata H. and histopathology of human meibomian gland. Cornea. 2002;21:S70–4. [DOI] [PubMed] [Google Scholar]
- [10].Reyes NJ, Yu C, Mathew R, Kunnen CM, Kalnitsky J, Redfern RL, et al. Neutrophils cause obstruction of eyelid sebaceous glands in inflammatory eye disease in mice. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baudouin C, Messmer EM, Aragona P, Geerling G, Akova YA, Benitez-del-Castillo J, et al. Revisiting the vicious circle of dry eye disease: a focus on the pathophysiology of meibomian gland dysfunction. Br J Ophthalmol. 2016;100:300–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Amano S, Shimazaki J, Yokoi N, Hori Y, Arita R, Obata H, et al. Meibomian Gland Dysfunction Clinical Practice Guidelines. Japanese Journal of Ophthalmology. 2023;67:448–539. [DOI] [PubMed] [Google Scholar]
- [13].Suzuki T, Teramukai S, Kinoshita S. Meibomian glands and ocular surface inflammation. Ocul Surf. 2015;13:133–49. [DOI] [PubMed] [Google Scholar]
- [14].Korb DR, Henriquez AS. Meibomian gland dysfunction and contact lens intolerance. J Am Optom Assoc. 1980;51:243–51. [PubMed] [Google Scholar]
- [15].Mathers WD, Shields WJ, Sachdev MS, Petroll WM, Jester JV. Meibomian gland dysfunction in chronic blepharitis. Cornea. 1991;10:277–85. [DOI] [PubMed] [Google Scholar]
- [16].Robin JB, Jester JV, Nobe J, Nicolaides N, Smith RE. In vivo transillumination biomicroscopy and photography of meibomian gland dysfunction. A clinical study. Ophthalmology. 1985;92:1423–6. [DOI] [PubMed] [Google Scholar]
- [17].Mathers WD. Ocular evaporation in meibomian gland dysfunction and dry eye. Ophthalmology. 1993;100:347–51. [DOI] [PubMed] [Google Scholar]
- [18].Ohnishi Y, Kohno T. Polychlorinated biphenyls poisoning in monkey eye. Invest Ophthalmol Vis Sci. 1979;18:981–4. [PubMed] [Google Scholar]
- [19].Reneker LW, Irlmeier RT, Shui YB, Liu Y, Huang AJW. Histopathology and selective biomarker expression in human meibomian glands. Br J Ophthalmol. 2020;104:999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Parfitt GJ, Xie Y, Geyfman M, Brown DJ, Jester JV. Absence of ductal hyper-keratinization in mouse age-related meibomian gland dysfunction (ARMGD). Aging (Albany NY). 2013;5:825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dursun D, Wang M, Monroy D, Li DQ, Lokeshwar BL, Stern ME, et al. A mouse model of keratoconjunctivitis sicca. Invest Ophthalmol Vis Sci. 2002;43:632–8. [PubMed] [Google Scholar]
- [22].Fabiani C, Barabino S, Rashid S, Dana MR. Corneal epithelial proliferation and thickness in a mouse model of dry eye. Exp Eye Res. 2009;89:166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stapleton F, Alves M, Bunya VY, Jalbert I, Lekhanont K, Malet F, et al. TFOS DEWS II Epidemiology Report. Ocul Surf. 2017;15:334–65. [DOI] [PubMed] [Google Scholar]
- [24].Nien CJ, Paugh JR, Massei S, Wahlert AJ, Kao WW, Jester JV. Age-related changes in the meibomian gland. Exp Eye Res. 2009;89:1021–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jester BE, Nien CJ, Winkler M, Brown DJ, Jester JV. Volumetric reconstruction of the mouse meibomian gland using high-resolution nonlinear optical imaging. Anat Rec (Hoboken). 2011;294:185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nien CJ, Massei S, Lin G, Nabavi C, Tao J, Brown DJ, et al. Effects of age and dysfunction on human meibomian glands. Arch Ophthalmol. 2011;129:462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Olami Y, Zajicek G, Cogan M, Gnessin H, Pe’er J. Turnover and migration of meibomian gland cells in rats’ eyelids. Ophthalmic Res. 2001;33:170–5. [DOI] [PubMed] [Google Scholar]
- [28].Jester JV, Potma E, Brown DJ. PPARgamma Regulates Mouse Meibocyte Differentiation and Lipid Synthesis. Ocul Surf. 2016;14:484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–7. [DOI] [PubMed] [Google Scholar]
- [30].Veniaminova NA, Jia Y, Hartigan AM, Huyge TJ, Tsai SY, Grachtchouk M, et al. Distinct mechanisms for sebaceous gland self-renewal and regeneration provide durability in response to injury. bioRxiv. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim SW, Xie Y, Nguyen PQ, Bui VT, Huynh K, Kang JS, et al. PPARgamma regulates meibocyte differentiation and lipid synthesis of cultured human meibomian gland epithelial cells (hMGEC). Ocul Surf. 2018;16:463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim SW, Rho CR, Kim J, Xie Y, Prince RC, Mustafa K, et al. Eicosapentaenoic acid (EPA) activates PPARgamma signaling leading to cell cycle exit, lipid accumulation, and autophagy in human meibomian gland epithelial cells (hMGEC). Ocul Surf. 2020;18:427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang X, Reneker LW, Zhong X, Huang AJW, Jester JV. Meibomian gland stem/progenitor cells: The hunt for gland renewal. Ocul Surf. 2023;29:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Parfitt GJ, Lewis PN, Young RD, Richardson A, Lyons JG, Di Girolamo N, et al. Renewal of the Holocrine Meibomian Glands by Label-Retaining, Unipotent Epithelial Progenitors. Stem Cell Reports. 2016;7:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Suhalim JL, Parfitt GJ, Xie Y, De Paiva CS, Pflugfelder SC, Shah TN, et al. Effect of desiccating stress on mouse meibomian gland function. Ocul Surf. 2014;12:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sun M, Puri S, Parfitt GJ, Mutoji N, Coulson-Thomas VJ. Hyaluronan Regulates Eyelid and Meibomian Gland Morphogenesis. Invest Ophthalmol Vis Sci. 2018;59:3713–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Otsuka K, Sawai-Ogawa M, Kihara A. Formation of fatty alcohols-components of meibum lipids-by the fatty acyl-CoA reductase FAR2 is essential for dry eye prevention. FASEB J. 2022;36:e22216. [DOI] [PubMed] [Google Scholar]
- [38].Widjaja-Adhi MAK, Silvaroli JA, Chelstowska S, Trischman T, Bederman I, Sayegh R, et al. Deficiency in Acyl-CoA:Wax Alcohol Acyltransferase 2 causes evaporative dry eye disease by abolishing biosynthesis of wax esters. FASEB J. 2020;34:13792–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Butovich IA, Lu H, McMahon A, Ketelson H, Senchyna M, Meadows D, et al. Biophysical and morphological evaluation of human normal and dry eye meibum using hot stage polarized light microscopy. Invest Ophthalmol Vis Sci. 2014;55:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Iyengar BT, Schlenk H. Melting points of synthetic wax esters. Lipids. 1969;4:28–30. [DOI] [PubMed] [Google Scholar]
- [41].Mahadevan V, Lundberg W. Preparation of cholesterol esters of long-chain fatty acids and characterization of cholesteryl arachidonate. Journal of Lipid Research. 1962;3:106–10. [Google Scholar]
- [42].Shrestha RK, Borchman D, Foulks GN, Yappert MC, Milliner SE. Analysis of the composition of lipid in human meibum from normal infants, children, adolescents, adults, and adults with meibomian gland dysfunction using (1)H-NMR spectroscopy. Invest Ophthalmol Vis Sci. 2011;52:7350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McCulley JP, Shine WE. Meibomian secretions in chronic blepharitis. Adv Exp Med Biol. 1998;438:319–26. [DOI] [PubMed] [Google Scholar]
- [44].Rosenfeld L, Cerretani C, Leiske DL, Toney MF, Radke CJ, Fuller GG. Structural and rheological properties of meibomian lipid. Invest Ophthalmol Vis Sci. 2013;54:2720–32. [DOI] [PubMed] [Google Scholar]
- [45].Luo S, Dzsotjan G, Joshi R, Juhasz T, Jester JV. Modeling Meibum Secretion: An Alternative Mechanisms for Obstructive Meibomian gland Dysfunction (MGD). Ocular Surface. (in Review). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dao AH, Spindle JD, Harp BA, Jacob A, Chuang AZ, Yee RW. Association of dyslipidemia in moderate to severe meibomian gland dysfunction. Am J Ophthalmol. 2010;150:371–5.e1. [DOI] [PubMed] [Google Scholar]
- [47].Pinna A, Blasetti F, Zinellu A, Carru C, Solinas G. Meibomian gland dysfunction and hypercholesterolemia. Ophthalmology. 2013;120:2385–9. [DOI] [PubMed] [Google Scholar]
- [48].Yamaguchi M, Shiraishi A. Relationship Between Eyelid Pressure and Ocular Surface Disorders in Patients With Healthy and Dry Eyes. Invest Ophth Vis Sci. 2018;59. [DOI] [PubMed] [Google Scholar]
- [49].Ruiz-Lozano RE, Hernandez-Camarena JC, Garza-Garza LA, Bustamante-Arias A, Colorado-Zavala MF, Cardenas-de la Garza JA. Isotretinoin and the eye: A review for the dermatologist. Dermatol Ther. 2020;33:e14029. [DOI] [PubMed] [Google Scholar]
- [50].Melnik BC. Isotretinoin and FoxO1: A scientific hypothesis. Dermatoendocrinol. 2011;3:141–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mathers WD, Shields WJ, Sachdev MS, Petroll WM, Jester JV. Meibomian gland morphology and tear osmolarity: changes with Accutane therapy. Cornea. 1991;10:286–90. [DOI] [PubMed] [Google Scholar]
- [52].Lambert RW, Smith RE. Pathogenesis of blepharoconjunctivitis complicating 13-cis-retinoic acid (isotretinoin) therapy in a laboratory model. Invest Ophthalmol Vis Sci. 1988;29:1559–64. [PubMed] [Google Scholar]
- [53].Lambert RW, Smith RE. Effects of 13-cis-retinoic acid on the hamster meibomian gland. J Invest Dermatol. 1989;92:321–5. [DOI] [PubMed] [Google Scholar]
- [54].Zhang P, Tian L, Bao J, Li S, Li A, Wen Y, et al. Isotretinoin Impairs the Secretory Function of Meibomian Gland Via the PPARγ Signaling Pathway. Invest Ophthalmol Vis Sci. 2022;63:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhu HY, Riau AK, Barathi VA, Chew J, Beuerman RW. Expression of neural receptors in mouse meibomian gland. Cornea. 2010;29:794–801. [DOI] [PubMed] [Google Scholar]
- [56].Alam J, Yu Z, de Paiva CS, Pflugfelder SC. Retinoid Regulation of Ocular Surface Innate Inflammation. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Blackie CA, Korb DR, Knop E, Bedi R, Knop N, Holland EJ. Nonobvious obstructive meibomian gland dysfunction. Cornea. 2010;29:1333–45. [DOI] [PubMed] [Google Scholar]
- [58].Qazi Y, Kheirkhah A, Blackie C, Cruzat A, Trinidad M, Williams C, et al. In vivo detection of clinically non-apparent ocular surface inflammation in patients with meibomian gland dysfunction-associated refractory dry eye symptoms: a pilot study. Eye (Lond). 2015;29:1099–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Reyes NJ, Mathew R, Saban DR. Induction and Characterization of the Allergic Eye Disease Mouse Model. Methods Mol Biol. 2018;1799:49–57. [DOI] [PubMed] [Google Scholar]
- [60].Ahadome SD, Mathew R, Reyes NJ, Mettu PS, Cousins SW, Calder VL, et al. Classical dendritic cells mediate fibrosis directly via the retinoic acid pathway in severe eye allergy. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Smith RE, Reyes NJ, Khandelwal P, Schlereth SL, Lee HS, Masli S, et al. Secondary allergic T cell responses are regulated by dendritic cell-derived thrombospondin-1 in the setting of allergic eye disease. J Leukoc Biol. 2016;100:371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lee HS, Hos D, Blanco T, Bock F, Reyes NJ, Mathew R, et al. Involvement of corneal lymphangiogenesis in a mouse model of allergic eye disease. Invest Ophthalmol Vis Sci. 2015;56:3140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Khandelwal P, Blanco-Mezquita T, Emami P, Lee HS, Reyes NJ, Mathew R, et al. Ocular mucosal CD11b+ and CD103+ mouse dendritic cells under normal conditions and in allergic immune responses. PLoS One. 2013;8:e64193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Schlereth S, Lee HS, Khandelwal P, Saban DR. Blocking CCR7 at the ocular surface impairs the pathogenic contribution of dendritic cells in allergic conjunctivitis. Am J Pathol. 2012;180:2351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mahajan A, Hasikova L, Hampel U, Gruneboom A, Shan X, Herrmann I, et al. Aggregated neutrophil extracellular traps occlude Meibomian glands during ocular surface inflammation. Ocul Surf. 2021;20:1–12. [DOI] [PubMed] [Google Scholar]
- [66].Lou B, Wu W, Zeng L, Zhou W, Zhang X, Zhou X, et al. Alleviating experimental allergic eye disease by inhibiting pro-lymphangiogenic VEGFR3 signal. Ocul Surf. 2022;26:1–12. [DOI] [PubMed] [Google Scholar]
- [67].Saban DR, Hodges RR, Mathew R, Reyes NJ, Yu C, Kaye R, et al. Resolvin D1 treatment on goblet cell mucin and immune responses in the chronic allergic eye disease (AED) model. Mucosal Immunol. 2019;12:145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. 2015;135:626–35. [DOI] [PubMed] [Google Scholar]
- [69].Pflugfelder SC, de Paiva CS. The Pathophysiology of Dry Eye Disease: What We Know and Future Directions for Research. Ophthalmology. 2017;124:S4–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2011;187:2213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Caspi RR, Roberge FG, McAllister CG, el-Saied M, Kuwabara T, Gery I, et al. T cell lines mediating experimental autoimmune uveoretinitis (EAU) in the rat. J Immunol. 1986;136:928–33. [PubMed] [Google Scholar]
- [72].Perez VL, Mousa HM, Soifer M, Beatty C, Sarantopoulos S, Saban DR, et al. Meibomian Gland Dysfunction: A Route of Ocular Graft-Versus-Host Disease Progression That Drives a Vicious Cycle of Ocular Surface Inflammatory Damage. Am J Ophthalmol. 2023;247:42–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chen Y, Dana R. Autoimmunity in dry eye disease - An updated review of evidence on effector and memory Th17 cells in disease pathogenicity. Autoimmun Rev. 2021;20:102933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].St Leger AJ, Desai JV, Drummond RA, Kugadas A, Almaghrabi F, Silver P, et al. An Ocular Commensal Protects against Corneal Infection by Driving an Interleukin-17 Response from Mucosal gammadelta T Cells. Immunity. 2017;47:148–58 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Alam J, Yazdanpanah G, Ratnapriya R, Borcherding N, de Paiva CS, Li D, et al. IL-17 Producing Lymphocytes Cause Dry Eye and Corneal Disease With Aging in RXRalpha Mutant Mouse. Front Med (Lausanne). 2022;9:849990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].De Paiva CS, Chotikavanich S, Pangelinan SB, Pitcher JD 3rd, Fang B, Zheng X, et al. IL-17 disrupts corneal barrier following desiccating stress. Mucosal Immunol. 2009;2:243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bustamante-Arias A, Ruiz Lozano RE, Rodriguez-Garcia A. Dry eye disease, a prominent manifestation of systemic autoimmune disorders. Eur J Ophthalmol. 2022:11206721221088259. [DOI] [PubMed] [Google Scholar]
- [78].Kang MH, Kim MK, Lee HJ, Lee HI, Wee WR, Lee JH. Interleukin-17 in various ocular surface inflammatory diseases. J Korean Med Sci. 2011;26:938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lee H, Min K, Kim EK, Kim TI. Minocycline controls clinical outcomes and inflammatory cytokines in moderate and severe meibomian gland dysfunction. Am J Ophthalmol. 2012;154:949–57 e1. [DOI] [PubMed] [Google Scholar]
- [80].Lee H, Chung B, Kim KS, Seo KY, Choi BJ, Kim TI. Effects of topical loteprednol etabonate on tear cytokines and clinical outcomes in moderate and severe meibomian gland dysfunction: randomized clinical trial. Am J Ophthalmol. 2014;158:1172–83 e1. [DOI] [PubMed] [Google Scholar]
- [81].Landsend ECS, Utheim OA, Pedersen HR, Aass HCD, Lagali N, Dartt DA, et al. The Level of Inflammatory Tear Cytokines is Elevated in Congenital Aniridia and Associated with Meibomian Gland Dysfunction. Invest Ophthalmol Vis Sci. 2018;59:2197–204. [DOI] [PubMed] [Google Scholar]
- [82].Gurumurthy S, Iyer G, Srinivasan B, Agarwal S, Angayarkanni N. Ocular surface cytokine profile in chronic Stevens-Johnson syndrome and its response to mucous membrane grafting for lid margin keratinisation. Br J Ophthalmol. 2018;102:169–76. [DOI] [PubMed] [Google Scholar]
- [83].Choi M, Han SJ, Ji YW, Choi YJ, Jun I, Alotaibi MH, et al. Meibum Expressibility Improvement as a Therapeutic Target of Intense Pulsed Light Treatment in Meibomian Gland Dysfunction and Its Association with Tear Inflammatory Cytokines. Sci Rep. 2019;9:7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Gao YF, Liu RJ, Li YX, Huang C, Liu YY, Hu CX, et al. Comparison of anti-inflammatory effects of intense pulsed light with tobramycin/dexamethasone plus warm compress on dry eye associated meibomian gland dysfunction. Int J Ophthalmol. 2019;12:1708–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Roy NS, Wei Y, Ying GS, Maguire MG, Asbell PA, Dry Eye A, et al. Association of Tear Cytokine Concentrations with Symptoms and Signs of Dry Eye Disease: Baseline Data from the Dry Eye Assessment and Management (DREAM) Study. Curr Eye Res. 2023;48:339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. [DOI] [PubMed] [Google Scholar]
- [87].Liu R, Lauridsen HM, Amezquita RA, Pierce RW, Jane-Wit D, Fang C, et al. IL-17 Promotes Neutrophil-Mediated Immunity by Activating Microvascular Pericytes and Not Endothelium. J Immunol. 2016;197:2400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zenobia C, Hajishengallis G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000. 2015;69:142–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 Family of Cytokines in Health and Disease. Immunity. 2019;50:892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Singh PP, Yu C, Mathew R, Perez VL, Saban DR. Meibomian gland dysfunction is suppressed via selective inhibition of immune responses by topical LFA-1/ICAM antagonism with lifitegrast in the allergic eye disease (AED) model. Ocul Surf. 2021;21:271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sun Y, Zhang R, Gadek TR, O’Neill CA, Pearlman E. Corneal inflammation is inhibited by the LFA-1 antagonist, lifitegrast (SAR 1118). J Ocul Pharmacol Ther. 2013;29:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tauber JA 6-Week, Prospective, Randomized, Single-Masked Study of Lifitegrast Ophthalmic Solution 5% Versus Thermal Pulsation Procedure for Treatment of Inflammatory Meibomian Gland Dysfunction. Cornea. 2020;39:403–7. [DOI] [PubMed] [Google Scholar]
- [93].Sonawane S, Khanolkar V, Namavari A, Chaudhary S, Gandhi S, Tibrewal S, et al. Ocular surface extracellular DNA and nuclease activity imbalance: a new paradigm for inflammation in dry eye disease. Invest Ophthalmol Vis Sci. 2012;53:8253–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Postnikoff CK, Held K, Viswanath V, Nichols KK. Enhanced closed eye neutrophil degranulation in dry eye disease. Ocul Surf. 2020;18:841–51. [DOI] [PubMed] [Google Scholar]
- [95].Nair AP, D’Souza S, Shetty R, Ahuja P, Kundu G, Khamar P, et al. Altered ocular surface immune cell profile in patients with dry eye disease. Ocul Surf. 2021;21:96–106. [DOI] [PubMed] [Google Scholar]
- [96].An S, Raju I, Surenkhuu B, Kwon JE, Gulati S, Karaman M, et al. Neutrophil extracellular traps (NETs) contribute to pathological changes of ocular graft-vs.-host disease (oGVHD) dry eye: Implications for novel biomarkers and therapeutic strategies. Ocul Surf. 2019;17:589–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Martin H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat Res. 2009;669:1–7. [DOI] [PubMed] [Google Scholar]
- [98].Qu JY, Xie HT, Xiao YT, Zhang YY, Hu ZX, Wang JS, et al. The inhibition of p38 MAPK blocked inflammation to restore the functions of rat meibomian gland epithelial cells. Exp Eye Res. 2023;231:109470. [DOI] [PubMed] [Google Scholar]