

Abstract
Purpose
6-O-(2-[18F]Fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN) is regarded as a non-selective opioid receptor radiotracer.
Procedure
Here, we report the first characterization of [18F]FE-DPN synthesized from the novel precursor, 6-O-(2-tosyloxyethoxy)-6-O-desmethyl-3-O-trityl-diprenorphine (TE-TDDPN), using a one-pot, two-step nucleophilic radiosynthesis to image opioid receptors in rats and mice using positron emission tomography.
Results
We also show that [18F]FE-DPN and [3H]DPN exhibit negligible brain uptake in mu opioid receptor (MOR) knockout mice.
Conclusions
Taken together with prior findings, our results suggest that [18F]FE-DPN and [3H]DPN preferentially bind to MOR in rodents in vivo.
Keywords: Diprenorphine, PET, Opioid, In vivo
Introduction
6-O-(2-[18F]Fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN) is used to measure opioid receptor density and endogenous opioid peptide release in humans and laboratory animals via positron emission tomography (PET) [1–3]. In vitro studies indicate that DPN binds to mu (MOR), kappa (KOR), and delta (DOR) opioid receptors with comparable affinity (KiMOR: ~ 0.07–0.2 nM; KiKOR: ~ 0.02–0.2 nM; KiDOR: ~ 0.18–0.25 nM) [4–7]. FE-DPN also binds to these receptors with comparable affinity in vitro for MOR and KOR (KiMOR: 0.24 nM; KiKOR: 0.2 nM) and lower affinity for DOR (KiDOR: 8 nM) [8]. Based on these observations, DPN and FE-DPN are widely regarded as non-selective opioid receptor ligands. However, the extent that these in vitro measures generalize to in vivo opioid receptor binding is unclear as discrepancies between in vitro and in vivo selectivity have been previously reported for opioid and other receptors [9, 10]. In particular, [3H]DPN has been previously reported to be selective for MOR in vivo in rats [11, 12] suggesting that [18F]FE-DPN may also show a similar selectivity profile. While [18F]FE-DPN was first synthesized decades ago, a new precursor was recently developed [13] which allows for simplified radiosynthesis. Given that no MOR-selective 18F-labeled PET radiotracer has been previously developed, our goals here were to confirm the utility of [18F]FE-DPN synthesized from this novel precursor to image opioid receptors in rats and mice, and to examine [18F]FE-DPN and [3H]DPN in vivo uptake and selectivity using wild-type and MOR knockout (KO) mice.
Materials and Methods
Experimental Subjects
We used Sprague–Dawley rats (males and females, 252 ± 52 g; strain #400, Charles River, 7–9 weeks) and wild-type (WT) (C57BL/6 J) or Oprm1 KO mice (males and female, 25 ± 4 g on C57BL/6 background; strain #007,559, Jackson, 8 weeks). Rats were maintained under a reverse 12 h light/dark cycle with ad libitum access to food and water and housed two per cage before surgery and individually after surgery. Mice were maintained under a normal 12 h light/dark cycle with ad libitum food and water. All experiments were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by NIDA’s Animal Care and Use Committee.
[18F]fluoride Production
Oxygen-18 enriched water (98%, Huayi Isotopes, Jiangsu, China, approximately 2 ml) was loaded into a niobium body, high-yield [18F]fluoride target of a General Electric Medical Systems (GEMS, Uppsala, Sweden) PETtrace cyclotron. The target was irradiated with a proton beam of 60 μA for 25 min to produce an average of 51 GBq (n = 6) of aqueous [18F]fluoride ion.
Radiochemicals
[3H]DPN (1169.2 GBq/mmol) was obtained from NIDA Drug Supply Program. [18F]FE-DPN radiosynthesis was performed using an in-house custom-made radiofluorination module (RFM) and LabView software. The cyclotron-produced [18F]fluoride ion was collected in a 5 ml V-vial (Wheaton) inside the hot cell, and assayed in a dose calibrator to obtain the initial starting radioactivity. The [18F]fluoride ion was then remotely transferred to the RFM where it was trapped on a Chromafix 30-PS-HCO3 solid-phase extraction (SPE) cartridge (ABX GmbH, Radeberg, Germany) earlier preconditioned using 1 ml of high-purity water (Honeywell). The [18O]Water was collected for recycling. The resin cartridge was eluted using 210 μl of a 1:1 acetonitrile/water mixture containing 13.9 μmol of potassium bicarbonate and 14.8 μmol of Kryptofix K222 into an empty 5 ml V-vial. The resin cartridge was then rinsed with 250 μl of acetonitrile into the same V-vial and the [18F]fluoride was dried at 110 °C with nitrogen flow (325 ml/min) for 150 s in a standard thermal heating block. Then, two separate additions of 250-μl acetonitrile were added with 150 s and 180 s of drying, respectively. The vial was cooled to 60 °C using compressed air. Precursor (TE-TDDPN, ABX GmbH, Germany) (1.75 mg in 0.5 ml acetonitrile) was added to the dried [18F]fluoride. The solution was heated at 100 °C for 12 min before being cooled to 60 °C; then, 0.25 ml of 1 M HCl was added and the solution heated to 80 °C for 12 min. Following the HCl deprotection, the reaction solution was cooled to 60 °C before being neutralized with 0.25 ml of 1 M NaOH, then buffered with 3 ml of 10 mM (+)-sodium L-ascorbate. The reaction solution was injected onto a Gemini 10 × 150 mm 10 μM semi-prep HPLC column (Phenomenex, Torrance CA) and eluted with 45/55 acetonitrile/10 mM (+)-sodium L-ascorbate at 10 ml/min. The [18F]FE-DPN fraction (RT ~ 16.5 min) was collected in 50 ml of 10 mM (+)-sodium L-ascorbate. The diluted product fraction was loaded onto a tC18 Sep Pak Plus short (Waters, Milford, MA). The tC18 Sep Pak Plus short was washed with 10 ml of 10 mM (+)-sodium L-ascorbate then manually eluted with 1 ml of ethanol (ABS), collected into 3 fractions (0.2 ml, 0.6 ml, 0.2 ml respectively). The majority of the [18F]FE-DPN was contained in the 0.6 ml fraction. Radiochemical purity and specific activity were determined by analytical HPLC using a Gemini 4.6 × 150 mm analytical HPLC column (Phenomenex, Torrance CA) eluted with 48/52 acetonitrile/triethylamine buffer pH 7.2 at 2 ml/min with UV set to 240 nm. An analytical sample of [18F]FE-DPN was injected onto the HPLC and the UV peak area was compared to an authentic FE-DPN standard calibration curve (seven concentration levels, linear r2 = 0.99979) to determine the specific activity.
Small Animal PET
Rats (n = 6; 252 ± 52 g body weight) were implanted with jugular vein catheters as previously described [14]. On the day of the scan, rats were injected subcutaneously (SC) with either saline (n = 3, 1 male and 2 female) or naltrexone (n = 3, 2 male and 1 female) (10 mg/kg) (Sigma-Aldrich (St. Louis, MO)). Fifteen minutes later, rats were anesthetized using isoflurane (5% induction, 2% maintenance), placed in a prone position on the bed of a nanoScan® PET/CT scanner (Mediso, Hungary), injected intravenously (IV) with 15.7 ± 1.1 MBq of [18F] FE-DPN (~ 0.2 ml) and scanned for 90 min, followed by a 2 min CT scan. WT (n = 4, 2 male and 2 female) and MOR KO (n = 4, 2 male and 2 female) mice (~ 30 g body weight) were injected SC with 7.8 ± 0.6 MBq [18F]FE-DPN (~ 0.2 ml). Sixty minutes after radiotracer injection, mice were anesthetized using isoflurane (5% induction, 2% maintenance) and placed in a prone position on the bed of a nanoScan® PET/CT scanner and scanned for 20 min followed by a 2-min CT scan. PET data were reconstructed and corrected for dead time and radioactive decay. PET image assessments were performed using PMOD (PMOD Technologies, Zurich Switzerland). Dynamic PET images were coregistered to MRI templates and time-activity curves were generated using PMOD’s built-in volume of interest (VOI) atlases. Standardized uptake value (SUV) was calculated using SUV = C/(dose/BW) where C = radiotracer tissue concentration (kBq/cc), dose =injected dose (MBq), and BW (kg) = subject body weight. BPND parametric maps were generated by pixel-based kinetic modeling using a multilinear reference tissue model [15] with the cerebellum as a reference region and the start time (t*) set to 5 min.
In Vivo [3H]DPN Uptake
WT (n = 6) and MOR KO (n = 6) mice were injected SC with [3H]DPN (37 kBq/g) with (n = 3 male and 3 female) or without (n = 3 male and 3 female) pretreatment with naltrexone (10 mg/kg, SC, 10 min before [3H]DPN), euthanized at 60 min, and brain and blood were collected. Brains were sectioned (20 μm) on a cryostat (Leica Biosystems), mounted onto glass microscope slides, and air-dried overnight at RT. Slides were placed into a Hypercassette™ covered by a BAS-TR2025 phosphor screen (FujiFilm; GE Healthcare) and exposed to a phosphor screen for 12 days. Slides were imaged using a phosphorimager (Typhoon FLA 7000; GE Healthcare). The digitized images were calibrated using 14C standard slides (ARC, Inc) and analyzed using Multigauge software (GE Healthcare).
Statistics
For [18F]FE-DPN experiments, we used two-way ANOVAs with VOI and treatment or genotype as factors. For [3H]DPN experiments, we used two-way ANOVAs with genotype and treatment as factors. Holm-Sidak post hoc tests were used for pairwise comparisons. All statistical tests were evaluated at P≤0.05.
Results
Validation of a Novel [18F]FE-DPN Radiosynthesis for Imaging Opioid Receptors Using PET
We used a novel precursor, TE-TDDPN [13], to perform a one-pot, two-step nucleophilic radiosynthesis of [18F]FE-DPN (Fig. 1). The average radiochemical yield was 10.0% ± 1.2% (non-decay corrected), producing 5.14 ± 0.87 GBq of final product starting from 51 GBq of fluoride in the 85-min synthesis. The average radiochemical purity was 98.0% with a specific activity of 2027 GBq/ μM ± 1075 GBq/μM. Stability analysis at 270 min after end-of-synthesis showed the radiochemical purity remained unchanged. This is the first time [18F]FE-DPN has been synthesized using this route and the resulting radiotracer has not yet been previously used for in vivo studies. Therefore, we first tested its ability to label opioid receptors using PET. Rats were implanted with jugular vein catheters as previously described [14]. On the day of the scan, rats were injected with either saline or naltrexone (10 mg/kg, SC). Fifteen minutes later, rats were injected IV with 15.7 ± 1.1 MBq of [18F]FE-DPN (~ 0.2 ml, 0.36 ± 0.05 nmol/kg) and scanned for 90 min. [18F]FE-DPN kinetics reached a steady state at ~ 30 min after injection (Fig. 2A–B). [18F]FE-DPN accumulated in opioid receptor-rich regions [16], such as the thalamus, periaqueductal gray, hypothalamus, striatum, superior colliculus, and brainstem, and had negligible accumulation in the cerebellum (Fig. 2C–D). Naltrexone fully blocked [18F]FE-DPN binding indicating that this new precursor and simplified radiosynthesis lead to a good quality radiotracer for in vivo opioid receptor imaging using PET (Fig. 2C–E).
Fig. 1.
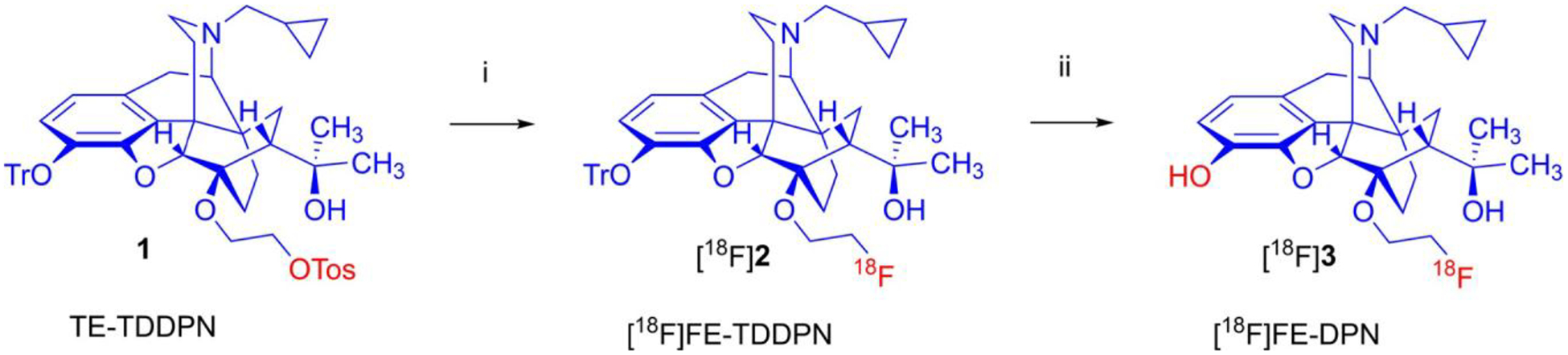
One-pot, two-step nucleophilic radiosynthesis of 6-O-(2-[18F]fluoroethyl-6-O-desmethyl-diprenorphine ([18F]FE-DPN). Reagents and conditions: (i): [K+K222]18F−, CH3CN, 100 °C, 12 min; (ii): 1 M HCl, 80 °C, 12 min.
Fig. 2.
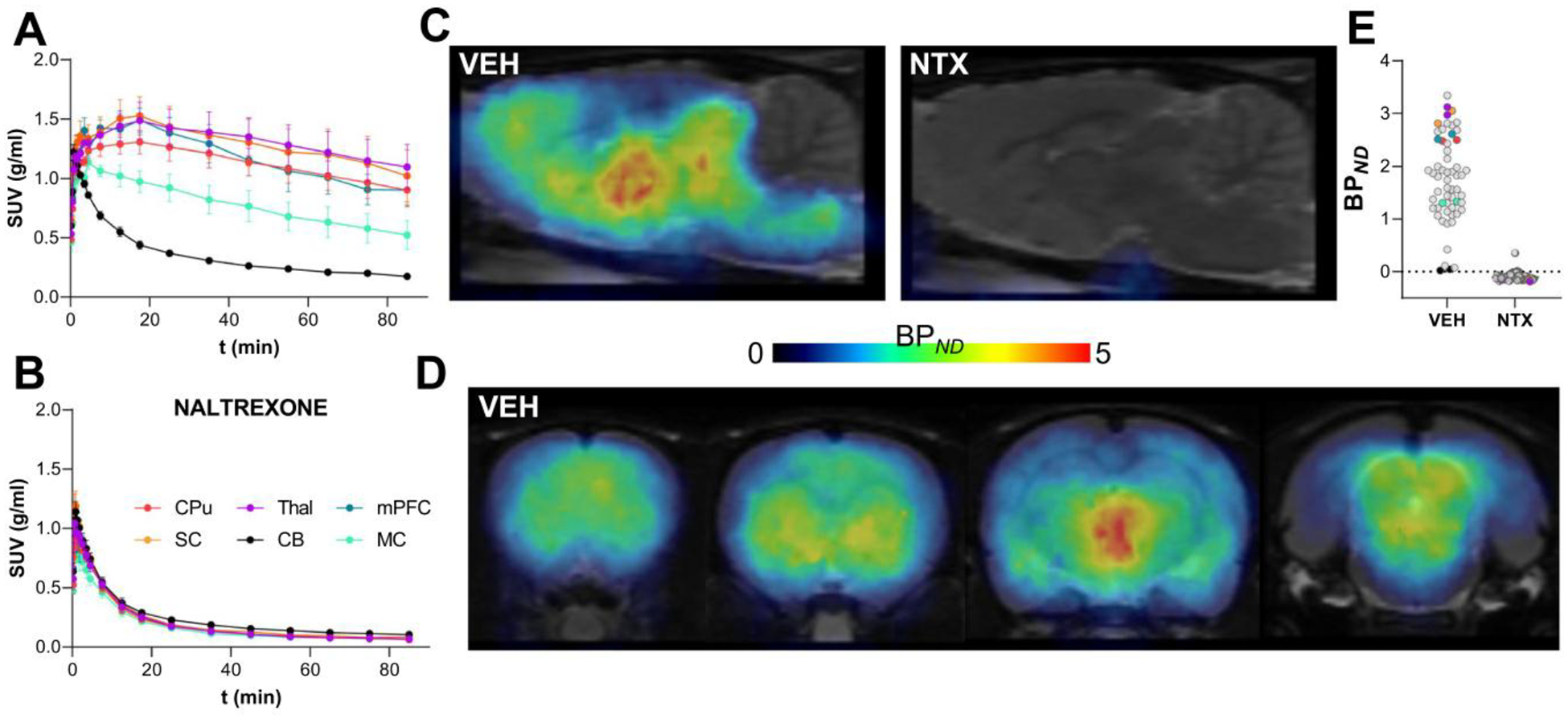
Validation of a novel [18F]FE-DPN radiosynthesis for imaging opioid receptors using PET. [18F]FE-DPN reached a steady state at around 50 min after injection in saline-treated (vehicle, VEH) rats (A), but was blocked in NTX pretreated rats (B). PET image of [18F] FE-DPN expression in saline pretreated rats shows expected high levels of binding in areas rich in opioid receptors as viewed in sagittal (C) and coronal slices (D); whereas NTX pretreated rats show no binding. NTX pretreatment fully blocked [18F]FE-DPN binding in all brain regions (E). [18F]FE-DPN = 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine; CPu =caudate putamen; CB =cerebellum; mPFC =medial prefrontal cortex; MC =motor cortex; NTX =naltrexone; PET =positron emission tomography; SC =superior colliculus; Thal =thalamus.
[18F]FE-DPN Preferentially Labels MORs In Vivo
To test whether [18F]FE-DPN preferentially labels MOR in vivo, we utilized mice devoid of MOR, but which have intact KOR and DOR levels [16]. WT and MOR KO mice were injected SC with 7.8 ± 0.6 MBq [18F]FE-DPN (~ 0.2 ml, 2.7 ± 0.6 nmol/kg). At 60 min after radiotracer injection, mice were anesthetized and scanned for 20 min. WT mice showed high [18F]FE-DPN accumulation in the brain (Fig. 3A). In contrast, MOR KO mice showed no discernible [18F]FE-DPN brain accumulation (Fig. 3B) with [18F]FE-DPN brain uptake comparable to the background (Fig. 3C).
Fig. 3.
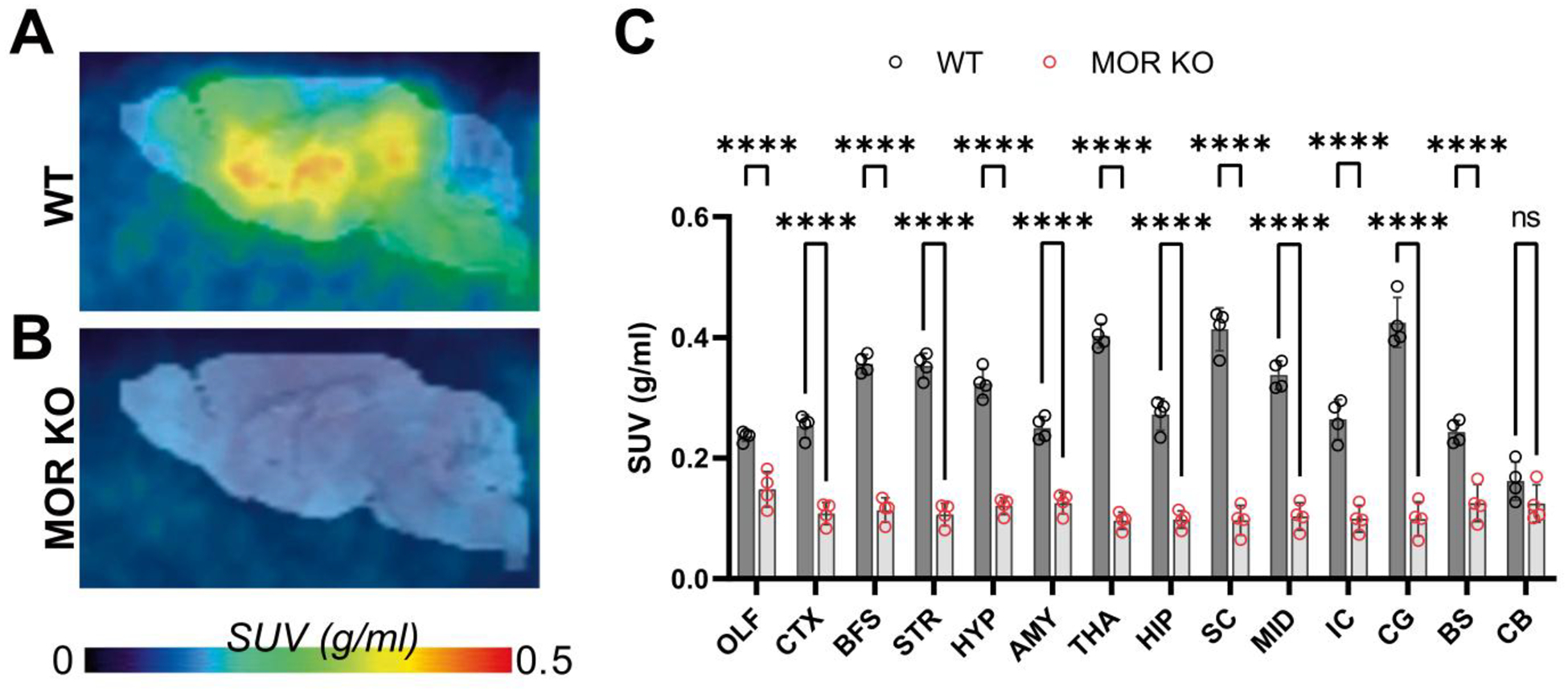
[18F]FE-DPN preferentially labels MORs in vivo. PET images displaying discrepancy of [18F]FE-DPN brain uptake between WT mice (A) and MOR KO mice (B). Comparison between [18F]FE-DPN signal in the brain of WT and MOR KO mice at regions throughout the brain (C). [18F]FE-DPN = 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine; AMY =amygdala; BFS =basal forebrain; BS =brain stem; CTX =cortex; CG =central gray; CB =cerebellum; HIP = hippocampus; HYP =hypothalamus; IC =inferior colliculus; MID =midbrain; MOR KO =μ opioid receptor knock out; OLF =olfactory bulb; PET =positron emission tomography; STR =striatum; SC =superior colliculus; THA =thalamus; WT =wild type.
[3H]DPN Preferentially Labels MORs In Vivo
To examine whether [3H]DPN shows the same in vivo properties as [18F]FE-DPN, we injected WT and MOR KO mice SC with [3H]DPN and euthanized them 60 min later. Additional WT and MOR KO mice were pretreated with naltrexone (10 mg/kg, SC) 10 min prior to [3H]DPN injection. As expected, [3H]DPN showed a distribution consistent with MOR expression in WT mice [16]; moreover, there was negligible [3H]DPN signal in the brains of MOR KO mice (Fig. 4A). In all brain regions, there was a significant genotype × treatment interaction (caudate putamen: F(1,8) = 19.08; P = 0.002, insula: F(1,8) = 9.95; P = 0.013, nucleus accumbens: F(1,8) = 20.46; P = 0.001, thalamus: F(1,8) = 22.38; P = 0.001). Multiple comparisons showed that pretreatment with naltrexone nearly abolished the signal in WT mice as non-MOR signal across these brain regions was approximately 4.4 ± 0.2% of the total (caudate putamen: P < 0.001; insula: P = 0.001; nucleus accumbens: P < 0.001; thalamus: P < 0.001). MOR KO mice had significantly lower [3H]DPN binding (~ 12.7 ± 3.8% of WT signal across brain regions) compared to WT mice (caudate putamen: P < 0.001; insula: P = 0.006; nucleus accumbens: P < 0.001; thalamus: P < 0.001). Naltrexone pretreatment did not significantly reduce the small amount of binding in the MOR KO mice in any region, suggesting negligible KOR or DOR binding (Fig. 4B).
Fig. 4.

[3H]DPN preferentially labels MORs in vivo. Representative brain slices of [3H]DPN ex vivo occupancy in WT and MOR KO mice (A). WT mice not pretreated with naltrexone showed significantly higher binding than MOR KO mice or WT mice pretreated with naltrexone in all brain regions (B). [3H]DPN =[3H]diprenorphine; CPu =caudate putamen; NAc =nucleus accumbens; MOR KO =μ opioid receptor knock out; NTX =naltrexone; WT =wild type.
Discussion
Here, we show that the TE-TDDPN precursor can be used to perform a one-pot, two-step nucleophilic radiosynthesis of [18F]FE-DPN with high yield, purity, and specific activity, leading to successful labeling of opioid receptors in rodent [18F]FE-DPN is regarded as a non-selective opioid radiotracer with similar in vitro potency at MOR, KOR, and DOR [7, 17]. Here, we used mice that lack MORs, but that have intact KOR and DOR [16], to demonstrate that in vivo administration of [3H]DPN leads to preferential MOR binding, with minimal binding to other opioid receptors (non-MOR signal was less than ~ 7% of the total). The MOR KO mice we used have unchanged levels of KOR and DOR, with DOR or KOR density (i.e., Bmax) equivalent to or higher than MOR in several brain regions (e.g., striatum/insula [16]), indicating that the lack of [18F]FE-DPN or [3H]DPN binding in MOR KO mice is mainly due to a lack of MOR. Although our mouse PET data were characterized by a relatively low SUV, likely due to the SC injection route, these findings are consistent with both the preferential [3H]DPN MOR labeling in vivo and results from prior studies in rats which showed that in vivo [3H]DPN administration leads to a radioligand brain distribution profile consistent with MOR but not KOR or DOR expression [11, 12].
Several reasons may explain the discrepancy observed between in vitro and in vivo binding of DPN and FE-DPN. These may include in vivo engagement of MOR-DOR heteromers, metal ion or nucleotide modulation of affinities, or differential diffusion barriers between opioid receptors [11]. Additionally, in vitro binding is performed using equilibrium binding conditions, whereas in vivo binding does not reach equilibrium. It may be that the residency time of each radioligand at each of the opioid receptors differs, which may become apparent under non-equilibrium conditions, thus contributing to the differences between in vivo and in vitro results.
Conclusions
Our results suggest that [18F]FE-DPN and [3H]DPN preferentially bind to MOR over other opioid receptors in vivo and may have important implications for prior PET studies in rodents that used [18F]FE-DPN and [11C]DPN and attributed radiotracer accumulation in the brain as reflecting non-selective binding to all three opioid receptors.
Funding
This work was supported by the NIDA Intramural Research Program (ZIA000069, MM) and by Grants RYC-2019-027371-I (JB) funded by MCIN/AEI/1013039/501100011033 and by “ESF Investing in your future,” Plan Nacional Sobre Drogas 2021I070 (JB) funded by the Spanish Ministerio de Sanidad, and (EB024495, MGP) funded by NIBIB.
Conflict of Interest
MM has received research funding from AstraZeneca, Redpin Therapeutics, and Attune Neurosciences. CAZ is listed as a coinventor on a patent for the use of ketamine in major depression and suicidal ideation. CAZ is listed as a coinventor on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydro and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain. CAZ is listed as coinventor on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post-traumatic stress disorders. CAZ has assigned his patent rights to the US government but will share a percentage of any royalties that may be received by the government. All other authors declare no conflicts of interest.
References
- 1.Wester HJ, Willoch F, Tolle TR et al. (2000) 6-O-(2-[18F] fluoroethyl)-6-O-desmethyldiprenorphine ([18F]DPN): synthesis, biologic evaluation, and comparison with [11C]DPN in humans. J Nucl Med 41:1279–1286 [PubMed] [Google Scholar]
- 2.Mueller C, Klega A, Buchholz HG et al. (2010) Basal opioid receptor binding is associated with differences in sensory perception in healthy human subjects: a [18F]diprenorphine PET study. Neuroimage 49:731–737 [DOI] [PubMed] [Google Scholar]
- 3.Thompson SJ, Pitcher MH, Stone LS et al. (2018) Chronic neuropathic pain reduces opioid receptor availability with associated anhedonia in rat. Pain 159:1856–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raynor K, Kong H, Chen Y et al. (1994) Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol 45:330–334 [PubMed] [Google Scholar]
- 5.Chang KJ, Hazum E, Cuatrecasas P (1980) Possible role of distinct morphine and enkephalin receptors in mediating actins of benzomorphan drugs (putative kappa and sigma agonists). Proc Natl Acad Sci U S A 77:4469–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang KJ, Hazum E, Cuatrecasas P (1981) Novel opiate binding sites selective for benzomorphan drugs. Proc Natl Acad Sci U S A 78:4141–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henriksen G, Willoch F, Talbot PS, Wester HJ (2006) Recent development and potential use of mu- and kappa-opioid receptor ligands in positron emission tomography studies. Drug Develop Res 67:890–904 [Google Scholar]
- 8.Schoultz BW, Hjornevik T, Reed BJ et al. (2014) Synthesis and evaluation of three structurally related (1)(8)F-labeled orvinols of different intrinsic activities: 6-O-[(1)(8)F]fluoro-ethyl-diprenorphine ([(1)(8)F]FDPN), 6-O-[(1)(8)F]fluoro-ethyl-buprenorphine ([(1)(8)F]FBPN), and 6-O-[(1)(8)F] fluoroethyl-phenethyl-orvinol ([(1)(8)F]FPEO). J Med Chem 57:5464–5469 [DOI] [PubMed] [Google Scholar]
- 9.Frost JJ, Dannals RF, Duelfer T et al. (1984) In vivo studies of opiate receptors. Ann Neurol 15(Suppl):S85–92 [DOI] [PubMed] [Google Scholar]
- 10.Zeeberg BR (1999) Pharmacokinetic computer simulations of the relationship between in vivo and in vitro neuroreceptor subtype selectivity of radioligands. Nucl Med Biol 26:803–809 [DOI] [PubMed] [Google Scholar]
- 11.Frost JJ, Smith AC, Wagner HN Jr (1986) 3H-diprenorphine is selective for mu opiate receptors in vivo. Life Sci 38:1597–1606 [DOI] [PubMed] [Google Scholar]
- 12.Atweh SF, Kuhar MJ (1977) Autoradiographic localization of opiate receptors in rat brain III. The telencephalon Brain Res 134:393–405 [DOI] [PubMed] [Google Scholar]
- 13.Marton J, Cumming P, Bauer B, Henriksen G (2021) A new precursor for the radiosynthesis of 6-O-(2-[(18) F]fluoroethyl)-6-O-desmethyl-diprenorphine ([F-18]FE-DPN) by nucleophilic radiofluorination. Lett Org Chem 18:344–352 [Google Scholar]
- 14.Bonaventura J, Lam S, Carlton M et al. (2021) Pharmacological and behavioral divergence of ketamine enantiomers: implications for abuse liability. Mol Psychiatry 26:6704–6722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichise M, Ballinger JR, Golan H et al. (1996) Noninvasive quantification of dopamine D2 receptors with iodine-123-IBF SPECT. J Nucl Med 37:513–520 [PubMed] [Google Scholar]
- 16.Kitchen I, Slowe SJ, Matthes HW, Kieffer B (1997) Quantitative autoradiographic mapping of mu-, delta- and kappa-opioid receptors in knockout mice lacking the mu-opioid receptor gene. Brain Res 778:73–88 [DOI] [PubMed] [Google Scholar]
- 17.Cumming P, Marton J, Lilius TO, Olberg DE, Rominger A (2019) A survey of molecular imaging of opioid receptors. Molecules 24:4190. [DOI] [PMC free article] [PubMed] [Google Scholar]