

Abstract
Non-directed regioselective activation of bis(boronic esters), followed by functionalization, is reported. A bulky activator is shown to selectively activate the less hindered boronic ester enabling it to undergo stereospecific cross-coupling to a variety of electrophiles. This steric-based regioselectivity provides a simple and efficient method to prepare highly functionalized, enantiomerically enriched products starting from simple alkenes.
Keywords: organoboronic esters, cross-coupling, enantioselective catalysis, difunctionalization
Graphical Abstract

Regio- and enantioselective catalytic difunctionalization of alkenes can be accomplished by a strategy involving alkene diboration followed by site-selective activation and copper-catalyzed coupling of the less-hindered boronic ester. This process allows a broad array of difunctional chiral products to be produced from simple olefin starting materials.
Propionate-derived natural products are an important class of compounds that often exhibit potent biological activity.[1] Structures such as discodermolide[2] and erythronolide A[3] (Figure 1a) are archetypal polypropionates that have captured the attention of synthetic chemists and have inspired the development of new catalytic enantioselective methods for the assembly of motifs bearing a methyl-containing stereocenter adjacent to an oxidized carbon center. Foremost among methods for catalytic propionate synthesis are aldol addition and crotylation reactions (Figure 1b), but a number of other approaches have also been devised to address these targets.[4] In relation to these processes, a complementary method could emerge from site-selective and enantioselective alkene difunctionalization (Figure 1c).[5] This method, specifically for propionate construction, would operate with 2-alkene feedstocks. Of note, 2-alkenes are easily prepared from α-olefin precursors by simple, scalable and stereoselective 1-alkene isomerization[6] or by stereoretentive, chain-extending catalytic cross-metathesis.[7] In this report, we describe a strategy for regio- and enantioselective difunctionalization of 2-alkenes and show that this process can be effective for the construction of propionate derivatives as well as other useful difunctional molecules.
Figure 1.

Polypropionates and strategies for their construction.
Despite the significant attention that has been paid to the development of enantioselective alkene transformations, significant methodological gaps limit our ability to transform these building blocks in an efficient and selective manner. For example, selective symmetric catalytic difunctionalization of trans alkenes (Figure 2a) can be accomplished but is largely limited to dihydroxylation,[8] dihalogenation,[9] diacetylation[10] and group transfer reactions (epoxidation, aziridination, cyclopropanation).[ 11] A more challenging task is the non-symmetric difunctionalization of disubstituted alkenes (Figure 2b), where a selective reaction outcome requires not only facial selectivity in the addition step, but also regiocontrolled attachment of different groups to each termini of the olefin. To accomplish regioselective non-symmetric difunctionalization of alkenes, a bias between the two alkene termini is required. This demand is most often met by employing alkenes bearing electronically differentiated substituents (styrenes, enones, enynes etc.) or by inclusion of a directing group in one alkene appendage.[12] Efforts in our laboratory and others have examined alkene difunctionalization by borylation-based strategies.[ 13] In one process, terminal alkenes undergo catalytic enantioselective diboration, followed by Pd or Cu-catalyzed cross-coupling (Figure 2c).[14] Site-selectivity in this sequence arises by steric effects that operate during the transmetalation: the primary carbon undergoes faster transfer from boron to the catalytic center and is thereby selectively functionalized. Relative to this process, use of diboration/cross-coupling for the difunctionalization of internal alkenes faces significant additional challenges: first, stereospecific cross-coupling of secondary alkylboronic esters is more challenging than reactions of primary boronates; second, with two secondary boronates in the intermediate structure, site-selectivity becomes inherently more difficult. With respect to the first challenge, we recently demonstrated that addition of sterically encumbered tertiary organolithium reagents to organoboronic esters enables efficient and stereospecific transmetallation of both secondary and tertiary carbons to copper, followed by efficient cross-coupling (Figure 2d).[ 15] It was of interest to determine if this activation mode could apply to compounds bearing vicinal bis(boronates) and whether the size of the tertiary alkyl activator might allow effective discrimination of the boronic esters based on the steric environment (Figure 2e).[16,17]
Figure 2.
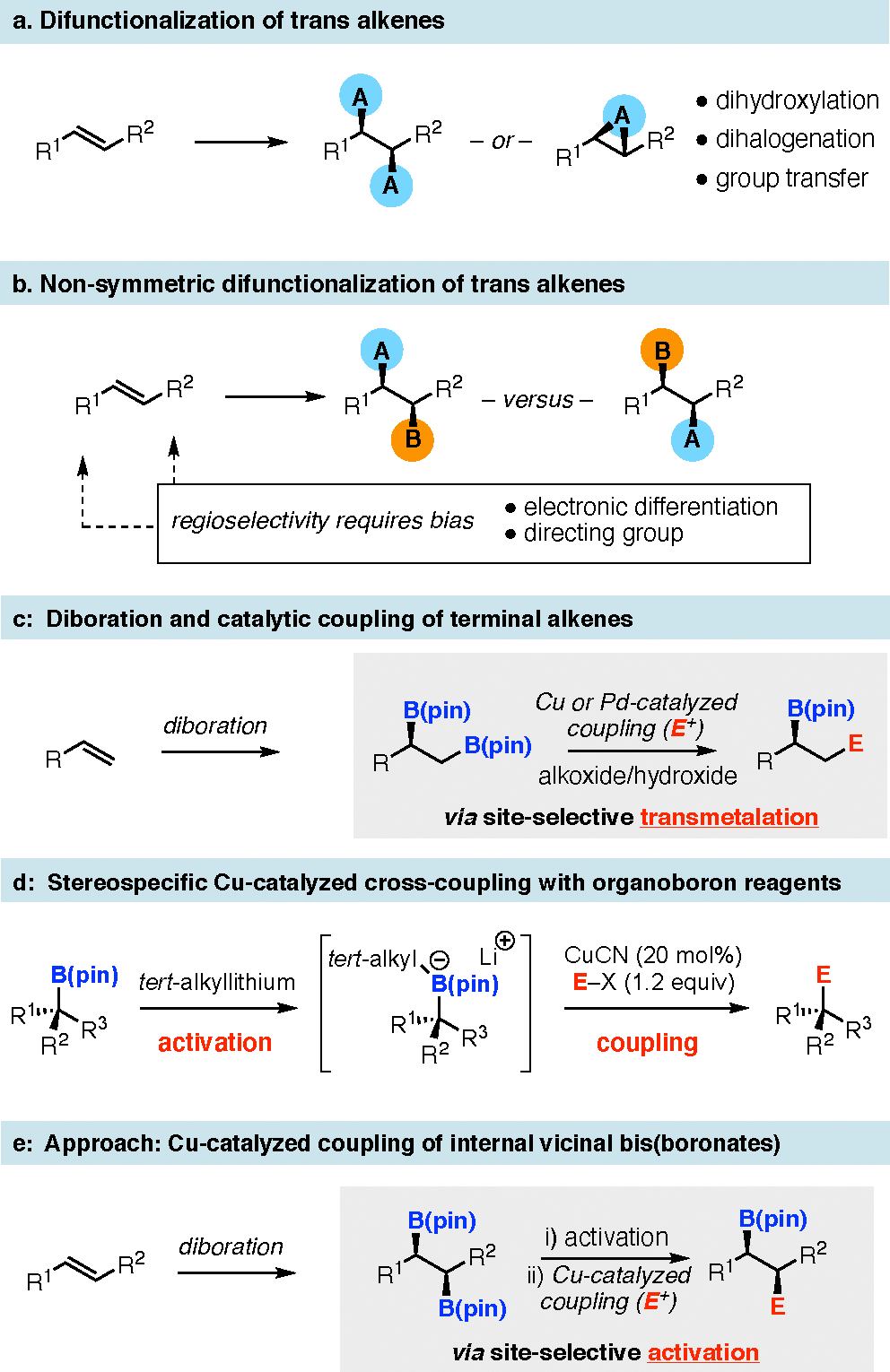
Enantioselective difunctionalization of alkenes. Abbreviations: pin = pinacolato.
To study the site selectivity in the addition of tert-butyllithium to vicinal bis(boronic esters), as well as learn about the stability of the derived ate complexes, isotopically labelled substrate 1 was prepared, subjected to activation, and the process followed by both 13C and 11B NMR. Of note, previous experiments in our laboratory showed that the chemical shift of carbon nuclei are sensitive to the coordination number of β boron groups, with shifts (Δδ) of 1–6 ppm occurring upon conversion of boronic esters to four-coordinate borates.[18] In line with these observations, when 1 was treated with tert-butyllithium at −78 °C in THF and warmed to 22 °C, the 13C resonance corresponding to 1 was replaced with a single new major species assigned to complex 2a. Along with shifts in the 13C NMR, the 11B NMR indicated 55% of the boron centers were four-coordinate (11B δ 10 ppm). Of note, integration of the 13C signal for 2a relative to minor species leads to the conclusion that selectivity in ate complex formation occurs with ≥9:1 site selectivity. Heating the mixture containing complex 2a to 60 °C for 3 hours did not lead to a substantial change in the NMR spectrum suggesting that not only is the ate complex formed selectively, but it is also not subject to significant decomposition or equilibration over the time course of a typical reaction. Of note, whereas Gao has shown19 that an aryl group of a boron ate complex may bridge to an adjacent boronic ester, we do not find evidence of such a process with tert-butyl-derived ate complexes (see Supporing Information for discussion).
To determine whether selective ate complex formation translates to selective coupling reaction, enantiomerically enriched substrate 3 (Figure 3c) was prepared, subjected to low-temperature activation, and then employed in catalytic coupling followed by oxidative work-up. Consistent with the NMR analysis in Figure 3b, the coupling product 4 was obtained with outstanding enantiospecificity and in 7:1 regioselectivity and 55% isolated yield.
Figure 3.

Analysis of site selective boron “ate” complex formation. Abbreviations: DMSO = dimethylsulfoxide.
With experiments suggesting that activation and coupling of vicinal bis(boronates) could be accomplished with site selectivity, several substrates were examined in subsequent copper-catalyzed cross-coupling reactions.[20] With 2,3-dichloropropene as an electrophile a variety of 2,3-bis(boronates) were studied (Figure 4) and revealed product regioisomer ratios ranging from 6:1 to 10:1 by 1H NMR analysis of unpurified reaction mixtures. For most substrates, the regioisomers were easily separable during purification and the yields in Figure 4a represent isolated yield of the major stereoisomer. In addition to aliphatic substrates that are devoid of any directing functionality (product 5), TBS-protected alcohol and arene-containing substrates (6, 7) were also effective. Noteworthy is that discrimination between a methyl and an ethyl group was possible and delivered product 8 (8:1 rr on crude NMR) from the cis diboration adduct of trans-2-pentene. Also of note, the substrate derived from diboration of a cis alkene provided convenient access to the opposite diastereoisomer of product (product 14). However, erosion in stereospecificity was observed in this reaction, an outcome that might be attributed to a transient anti boratirane ion intermediate.[21]
Figure 4.
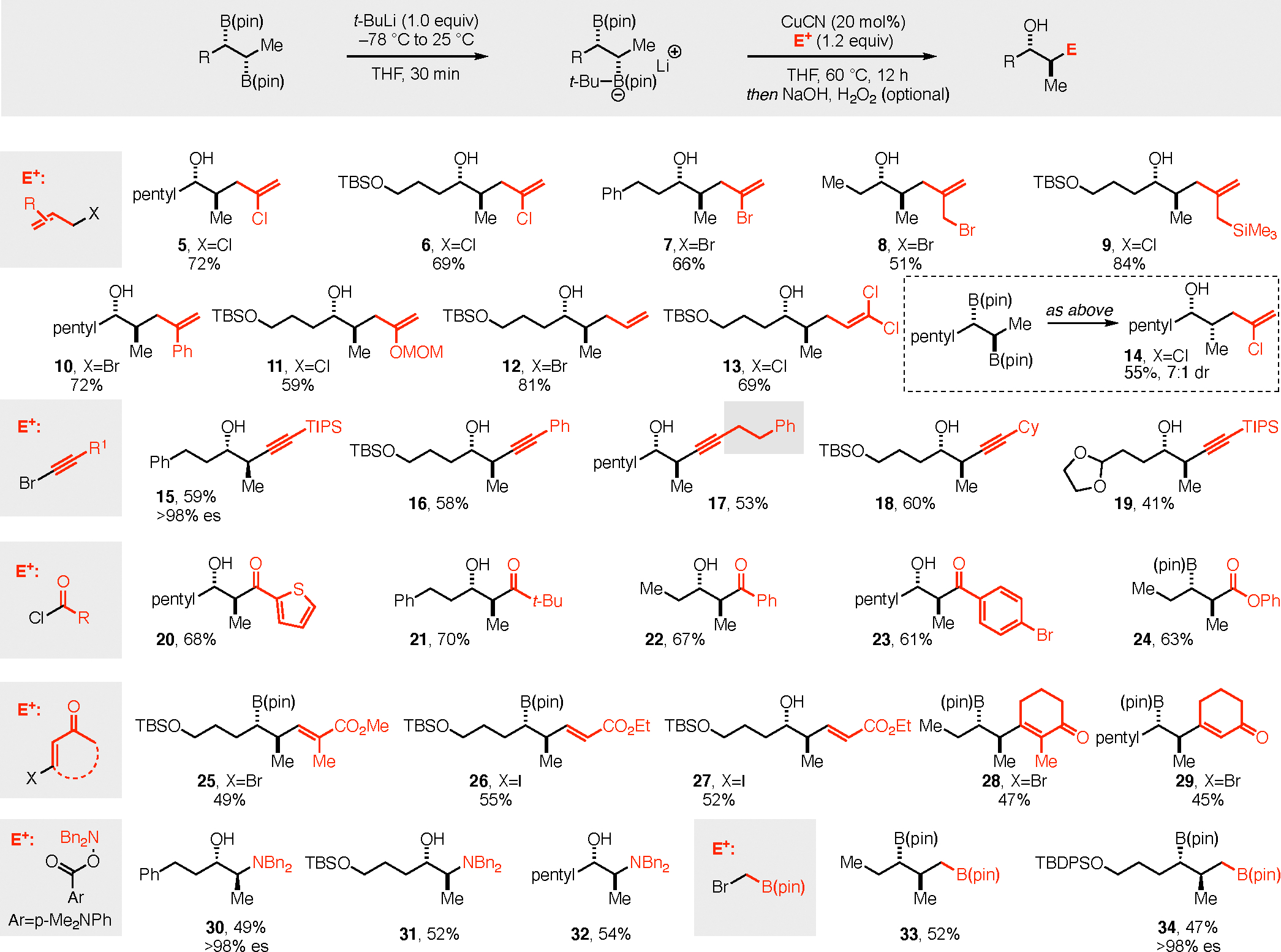
Substrate survey for the Cu-catalyzed site-selective coupling of vicinal bis(boronates). Abbreviations: Bn = benzyl; TIPS = triisopropylsilyl; MOM = methoxymethyl.
In addition to dichloropropene, other allylic electrophiles engaged in the reaction and delivered useful homoallylic alcohols bearing a range of functional groups (product 8–12). Other electrophile classes also participated in the reaction, with alkynyl bromides (15–19), acyl chlorides (20–24), β-halo enones and enoates (25–29), and amine electrophiles (30–32) proving competent in the process and providing a convenient route to an array of different difunctional materials. Lastly, site-selective activation followed by catalytic coupling to (halomethyl)boronic esters provides an effective synthesis of 1,3-bis(boronates)[ 22] (33, 34) by the selective single-carbon homologation of the less-hindered boronic ester.
Of note, several electrophiles in Figure 4 that undergo effective catalytic coupling with tert-butyllithium activated ate complexes are incompatible with previously described hydroxide or methoxide-promoted catalytic coupling[14e] of terminal 1,2-bis(boronates) (Figure 2c). For example, BrCH2B(pin), β-halo enones, acyl chlorides, and O-acylhydroxylamines either do not react with methoxide-activated vicinal bis(boronates), or are decomposed by the methoxide activator. Thus, use of tert–butyllithium as an activator provides a broader scope of couplings and it was of interest to examine this process with other bis(boronate) substrates. As depicted in Figure 5, when vicinal bis(boronates) derived from terminal alkenes were subjected to activation and copper-catalyzed coupling, discrimination between the primary and secondary boronate groups was efficient and good yields of reaction products 35–43 (Figure 5a) were obtained from acyl, enone, amine, and bromomethylboronate electrophiles. Diboration of 1,1-disubstituted alkenes followed by the coupling process delivered 1,3-bis(boronates) bearing a tertiary boronic esters (44, 45). That the Cu-catalyzed coupling with enoates is stereospecific with respect to the electrophile is demonstrated by coupling of a cis β-bromoenoate to give 46, a compound that was readily converted to massoia lactone (47). With the reaction of (halomethyl)boronates giving ready access 1,3-bis(boronates), these compounds were examined as substrates for subsequent activation/coupling. Whereas methoxide activation is ineffective for copper-catalyzed coupling of 1,3-bis(boronates)[14e], with t-BuLi activation, these substrates and were found to react with good efficiency and site selectivity (Figure 5b, 48–52). Encouraged by this observation, sequential homologations were examined and found to provide a route to 1,4 and 1,5 bis(boronates) (55, 56) from the 1,2-bis(boronate) precursor (53) (Figure 5c). Lastly, because directed diboration provides a simple mechanism to functionalize alkenes[23], we examined connecting this process to site selective couplings. As depicted in Figure 5d, substrate 57 was converted to diboron 58, which was an excellent substate for selective activation and coupling to give ketide derivative 59.
Figure 5.

t-Butyllithium activation and coupling with various bis(boronates). Abbreviations: TBDPS = tert-butyldiphenylsilyl.
With a sense for the efficacy of activation-based selective coupling reactions, we examined practical aspects of this process. In one experiment (Figure 6a, eq. 1), it was determined that tert-butyllithium could be replaced with non-pyrophoric lithium–biphenylide and adamantyl chloride as the activator for the process. This activation could be conducted at −78 °C by addition of pre-mixed lithium metal and 4,4’-di-tert-butylbiphenyl to a mixture of adamantyl chloride and substrate 60; warming to room temperature followed by coupling provided 61 as a single regioisomer. A second experiment (eq. 2) was conducted on preparative scale. In this case, Rh-catalyzed enantioselective diboration[ 24] converted 62 to 60 with 98:2 er. Subsequent activation and coupling delivered 63 in excellent yield and selectivity. Lastly, we considered that Pt-catalyzed diboration/activation/coupling could operate enantioselectively and provide a useful method to transform terminal alkenes into targets that were heretofore inaccessible by single-flask reaction. As depicted in Figure 6b, this sequence proved effective on >1 gram scale and delivered 1,2-aminoalcohol 64, boron-containing enoate 65, and 1,3-diboron 66.
Figure 6.

Practical aspects of cross-coupling with alkyllithium-activated boronate complexes. Abbreviations: DBB = 4,4’-di-tert-butyl-1,1’-biphenyl.
In summary, steric-based regioselective activation of bis(boronic esters), followed by Cu-catalyzed stereospecific cross-coupling can operate with a wide selection of electrophiles and provides simple routes to 1,2-funtional compounds. This method allows functionalization of each boronic ester regioselectively, independently, and enantiospecifically, thereby enabling enantioselective difunctionalization of unactivated alkenes.
Supplementary Material
Acknowledgements
This work was supported by the NIH (R35-GM1217140). We thank Hao Liang for DFT calculations that appear in the Supporting Information.
Footnotes
Supporting Information
The authors have cited additional references within the Supporting Information.[23–41]
References
- [1].Liu Z, Liu H, Zhang W, Marine Drugs 2020, 18, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].(a) Gunasekera SP, Gunasekera M, Longley RE, Schulte GK, J. Org. Chem. 1990, 55, 4912–4915. For review, see: [Google Scholar]; (b) Paterson I, Florence GJ, Eur. J. Org. Chem. 2003, 12, 2193–2208. [Google Scholar]
- [3].(a) McGuire JM, Bunch RL, Anderson RC, Boaz HE, Flynn EH, Powell HM, Smith JW, Antibiotics and Chemotherapy 1952, 2, 281–283. For review, see: [PubMed] [Google Scholar]; (b) Mulzer J, Angew. Chem. Int. Ed. Engl. 1991, 30, 1452–1454. [Google Scholar]
- [4]:(a) Turks M, Laclef S, Vogel P, in Stereoselective Synthesis of Drugs and Natural Products (Eds.: Andrushko V, Andrushko N), Wiley, 2013, pp. 1–48. [Google Scholar]; (b) Li J, Menche D, Synthesis 2009, 14, 2293–2315. [Google Scholar]
- [5]:(a) Giofrè S, Molteni L, Beccalli EM, Eur. J. Org. Chem. 2023, 26, e202200976. [Google Scholar]; (b) Bahamonde A, Trends in Chemistry 2021, 3, 863–876. [Google Scholar]
- [6].(a) Larsen CR, Erdogan G, Grotjahn DB, J. Am. Chem. Soc. 2014, 136, 1226–1229. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Qin C, Jia X, Leng X, Huang Z, Angew. Chem. Int. Ed. 2017, 56, 1614–1618. [DOI] [PubMed] [Google Scholar]; (c) Paulson ER, Moore CE, Rheingold AL, Pullman DP, Sindewald RW, Cooksy AL, Grotjahn DB, ACS Catal. 2019, 9, 7217–7231. [Google Scholar]
- [7].(a) Xu C, Shen X, Hoveyda AH, J. Am. Chem. Soc. 2017, 139, 10919–10928. [DOI] [PubMed] [Google Scholar]; (b) Ahmed TS, Grubbs RH, J. Am. Chem. Soc. 2017, 139, 1532–1537. [DOI] [PubMed] [Google Scholar]
- [8]:(a) Kolb HC, Van Nieuwenhze MS, Sharpless KB, Chem. Rev. 1994, 94, 2483–2547. [Google Scholar]; (b) Bataille CJR, Donohoe TJ, Chem. Soc. Rev. 2011, 40, 114–128. [DOI] [PubMed] [Google Scholar]; (c) Tao Z-L, Denmark SE, Synthesis 2021, 53, 3951–3962. [Google Scholar]
- [9].Cresswell AJ, Eey ST-C, Denmark SE, Angew. Chem. Int. Ed. 2015, 54, 15642–15682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tian B, Chen P, Leng X, Liu G, Nat. Catal. 2021, 4, 172–179. [Google Scholar]
- [11]:(a) Lebel H, Marcoux J-F, Molinaro C, Charette AB, Chem. Rev. 2003, 103, 977–1050. [DOI] [PubMed] [Google Scholar]; (b) Bartoli G, Bencivenni G, Dalpozzo R, Synthesis 2014, 46, 979–1029 [Google Scholar]; (c) Xia Q-H, Ge H-Q, Ye C-P, Liu Z-M, Su K-X, Chem. Rev. 2005, 105, 1603–1662. [DOI] [PubMed] [Google Scholar]; (d) Rose E, Andrioletti B, Zrig S, Quelquejeu-Ethève M, Chem. Soc. Rev. 2005, 34, 573. [DOI] [PubMed] [Google Scholar]; (e) Pellissier H, Adv. Synth. Catal. 2014, 356, 1899–1935. [Google Scholar]; (f) Zhu Y, Wang Q, Cornwall RG, Shi Y, Chem. Rev. 2014, 114, 8199–8256. [DOI] [PubMed] [Google Scholar]
- [12]:(a) Kang T, Apolinar O, Engle KM, Synthesis 2023, 56, 1–15. [Google Scholar]; (b) Dong Z, Song L, Chen L, ChemCatChem 2023, 15, e202300803. [Google Scholar]
- [13].For a recent overview of synthesis with organoboron compounds, see: Israel EM, Fyfe JWB, Watson AJB, In Comprehensive. Organometallic Chemistry IV: Volume 11 Elsevier, Inc. 2022, 305–334. [Google Scholar]
- [14].(a) Miller SP, Morgan JB, Nepveux J. P. Morken, Org. Lett. 2004, 6, 131–133. [DOI] [PubMed] [Google Scholar]; (b) Mlynarski SN, Schuster CH, Morken JP, Nature 2014, 505, 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Willems S, Toupalas G, Reisenbauer JC, Morandi B, Chem. Commun. 2021, 57, 3909–3912. [DOI] [PubMed] [Google Scholar]; (d) Lee Y, Jang H, Hoveyda AH, J. Am. Chem. Soc. 2009, 131, 18234–18235. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xu N, Kong Z, Wang JZ, Lovinger GJ, Morken JP, J. Am. Chem. Soc. 2022, 144, 17815–17823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].(a) Xu N, Liang H, Morken JP, J. Am. Chem. Soc. 2022, 144, 11546–11552. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang H, Morken JP, J. Am. Chem. Soc. 2023, 145, 20755–20760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]:(a) Fawcett A, Nitsch D, Ali M, Bateman JM, Myers EL, Aggarwal VK, Angew. Chem. Int. Ed. 2016, 55, 14663–14667. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kaiser D, Noble A, Fasano V, Aggarwal VK, J. Am. Chem. Soc. 2019, 141, 14104–14109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang H, Han W, Noble A, Aggarwal VK, Angew. Chem. Int. Ed. 2022, 61, e202207988. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang H, Wu J, Noble A, Aggarwal VK, Angew. Chem., Int. Ed. 2022, 61, e202202061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Aiken SG, Bateman JM, Liao H-H, Fawcett A, Bootwicha T, Vincetti P, Myers EL, Noble A, Aggarwal VK, Nat. Chem. 2023, 15, 248–256. [DOI] [PubMed] [Google Scholar]
- [17].For a review on site-selective reactions of vicinal bis(boronates), see: Viso A, de la Pradilla RF, Tortosa M ACS Catal. 2022, 12, 10603–10620. [Google Scholar]
- [18].For 13C NMR of “ate” complex formation: Liang H, Morken JP, J. Am. Chem. Soc. 2023, 145, 9976–9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen A, Qiao Y, Gao D-W Angew. Chem 2023, e202312605. [DOI] [PubMed] [Google Scholar]
- [20]:(a) Moriya K, Simon M, Mose R, Karaghiosoff K, Knochel P,Angew. Chem. Int. Ed. 2015, 54, 10963–10967. [DOI] [PubMed] [Google Scholar]; (b) Morozova V, Skotnitzki J, Moriya K, Karaghiosoff K, Knochel P, Angew. Chem. Int. Ed. 2018, 57, 5516–5519. For review, see: [DOI] [PubMed] [Google Scholar]; (c) Skotnitzki J, Kremsmair A, Knochel P, Synthesis 2020, 52, 189–196. [Google Scholar]
- [21].Tao Z, Robb KA, Panger JL, Denmark SE, J. Am. Chem. Soc. 2018, 140, 15621–15625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]:(a) You C, Studer A, Angew. Chem. Int. Ed. 2020, 59, 17245–17249. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blair DJ, Tanini D, Bateman JM, Scott HK, Myers EL, Aggarwal VK, Chem. Sci. 2017, 8, 2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li S, Hu C, Cui X, Zhang J, Liu LL, Wu L, Angew. Chem. Int. Ed. 2021, 60, 26238–26245. [DOI] [PubMed] [Google Scholar]
- [23].Blaisdell TP, Morken JP, J. Am. Chem. Soc. 2015, 137, 8712–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Morgan JB, Miller SP, Morken JP, J. Am. Chem. Soc. 2003, 125, 8702–8703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.