

Abstract
Most physiological agonists increase cytosolic free [Ca2+]c (cytosolic free Ca2+ concentration) to regulate a variety of cellular processes. How different stimuli evoke distinct spatiotemporal Ca2+ responses remains unclear, and the presence of separate intracellular Ca2+ stores might be of great functional relevance. Ca2+ accumulation into intracellular compartments mainly depends on the activity of Ca2+- and H+-ATPases. Platelets present two separate Ca2+ stores differentiated by the distinct sensitivity to thapsigargin and TBHQ [2,5-di-(t-butyl)-1,4-hydroquinone]. Although one store has long been identified as the dense tubular system, the nature of the TBHQ-sensitive store remains uncertain. Treatment of platelets with GPN (glycylphenylalanine-2-naphthylamide) impaired Ca2+ release by TBHQ and reduced that evoked by thrombin. In contrast, GPN did not modify Ca2+ mobilization stimulated by ADP or AVP ([arginine]vasopressin). Treatment with nigericin, a proton carrier, and bafilomycin A1, an inhibitor of the vacuolar H+-ATPase, to dissipate the proton gradient into acidic organelles induces a transient increase in [Ca2+]c that was abolished by previous treatment with the SERCA (sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase) 3 inhibitor TBHQ. Depleted acidic stores after nigericin or bafilomycin A1 were refilled by SERCA 3. Thrombin, but not ADP or AVP, reduces the rise in [Ca2+]c evoked by nigericin and bafilomycin A1. Our results indicate that the TBHQ-sensitive store in human platelets is an acidic organelle whose Ca2+ accumulation is regulated by both Ca2+- and vacuolar H+-ATPases.
Keywords: acidic organelles, bafilomycin, calcium stores, nigericin, platelets, thrombin
Abbreviations: AVP, [arginine]vasopressin; [Ca2+]c, cytosolic free calcium concentration; ER, endoplasmic reticulum; GPN, glycylphenylalanine 2-naphthylamide; HBS, Hepes-buffered saline; PMCA, plasma-membrane Ca2+ ATPase; SERCA 3, sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase; TBHQ, 2,5-di-(t-butyl)-1,4-hydroquinone; TG, thapsigargin
INTRODUCTION
Increases in [Ca2+]c (cytosolic free Ca2+ concentration) is one of the most widely occuring transduction mechanisms in response to stimuli regulating a large number of cellular processes, including cell growth, muscle contraction, secretion and platelet aggregation [1–4]. Ca2+-mobilizing cellular agonists increase [Ca2+]c by two mechanisms: the release of Ca2+ from intracellular stores and the entry of extracellular Ca2+ through plasma-membrane channels. The ER (endoplasmic reticulum) is the most investigated organelle and probably represents the major Ca2+ store in most cell types. The ER expresses two different types of Ca2+-release channels, namely Ins(1,4,5)P3 and ryanodine receptors [5–7]. In addition, several other cellular organelles also store Ca2+ and act as physiological agonist-releasable Ca2+ compartments. A number of studies have provided evidence of the importance of mitochondria in cellular Ca2+ homoeostasis [8–11]. In addition, a role for the nuclear envelope, the Golgi apparatus, secretory granules and lysosomes has received support [12–15].
In this context, previous studies have reported the presence of a Ca2+ pool in lysosome-related (acidic) organelles in different cell types, MDCK (Madin–Darby canine kidney) cells [16], pancreatic acinar and β-cells [17], and pulmonary arterial smooth-muscle cells [15]. The acidic Ca2+ store has been presented as the point of origin of NAADP (nicotinic acid–adenine dinucleotide phosphate)-mediated Ca2+ signalling [15,17]. It has been demonstrated that Ca2+ uptake into acidic organelles is driven by proton gradients maintained by vacuolar proton pumps (H+-ATPase) [18,19].
Of all the mammalian cell models used for the study of intracellular Ca2+ homoeostasis, human platelets are one of the most extensively investigated. In these cells, two different isoforms of SERCA (sarcoplasmic/endoplasmic-reticulum Ca2+-ATPase), with molecular masses of 100 and 97 kDa, have been shown to be distributed separately in two distinct Ca2+ stores [20–22]. The dense tubular system, the equivalent to the ER in other mammalian cells, has been presented as the main intracellular Ca2+ store in platelets [23]; however, the nature of the second Ca2+ pool is still uncertain. The 100 kDa isoform has been identified as SERCA 2b, and is inhibited by low concentrations of TG (thapsigargin) [21]. In contrast, the 97 kDa isoform, identified as SERCA 3, is inhibited only by high concentrations of TG [24,25]. These SERCA isoforms can also be differentiated by the distinct sensitivity to TBHQ [2,5-di-(t-butyl)-1,4-hydroquinone], a specific inhibitor of certain SERCA isoforms, so that SERCA 3, but not SERCA 2b, is sensitive to TBHQ [26]. In the present study, we investigated the nature of the Ca2+ compartments openable by physiological agonists, such as thrombin, ADP and AVP ([arginine]vasopressin), in human platelets; paying particular attention to the existence of acidic organelles in these cells.
MATERIAL AND METHODS
Materials
fura 2/AM (fura 2 acetoxymethyl ester), Lysosensor Green DND-189 and calcein were from Molecular Probes (Leiden, The Netherlands). Apyrase (grade V), aspirin, thrombin, ADP, AVP, BSA, ionomycin, bafilomycin A1 (an inhibitor of the vacuolar H+-ATPase), nigercin (a proton carrier), GPN (glycylphenylalanine 2-naphthylamide) and TG were from Sigma (Madrid, Spain). TBHQ was from Alexis (Nottingham, U.K.). All other reagents were of analytical grade.
Platelet preparation
fura 2-loaded human platelets were prepared as described previously [27], and approved by Local Ethical Committees. Briefly, blood was obtained with informed consent from healthy volunteers and mixed with one-sixth volume of acid/citrate dextrose anti-coagulant containing (in mM): 85 sodium citrate, 78 citric acid and 111 D-glucose. Platelet-rich plasma was then prepared by centrifugation for 5 min at 700 g, and aspirin (100 μM) and apyrase (40 μg/ml) added. Platelet-rich plasma was incubated at 37 °C with 2 μM fura-2/AM for 45 min. Cells were then collected by centrifugation at 350 g for 20 min and resuspended in HBS (Hepes-buffered saline) containing (in mM): 145 NaCl, 10 Hepes, 10 D-glucose, 5 KCl, 1 MgSO4, pH 7.45, and supplemented with 0.1% (w/v) BSA and 40 μg/ml apyrase.
Removal of nigericin and bafilomycin A1 to allow refilling of the acidic Ca2+ stores was performed as described previously [27]. Briefly, fura 2-loaded platelets were incubated in a Ca2+-free medium with 10 μM nigericin or 1 μM bafilomycin A1 in the absence or presence of 20 μM TBHQ for 5 min at 37 °C. Cells were then washed with HBS to remove nigericin or bafilomycin A1, and resuspended in nominally Ca2+-free HBS, with or without TBHQ as described above, to be treated with nigericin or bafilomycin A1 again.
Cell viability
Calcein and Trypan Blue were used to assess cell viability. For calcein loading, resting cells, or cells treated with inhibitors for the time indicated, were incubated for 30 min with 5 μM calcein/AM (calcein acetoyxymethyl ester) at 37 °C, centrifuged and the pellet resuspended in fresh HBS. Fluorescence was recorded in 2 ml aliquots using a Shimadzu (Kyoto, Japan) spectrophotometer. Samples were excited at 494 nm and the resulting fluorescence was measured at 535 nm. The calcein fluorescence remaining in cells after treatment with inhibitors used was the same as in controls, suggesting that, under our conditions, there was no cellular damage. The results obtained with calcein were confirmed using the Trypan Blue exclusion technique; 95% of cells were viable after treatment with the inhibitors, similar to results observed in our resting platelet suspensions.
Measurement of [Ca2+]c
Fluorescence was recorded from 2 ml aliquots of magnetically stirred cell suspensions (108 cells/ml) at 37 °C using a fluorescence spectrophotometer (Varian Ltd, Madrid, Spain) with excitation wavelengths of 340 nm and 380 nm, and emission at 505 nm. Changes in [Ca2+]c were monitored using the fura 2 340 nm/380 nm fluorescence ratio and calibrated by the method of Grynkiewicz et al. [28]. TBHQ, thrombin, ADP or AVP-induced Ca2+ release was estimated using the initial peak [Ca2+]c elevation above basal after agonist stimulation. Nigericin- and bafilomycin A1-induced Ca2+ release was estimated using the initial peak [Ca2+]c elevation above basal, after their addition, and by the integral of the rise in [Ca2+]c for 1.5 min, after their addition, taking a sample every second and expressed as nM/s [29].
To compare the rate of decay of [Ca2+]c with basal values, after treatment of platelets with thrombin, ADP or AVP in the absence or presence of GPN, we used the constant of the exponential decay as described previously [30]. Traces were fitted to the equation:
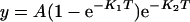 |
where K1 and K2 are the constants of the exponential increase and decay respectively, T is time and A is the span.
Staining of acidic organelles in platelets
Platelet-rich plasma was incubated at 37 °C with 100 nM Lysosensor Green DND-189 for 1 h. Cells were then collected by centrifugation at 350 g for 20 min and resuspended in HBS. Aliquots of Lysosensor Green-loaded cells were placed on to a coverslip attached to the bottom of a perfusion chamber on the stage of a confocal microscope (Nikon Eclipse TE300) using a ×60 oil-immersion objective. Cells were studied with a confocal laser-scanning system (MRC 1024, Bio-Rad) using a 488 nm laser line as excitation source and a 515 nm long-pass filter to collect the emitted fluorescence. Lysosensor Green fluorescence was quantified using a fluorescence spectrophotometer with an excitation wavelength of 445 nm and an emission wavelength at 505 nm.
Statistical analysis
Analysis of statistical significance was performed using Student's t test and only values with P<0.05 were accepted as significant.
RESULTS AND DISCUSSION
GPN decreases TBHQ- and thrombin-induced Ca2+ release
In human platelets two Ca2+ stores have been described on the basis of immunolocalization studies [22], different sensitivities to TG and TBHQ [24–26], and distinct cellular properties [31]. In the present study we used TBHQ to selectively deplete one of these stores, which we have previously found to be about five times smaller, in terms of Ca2+ storage, than the TBHQ-insensitive compartment [31]. In order to investigate the nature of the TBHQ-sensitive store we used inhibitors that selectively abrogate Ca2+ storage into acidic organelles, such as GPN, a substrate of lysosomal cathepsin C whose cleavage results in lysis of lysosomes by osmotic swelling [17,32,33]. Figure 1(A) shows a typical staining pattern of platelets loaded with Lysosensor Green DND-189 observed by confocal microscopy. Lysosensor Green is a fluorescent pH indicator that partitions into acidic organelles and is highly fluorescent at acidic pH.
Figure 1. Effect of GPN on agonist-dependent Ca2+ release from intracellular stores.

Resting human platelets (A) or platelets incubated for 10 min at 37 °C in the presence of 50 μM GPN (B) were loaded with Lysosensor Green DND-189 to stain the acidic organelles and fluorescence was detected using a confocal microscope as described in the Materials and methods section. Images shown are representative of six separate experiments. (C) Human platelets were loaded either with Lysosensor Green DND-189 (thick line) or with fura 2 (thin line) and then treated with 50 μM GPN as indicated in a Ca2+-free medium. Fluorescence was detected using a fluorescence spectrophotometer as described in the Materials and methods section. Results shown are representative of four to six experiments, which, in the case of loading with Lysosensor Green, were also used to obtain the images shown in (A) and (B). (D–G) fura 2-loaded human platelets were incubated for 10 min at 37 °C in the presence of 50 μM GPN or the vehicle (DMSO as control). At the time of experiment 100 μM EGTA was added. Cells were then stimulated with TBHQ (20 μM; D), thrombin (0.1 unit/ml; E), ADP (10 μM; F), or AVP (0.1 μM; G). Changes in [Ca2+]c were monitored as described in the Materials and methods section and traces are representative of four to six independent experiments.
Despite the limited resolution of platelets achieved by confocal microscopy due to their small size, we observed that treatment of human platelets for 10 min at 37 °C with 50 μM GPN decreased the fluorescence detected in the cells (Figure 1B). However, after treatment with GPN, some fluorescence still remained in the platelet preparation and may be emitted by acidic organelles distinct from lysosomes therefore we have used the term ‘acidic organelle’ throughout the text to describe the origin of the Lysosensor Green fluorescence. These findings were confirmed by quantification of the Lysosensor Green-derived fluorescence using a fluorescence spectrophotometer. Our results revealed that GPN induces a slow and time-dependent reduction in fluorescence, reaching a maximum inhibitory effect of 58.6±3.2% after 9 min of treatment (from 2.9±0.1 arbitrary units at resting conditions to 1.2±0.1 arbitrary units; Figure 1C; P<0.05; n=6), therefore, platelets contain acidic compartments, including lysosomes and other organelles.
In order to investigate whether acidic organelles might be a Ca2+ store we tested the effect of 50 μM GPN on Ca2+ mobilization in platelets suspended in a Ca2+-free medium. As shown in Figure 1(C), GPN induced a slow and small increase in [Ca2+]c consistent with the slow rate of decrease in Lysosensor Green-derived fluorescence. GPN induced a maximum increase in [Ca2+]c over basal of 43±5 nM (n=4). A similar effect using GPN has been previously reported in endothelial and pancreatic acinar cells [17,34]. The increase in [Ca2+]c, despite the small size of this Ca2+ pool, and that SERCA and PMCA (plasma-membrane Ca2+-ATPase) are operative, might be attributed to the slow release of Ca2+ into the cytosol from the swelling lysosomes. We have recently described for platelets that the rate of activation of SERCA and PMCA depends on the rate of increase in [Ca2+]c, so that at high rates of increase in [Ca2+]c the activation of SERCA and PMCA is rapid and vice versa [35]. In agreement with this, we have found that GPN induced a rise in [Ca2+]c that was returned to basal level after 30 min of stimulation (results not shown).
Treatment of human platelets with 50 μM GPN reduced Ca2+ release induced by TBHQ by 88±5%, suggesting that TBHQ selectively releases Ca2+ from acidic organelles ([Ca2+]c elevation above basal was 4±5 nM in the presence of GPN and 44±6 nM in controls; Figure 1D; n=6).
We have recently reported that thrombin releases Ca2+ from TBHQ-insensitive and -sensitive compartments in platelets through the synthesis of different second messengers, Ins(1,4,5)P3 and NAADP respectively; in contrast, ADP and AVP only evoke Ins(1,4,5)P3-dependent Ca2+ release from the TBHQ-insensitive store [36,37]. Therefore thrombin is the only agonist of the three studied that is able to release Ca2+ from the acidic store through the generation of NAADP [36,37]. Consistent with this finding, GPN significantly decreased thrombin-induced Ca2+ release by 22.2±3.1%, which is the expected fraction of Ca2+ release by thrombin-dependent action on the TBHQ-sensitive store [31,36]. The initial peak [Ca2+]c elevation above basal after thrombin was significantly reduced from 297±12 to 228±9 nM (Figure 1E; P<0.05; n=6). However, GPN was without effect on ADP- or AVP-stimulated Ca2+ release (the initial peak [Ca2+]c elevation was 135±7 and 119±6 nM in controls versus 136±9 and 117±7 nM in the presence of GPN in platelets treated with ADP or AVP, respectively (see Figures 1F and 1G; n=6), and is consistent with the lack of dependence of the TBHQ-sensitive compartments [36].
GPN had no effect on Ca2+ removal from the cytosol after platelet stimulation with thrombin, ADP or AVP (the decay constants were 0.0156±0.0005, 0.0174±0.0008 and 0.0168±0.0006 for thrombin, ADP and AVP, respectively, in the presence of GPN and 0.0159±0.0007, 0.0175±0.0008 and 0.0165±0.0006 for these agonists in the absence of GPN; Figures 1E–1G; n=6), which confirms that GPN did not alter the activity of SERCA or PMCA.
To further assess whether GPN or TBHQ are able to release Ca2+ from the TBHQ-insensitive Ca2+ store we performed a series of experiments shown in Figures 2(A)–2(C). Our results indicate that preincubation with GPN did not alter Ca2+ release induced by a low concentration of TG (10 nM), which has been shown to selectively inhibit SERCA 2b, the isoform present in the TBHQ-insensitive pool [21] ([Ca2+]c elevation above basal evoked by 10 nM TG was 52±7 nM in the presence of GPN and 50±6 nM in controls; Figure 2A; n=4). In addition, treatment of platelets with TBHQ (20 μM) did not modify Ca2+ release induced either by 10 nM TG ([Ca2+]c elevation was 53±5 and 51±6 nM in the absence and presence of TBHQ; Figure 2B; n=4) or by 10 μM ADP (the initial peak [Ca2+]c elevation above basal after ADP was 132±10 and 130±9 nM in the absence and presence of TBHQ; Figure 2C; n=4). These findings indicate that the TBHQ-insensitive store is not an acidic organelle and provide evidence that TBHQ does not mobilize Ca2+ from the TBHQ-insensitive Ca2+ compartment.
Figure 2. GPN and TBHQ release Ca2+ from acidic TBHQ-sensitive intracellular stores.

(A) fura 2-loaded human platelets were incubated for 10 min at 37 °C in the presence of 50 μM GPN or the vehicle (Control). At the time of experiment 100 μM EGTA was added. Cells were then stimulated with TG (10 nM). (B and C) fura 2-loaded human platelets were treated in a Ca2+-free medium (100 μM EGTA was added) with 20 μM TBHQ or the vehicle (DMSO), as control, followed by stimulation with either TG (10 nM; B) or ADP (10 μM; C). (D) Human platelets were stimulated in a Ca2+-free medium (100 μM EGTA was added) with 20 μM TBHQ in the absence (top panel) or presence of 1 mM LaCl3 (middle and bottom panels). In the bottom panel, cells were treated with 10 nM TG 4 min later. (E) Human platelets were pretreated in a Ca2+-free medium (100 μM EGTA was added) with 20 μM TBHQ and 30 min later were stimulated with 0.1 unit/ml thrombin. Changes in [Ca2+]c were monitored as described in the Materials and methods section and traces are representative of four to six independent experiments.
We have investigated the Ca2+ mobilization induced by TBHQ in platelets. The acidic organelles are a small and finite Ca2+ store, and as expected TBHQ induced a transient increase in [Ca2+]c (Figure 2D, top panel; n=4). Since SERCA 3 is inhibited by TBHQ we have investigated the role of PMCA and SERCA 2b in the removal of Ca2+ from the cytosol. In the presence of 1 mM LaCl3, which prevents Ca2+ extrusion mediated by PMCA in platelets [38], the rate of decay of [Ca2+]c to the basal level was similar to the control (the decay constants were 0.0038±0.0002 and 0.0036±0.0003 in the absence and presence of LaCl3; Figure 2D, top and middle panels). In contrast, inhibition of SERCA 2b with 10 nM TG induced a further release of Ca2+ from the TBHQ-insensitive store, which significantly reduced the rate of the return of [Ca2+]c to basal levels (the decay constant was 0.0009±0.0001; Figure 2D, bottom panel). These findings suggest that Ca2+ removal from the cytosol after treatment with TBHQ is mostly mediated by SERCA 2b. These observations were further confirmed by a similar response to thrombin, of platelets pretreated for 30 min in the absence and presence of TBHQ, which releases Ca2+ from both stores [36,37]. If Ca2+ was pumped out of the cell it would be expected to find some inhibition in thrombin-induced response (Figure 2E; n=4). Results shown in Figure 2(E) support that TBHQ had no effect either on PMCA or SERCA 2b activities, the main mechanisms responsible for Ca2+ removal after platelet stimulation with thrombin in the presence of TBHQ.
Effect of nigericin and bafilomycin A1 on Ca2+ release from intracellular stores
Previous studies have presented acidic compartments as physiological sources of Ca2+. Acidic organelles contain a high Ca2+ concentration that is maintained in part by the proton gradient across their membranes [19,39]. In order to investigate the role of proton gradients in Ca2+ release in platelets we used nigericin, a proton carrier, and bafilomycin A1, an inhibitor of the vacuolar H+-ATPase [40]. We confirmed that nigericin impairs the pH gradient of acidic organelles by using confocal microscopy to analyse its effect on the fluorescence of Lysosensor Green DND-189 (Figures 3A and 3B). Quantification of the Lysosensor Green-derived fluorescence revealed that nigericin induces a rapid decrease in the fluorescence intensity, which was reduced by 74.2±6.2% after 2 min of treatment (from 3.1±0.1 arbitrary units at resting conditions to 0.8±0.1 arbitrary unit; Figure 3C; n=6).
Figure 3. Effect of nigericin on TBHQ-induced Ca2+ release in human platelets.

Resting human platelets (A) or platelets treated for 4 min at 37 °C with 10 μM nigericin (B) were loaded with Lysosensor Green DND-189 to stain the acidic organelles. Fluorescence was detected using a confocal microscope as described in the Materials and methods section. Images shown are representative of six separate experiments. (C) Human platelets were loaded with Lysosensor Green DND-189 and then 10 μM nigericin was added to the suspension. Fluorescence was detected using a fluorescence spectrophotometer as described in the Materials and methods section. Results shown are representative of six experiments also used to obtain the images shown in (A) and (B). (D) fura 2-loaded human platelets, suspended in a Ca2+-free medium (100 μM EGTA was added at the time of experiment), were treated with rotenone (10 μM) and 4 min later TBHQ (20 μM) was added to release Ca2+ from the stores. Nigericin (10 μM) was added to the platelet suspension 4 min later. (E) fura 2-loaded human platelets were treated with rotenone (10 μM) in a Ca2+-free medium (100 μM EGTA was added) and 4 min later nigericin (10 μM) was added. TBHQ (20 μM) was added to the platelet suspension 4 min later to discharge the stores. (F) Human platelets were suspended in a Ca2+-free medium (100 μM EGTA was added), treated with rotenone (10 μM), and 4 min later TBHQ (20 μM) combined with nigericin (10 μM) were added. (G) Cells, suspended in a Ca2+-free medium (100 μM EGTA was added), were pre-treated with rotenone (10 μM) followed by 10 μM nigericin alone (top panel) or in combination with 20 μM TBHQ (bottom panel). Nigericin was removed (where shown by the break in the trace) as described in the Materials and methods section, cells were suspended in nominally Ca2+-free HBS and stimulated again with 10 μM nigericin. (H) Human platelets were suspended in a Ca2+-free medium (100 μM EGTA was added) and pre-treated with rotenone (10 μM). Cells were then stimulated with nigericin (10 μM) and 1.5 min later treated with either 50 μM GPN (top panel) or 20 μM TBHQ followed by addition of GPN 2 min later (bottom panel). Changes in [Ca2+]c were monitored as described in the Materials and methods section and traces are representative of six independent experiments.
During the performance of the experiments, rotenone (10 μM), an inhibitor of complex I of the respiratory chain that dissipates the membrane potential [41], was added to the platelet suspension to release mitochondrial Ca2+ and avoid interference with this organelle during the use of nigericin. Rotenone did not significantly alter Ca2+ release induced by 20 μM TBHQ ([Ca2+]c elevation above basal was 43±5 nM in the presence of rotenone and 46±5 nM in controls) or the physiological agonists thrombin (0.1 unit/ml), ADP (10 μM) or AVP (0.1 μM) ([Ca2+]c elevation above basal was 270±17, 130±9 and 117±11 nM in the presence of rotenone, and 272±15, 137±10 and 125±9 nM in controls for thrombin, ADP and AVP respectively). These findings are consistent with the reportedly reduced role of mitochondria in intracellular Ca2+ homoeostasis in human platelets [38].
As shown in Figure 3(D), treatment of platelets with 20 μM TBHQ in a Ca2+-free medium induces a small increase in [Ca2+]c, reaching a maximum [Ca2+]c elevation above basal of 41±6 nM; subsequent addition of nigericin (10 μM) was unable to release further Ca2+ from the acidic compartment. This finding suggests that nigericin releases Ca2+ from TBHQ-sensitive stores and confirms that TBHQ releases Ca2+ from acidic stores. We conducted converse experiments where cells were treated with nigericin prior to stimulation with TBHQ. As shown in Figure 3(E), platelet treatment with nigericin induced a rapid and transient increase in [Ca2+]c, the initial peak [Ca2+]c elevation above basal being 41±5 nM. The integral of the rise in [Ca2+]c above basal for 1.5 min after the addition of nigericin, taking data every 1 s, was 1752±260 nM·s−1. Subsequent addition of TBHQ, once [Ca2+]c had returned to basal levels, induced an increase in [Ca2+]c that was found to be identical with that observed in the absence of prior treatment with nigericin (TBHQ-induced [Ca2+]c elevation above basal was 40±4 nM; Figure 3E). To further investigate whether TBHQ and nigericin release Ca2+ from the same compartment, platelets were stimulated in a Ca2+-free medium, with a combination of both agents. As shown in Figure 3(F) Ca2+ release, induced by treatment of platelets with TBHQ in combination with nigericin, was similar to that induced by TBHQ alone ([Ca2+]c elevation above basal induced by TBHQ+nigericin was 39±6 nM; Figure 3F).
The results shown suggest that nigericin and TBHQ release Ca2+ from the same Ca2+ compartment, since TBHQ abolishes the response to nigericin. However, TBHQ induces a similar release of Ca2+ in the absence or presence of nigericin, which suggests that either TBHQ does not release Ca2+ from the acidic compartment, which is not supported by the results presented in Figures 3(D) and 3(F), or SERCA 3 is refilling the acidic stores after treatment with nigericin. To investigate this possibility, 10 μM nigericin was added to platelets suspended in a Ca2+-free medium, and then removed, to allow platelets to recover the proton gradient in the acidic organelles. Subsequent addition of nigericin induced a rise in [Ca2+]c that was comparable with that obtained in control conditions (Figure 3G, top panel; the initial peak [Ca2+]c elevation above basal was 40±4 nM). When the experiments were performed in the presence of 20 μM TBHQ the second addition of nigericin resulted in a significantly smaller increase in [Ca2+]c (Figure 3G, bottom panel; the initial peak [Ca2+]c elevation above basal was 12±3 nM and the integral of the rise in [Ca2+]c above basal for 1.5 min after the addition of nigericin, taking data every 1 s, was 392±80 nM·s−1). These findings suggest that, after depletion of the acidic stores by disruption of the proton gradient, these stores are refilled by a mechanism partially dependent on SERCA 3 activity. Partial refilling of the acidic pool when SERCA 3 is inhibited by TBHQ might be mediated by Ca2+–H+ exchange, activated by the restored proton gradient as previously described [42,43], although this seems to be less effective than SERCA 3 in platelets.
The refilling of the acidic stores after treatment with nigericin is demonstrated further by the results showed in Figure 3(H). Our results demonstrate that GPN, which induces lysosome lysis, causes an increase in [Ca2+]c after platelet treatment with nigericin, confirming that lysosomal related stores were refilled. The effect of GPN was impaired by treatment with TBHQ, which offers further support that GPN and TBHQ release Ca2+ from the nigericin-releasable stores and for the role of SERCA 3 in store refilling after disruption of the proton gradient.
In the presence of rotenone (Figure 4A), treatment of platelets in a Ca2+-free medium with thrombin (0.1 unit/ml) significantly reduces Ca2+ release by nigericin by 44±2% (the initial peak [Ca2+]c elevation above basal was 23±4 nM and the integral of the rise in [Ca2+]c above basal for 1.5 min after the addition of nigericin, taking data every 1 s, was 948±189 nM·s−1; P<0.05; n=6). The lack of complete inhibition of nigericin-induced Ca2+ mobilization by thrombin can be explain by the activity of SERCA during platelet activation with thrombin, which might reload, at least partially, the TBHQ-sensitive store. Alternatively, thrombin at 0.1 unit/ml might be unable to fully deplete the acidic store. On the other hand, platelet treatment with ADP or AVP did not modify nigericin-induced Ca2+ mobilization (the mean initial peak [Ca2+]c elevation above basal was 40±5 nM and the integral of the rise in [Ca2+]c above basal for 1.5 min after the addition of nigericin, taking data every 1 s, was 1802±204 nM·s−1; Figures 4B and 4C). These results support our previous observations [36,37]. As observed with TBHQ, Ca2+ release by treatment of platelets with thrombin after nigericin was identical with that observed in the absence of nigericin (compare Figure 4D with Figure 4A). The initial peak [Ca2+]c elevation above basal after thrombin was 292±11 nM (Figure 1E; n=6). These observations suggest that released Ca2+ was returned back into the acidic organelles before stimulation with thrombin. As expected, no effect of nigericin on ADP or AVP-induced Ca2+ release was detected ([Ca2+]c elevation above basal was 134±10 and 122±8 nM; Figures 4E and 4F; n=6).
Figure 4. Effect of nigericin on agonist-stimulated Ca2+ release in human platelets.

(A–C) fura 2-loaded human platelets, suspended in a Ca2+-free medium (100 μM EGTA was added at the time of experiment), were treated with rotenone (10 μM) and 4 min later cells were stimulated either with thrombin (0.1 unit/ml; A), ADP (10 μM; B) or AVP (0.1 μM; C). Nigericin (10 μM) was added to the platelet suspension 4 min later. (D–F) fura 2-loaded human platelets were treated with rotenone (10 μM) in a Ca2+-free medium (100 μM EGTA was added) and 4 min later nigericin (10 μM) was added. After 4 min, cells were stimulated either with thrombin (0.1 unit/ml; D), ADP (10 μM; E) or AVP (0.1 μM; F) to discharge the stores. Changes in [Ca2+]c were monitored as described in the Materials and methods section. Traces are representative of six independent experiments.
To further investigate whether nigericin was indeed acting at the acidic organelles, we also used bafilomycin A1, an inhibitor of the vacuolar H+-ATPase responsible for the proton gradient [17,40]. This gradient is involved in Ca2+ uptake into the ‘acidic organelles’ [17]. Under our conditions, bafilomycin A1 (1 μM) impaired the pH gradient in acidic compartments, as observed by confocal microscopy in platelets loaded with the fluorescent marker Lysosensor Green DND 189 (compare Figures 5A and 5B).
Figure 5. Effect of bafilomycin A1 on TBHQ-induced Ca2+ release in human platelets.

Resting human platelets (A) or platelets treated for 4 min at 37 °C with 1 μM bafilomycin A1 (B) were loaded with Lysosensor Green DND-189 to stain the acidic organelles; fluorescence was detected using a confocal microscope, as described in the Materials and methods section. Images shown are representative of six separate experiments. (C) Human platelets were loaded with Lysosensor Green DND-189 and then 1 μM bafilomycin A1 was added to the suspension. Fluorescence was detected using a fluorescence spectrophotometer as described in the Materials and methods section. Results shown are representative of six experiments also used to obtain the images shown in (A) and (B). (D) fura 2-loaded human platelets, suspended in a Ca2+-free medium (100 μM EGTA was added at the time of experiment), were treated with TBHQ (20 μM) to release Ca2+ from the stores and 4 min later bafilomycin A1 (1 μM) was added to the platelet suspension. (E) fura 2-loaded human platelets, suspended in a Ca2+-free medium (100 μM EGTA was added at the time of experiment), were treated with bafilomycin A1 (1 μM) and 4 min later TBHQ (20 μM) was added to the platelet suspension. (F) Cells, suspended in a Ca2+-free medium (100 μM EGTA was added), were treated with 1 μM bafilomycin A1 alone (top panel) or in combination with 20 μM TBHQ (bottom panel). Bafilomycin A1 was removed (shown by a break in the trace) as described in the Materials and methods section, and platelets were suspended in nominally Ca2+-free HBS and stimulated again with 1 μM bafilomycin A1. (G) Human platelets, suspended in a Ca2+-free medium (100 μM EGTA was added), were treated with 1 μM bafilomycin A1 and 1.5 min later treated with either 50 μM GPN (top panel) or 20 μM TBHQ, followed by addition of GPN 2 min later (bottom panel). Changes in [Ca2+]c were monitored as described in the Materials and methods section. Traces are representative of six independent experiments.
Quantification of the Lysosensor Green-derived fluorescence revealed that, as for nigericin, bafilomycin A1 induces a rapid decrease in the fluorescence intensity that was reduced by 71.3±5.2% after 2 min of treatment (from 3.0±0.1 arbitrary units at resting state to 0.86±0.1 arbitrary unit; Figure 5C; n=6). As shown in Figure 5(D), treatment of platelets in a Ca2+-free medium with 20 μM TBHQ induces a sustained increase in [Ca2+]c; the subsequent addition of bafilomycin A1 (1 μM) had no effect on [Ca2+]c, suggesting that TBHQ depleted the acidic stores. As observed using nigericin, platelet treatment with bafilomycin A1 induced a rapid and transient increase in [Ca2+]c, the initial peak [Ca2+]c elevation above basal was 30±3 nM. The integral of the rise in [Ca2+]c above basal for 1.5 min after the addition of bafilomycin A1, taking data every 1 s, was 1301±198 nM·s−1. Addition of TBHQ once [Ca2+]c had returned to basal levels induced an increase in [Ca2+]c similar to that observed in the absence of bafilomycin A1 (TBHQ-induced [Ca2+]c elevation above basal was 43±4 nM in the presence of bafilomycin A1 and 47±6 nM in control; compare Figure 5E with 5D; n=6). As with nigericin, we further investigated whether the lack of an effect following treatment with bafilomycin on the subsequent TBHQ-induced Ca2+ mobilization was due to refilling of the acidic store. Consistent with the results obtained for nigericin, after depletion of the acidic stores using bafilomycin A1, the stores are refilled by a mechanism mainly dependent on SERCA 3. Thus removal of bafilomycin A1 from the cell suspension, in order to allow platelets to restore the proton gradient, and subsequent stimulation with bafilomycin A1 shows a similar increase in [Ca2+]c as shown in Figure 5(F) (the initial peak [Ca2+]c elevation above basal was 36±3 nM in the first treatment and 34±4 nM in the second addition of bafilomycin A1). As observed following treatment with nigericin, addition of 50 μM GPN, after bafilomycin A1, resulted in an elevation in [Ca2+]c similar to that found under control conditions (Figure 5G, top panel; the [Ca2+]c elevation above basal was 42±4 nM). Refilling of the acidic stores was prevented in the presence of TBHQ (Figures 5F and 5G, bottom panels).
Finally, treatment of platelets in a Ca2+-free medium with thrombin (0.1 unit/ml) significantly decreases Ca2+ release induced by bafilomycin A1 by 37±5% (after treatment with thrombin, bafilomycin A1 induced an initial peak [Ca2+]c elevation above basal of only 19±3 nM, the integral of the rise in [Ca2+]c above basal was 799±128 nM·s−1; Figure 6A; P<0.05; n=6). In contrast, treatment with bafilomycin A1 did not modify Ca2+ release by thrombin ([Ca2+]c elevation above basal 267±12 nM in the presence of bafilomycin A1 and 271±11 nM in controls; compare Figure 6B with Figure 6A; n=6), which further supports the notion that Ca2+ released by bafilomycin A1 is taken up again by the acidic store by a mechanism mainly dependent on SERCA 3.
Figure 6. Effect of bafilomycin A1 on thrombin-stimulated Ca2+ release in human platelets.

(A) fura 2-loaded human platelets were stimulated with thrombin (0.1 unit/ml) in a Ca2+-free medium (100 μM EGTA was added at the time of experiment) and 4 min later bafilomycin A1 (1 μM) was added to the platelet suspension. (B) fura 2-loaded human platelets, suspended in a Ca2+-free medium (100 μM EGTA was added at the time of experiment), were treated with bafilomycin A1 (1 μM) and 4 min later thrombin (0.1 unit/ml) was added to the platelet suspension. Changes in [Ca2+]c were monitored as described in the Materials and methods section. Traces are representative of six independent experiments.
Store overlapping has been described in different cell types, such as pancreatic acinar cells, where reloading of the ER or the nuclear envelope is based on the activity of SERCA, and that of the luminal Ca2+ compartments depends solely on the pH gradient generated by the vacuolar H+-ATPase [12,39,44]. In the latter case, treatment with nigericin has been shown to induce a sustained increase in [Ca2+]c as the driving force that maintains Ca2+ accumulated into the acidic organelles is dissipated [19,45]. A similar effect has been observed in other cell types, such as macrophages [46]. However, our results indicate for the first time that the acidic Ca2+ pool in platelets is regulated by two mechanisms: SERCA 3 and the proton gradient, such that elimination of this gradient by nigericin or bafilomycin A1 or inhibition of SERCA 3 by TBHQ results in discharge of the acidic store. In both mechanisms, SERCA 3 and the proton gradient, work together and cooperate in the accumulation of Ca2+ into the acidic organelles.
Our findings support the universality of the existence of physiological intracellular Ca2+ stores in acidic organelles. In human platelets this Ca2+ pool is mainly a lysosomal store, although our results do not rule out the possibility that other acidic organelles, such as dense granules [43], are also physiological acidic Ca2+ stores. The existence of separate Ca2+ pools, whose Ca2+ concentration might be differentially regulated, by either Ca2+- or vacuolar H+-ATPases, is essential for the great variability in spatiotemporal Ca2+ signals that precisely regulate a wide variety of cellular functions.
Acknowledgments
We thank Dr Stewart Sage, Deparment of Physiology, University of Cambridge, Cambridge, U.K., for helpful suggestions and discussion, and Mercedes Gómez Blázquez for technical assistance. This work was supported by DGI-MEC (Dirección General de Investigación del Ministerio de Educación y Ciencia) Grants BFI2001–0624 and BFU2004–00165 and by a grant from the Consejería de Sanidad y Consumo–Junta de Extremadura (SCSS0405).
References
- 1.Reembold C. M. Regulation of contraction and relaxation in arterial smooth muscle. Hypertension. 1992;20:129–137. doi: 10.1161/01.hyp.20.2.129. [DOI] [PubMed] [Google Scholar]
- 2.Brown B. L., Walker S. W., Tomlinson S. Calcium calmodulin and hormone secretion. Clin. Endocrinol. 1985;23:201–218. doi: 10.1111/j.1365-2265.1985.tb00216.x. [DOI] [PubMed] [Google Scholar]
- 3.Salzman E. W., Ware J. A. Ionized calcium as an intracellular messenger in blood platelets. Prog. Hemostasis Thromb. 1989;9:177–202. [PubMed] [Google Scholar]
- 4.Means A. R. Calcium, calmodulin and cell cycle regulation. FEBS Lett. 1994;347:1–4. doi: 10.1016/0014-5793(94)00492-7. [DOI] [PubMed] [Google Scholar]
- 5.Pozzan T., Rizzuto R., Volpe P., Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- 6.Ashby M. C., Tepikin A. V. Polarized calcium and calmodulin signalling in secretory epithelia. Physiol. Rev. 2002;82:701–734. doi: 10.1152/physrev.00006.2002. [DOI] [PubMed] [Google Scholar]
- 7.Bootman M. D., Berridge M. J., Roderick H. L. Calcium signalling: more messengers, more channels, more complexity. Curr. Biol. 2002;12:R563–R565. doi: 10.1016/s0960-9822(02)01055-2. [DOI] [PubMed] [Google Scholar]
- 8.Collins T. J., Berridge M. J., Lipp P., Bootman M. D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilabert J. A., Bakowski D., Parekh A. Energized mitochondria increase the dynamic range over which inositol 1,4,5-trisphosphate activates store-operated calcium influx. EMBO J. 2001;20:2672–2679. doi: 10.1093/emboj/20.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villalobos C., Nunez L., Montero M., Garcia A. G., Alonso M. T., Chamero P., Alvarez J., Garcia-Sancho J. Redistribution of Ca2+ among cytosol and organella during stimulation of bovine chromafin cells. FASEB J. 2002;16:343–353. doi: 10.1096/fj.01-0630com. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez A., Granados M. P., Salido G. M., Pariente J. A. Changes in mitochondrial activity evoked by cholecystokinin in isolated mouse pancreatic acinar cells. Cell. Signalling. 2003;15:1039–1048. doi: 10.1016/s0898-6568(03)00067-6. [DOI] [PubMed] [Google Scholar]
- 12.Gerasimenko J. V., Maruyama Y., Yano K., Dolman N. J., Tepikin A. V., Petersen O. H., Gerasimenko O. V. NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J. Cell Biol. 2003;163:271–282. doi: 10.1083/jcb.200306134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinton P., Pozzan T., Rizzuto R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 1998;17:5298–5308. doi: 10.1093/emboj/17.18.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoo S. H. Coupling of the IP3 receptor/Ca2+ channel with Ca2+ storage proteins chromogranins A and B in secretory granules. Trends Neurosci. 2000;23:424–428. doi: 10.1016/s0166-2236(00)01621-0. [DOI] [PubMed] [Google Scholar]
- 15.Kinnear N. P., Boittin F. X., Thomas J. M., Galione A., Evans A. M. Lysosome–sarcoplasmic reticulum junctions: a trigger zone for calcium signalling by nicotinic acid adenine dinucleotide phosphate and endothelin-1. J. Biol. Chem. 2004;279:54319–54326. doi: 10.1074/jbc.M406132200. [DOI] [PubMed] [Google Scholar]
- 16.Haller T., Völkl H., Deetjen P., Dietl P. The lysosomal Ca2+ pool in MDCK cells can be released by Ins(1,4,5)P3-dependent hormones or thapsigargin but does not activate store-operated Ca2+ entry. Biochem. J. 1996;319:909–912. doi: 10.1042/bj3190909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamasaki M., Masgrau R., Morgan A. J., Churchill G. C., Patel S., Ashcroft S. J., Galione A. Organelle selection determines agonist-specific Ca2+ signals in pancreatic acinar and beta cells. J. Biol. Chem. 2004;279:7234–7240. doi: 10.1074/jbc.M311088200. [DOI] [PubMed] [Google Scholar]
- 18.Haller T., Dietl P., Deetjen P., Volkl H. The lysosomal compartment as intracellular calcium store in MDCK cells: a possible involvement in InsP3-mediated Ca2+ release. Cell Calcium. 1996;19:157–165. doi: 10.1016/s0143-4160(96)90084-6. [DOI] [PubMed] [Google Scholar]
- 19.Christensen K. A., Myers J. T., Swanson J. A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002;115:599–607. doi: 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- 20.Papp B., Enyedi A., Kovacs T., Sarkadi B., Wuytack F., Thastrup O., Gardos G., Bredoux R., Levy-Toledano S., Enouf J. Demonstration of two forms of calcium pumps by thapsigargin inhibition and radioimmunoblotting in platelet membrane vesicles. J. Biol. Chem. 1991;266:14593–14596. [PubMed] [Google Scholar]
- 21.Cavallini L., Coassin M., Alexandre A. Two classes of agonist-sensitive Ca2+ stores in platelets, as identified by their differential sensitivity to 2,5-di-(tert-butyl)-1,4-benzohydroquinone and thapsigargin. Biochem. J. 1995;310:449–452. doi: 10.1042/bj3100449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovacs T., Berger G., Corvazier E., Paszty K., Brown A., Bobe R., Papp B., Wuytack F., Cramer E. M., Enouf J. Immunolocalization of the multi-sarco/endoplasmic reticulum Ca2+ ATPase system in human platelets. Br. J. Haematol. 1997;97:192–203. doi: 10.1046/j.1365-2141.1997.9982639.x. [DOI] [PubMed] [Google Scholar]
- 23.Gerrard J. M., White J. G., Peterson D. A. The platelet dense tubular system: its relationship to prostaglandin synthesis and calcium flux. Thromb. Haemostasis. 1978;40:224–231. [PubMed] [Google Scholar]
- 24.Wuytack F., Papp B., Verboomen H., Raeymaekers L., Dode L., Bobe R., Enouf J., Bokkala S., Authi K. S., Casteels R. A sarco/endoplasmic reticulum Ca2+-ATPase 3-type Ca2+ pump is expressed in platelets, in lymphoid cells, and in mast cells. J. Biol. Chem. 1994;269:1410–1416. [PubMed] [Google Scholar]
- 25.Bobe R., Bredoux R., Wuytack F., Quarck R., Kovacs T., Papp B., Corvazier E., Magnier C., Enouf J. The rat platelet 97-kDa Ca2+ ATPase isoform is the sarcoendoplasmic reticulum Ca2+ ATPase 3 protein. J. Biol. Chem. 1994;269:1417–1424. [PubMed] [Google Scholar]
- 26.Enyedi A., Paszty K., Kovacs T., Sarkadi B., Gardos G., Magnier C., Wuytack F., Enouf J. Simultaneous presence of two distinct endoplasmic-reticulum-type calcium-pump isoforms in human cells. Characterization by radio-immunoblotting and inhibition by 2,5-di-(t-butyl)-1,4-benzohydroquinone. Biochem. J. 1992;288:297–302. doi: 10.1042/bj2880297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosado J. A., Sage S. O. Activation of store-mediated calcium entry by secretion-like coupling between the inositol 1,4,5-trisphosphate receptor type II and human transient receptor potential (hTrp1) channels in human platelets. Biochem. J. 2001;356:191–198. doi: 10.1042/0264-6021:3560191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grynkiewicz G., Poenie M., Tsien R. Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 29.Rosado J. A., Sage S. O. Farnesylcysteine analogues inhibit store-regulated Ca2+ entry in human platelets: evidence for involvement of small GTP-binding proteins and actin cytoskeleton. Biochem. J. 2000;347:183–192. [PMC free article] [PubMed] [Google Scholar]
- 30.Redondo P. C., Lajas A. I., Salido G. M., Gonzalez A., Rosado J. A., Pariente J. A. Evidence for secretion-like coupling involving pp60src in the activation and maintenance of store-mediated Ca2+ entry in mouse pancreatic acinar cells. Biochem. J. 2003;370:255–263. doi: 10.1042/BJ20021505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosado J. A., Lopez J. J., Harper A. G., Harper M. T., Redondo P. C., Pariente J. A., Sage S. O., Salido G. M. Two pathways for store-mediated calcium entry differentially dependent on the actin cytoskeleton in human platelets. J. Biol. Chem. 2004;279:29231–29235. doi: 10.1074/jbc.M403509200. [DOI] [PubMed] [Google Scholar]
- 32.Jadot M., Colmant C., Wattiaux-De Coninck S., Wattiaux R. Intralysosomal hydrolysis of glycyl-L-phenylalanine 2-naphthylamide. Biochem. J. 1984;219:965–970. doi: 10.1042/bj2190965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Churchill G. C., Okada Y., Thomas J. M., Genazzani A. A., Patel S., Galione A. NAADP mobilizes Ca2+ from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111:703–708. doi: 10.1016/s0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- 34.Srinivas S. P., Ong A., Goon L., Goon L., Bonanno J. A. Lysosomal Ca2+ stores in bovine corneal endothelium. Invest. Ophthalmol. Visual Sci. 2002;43:2341–2350. [PubMed] [Google Scholar]
- 35.Juska A., Redondo P. C., Salido G. M., Rosado J. A. Dynamics of SERCA and PMCA activities depend on the inicial rate of calcium influx from the stores in human platelets. J. Physiol. Biochem. 2005;61:251–252. doi: 10.1007/BF03168376. [DOI] [PubMed] [Google Scholar]
- 36.López J. J., Redondo P. C., Salido G. M., Pariente J. A., Rosado J. A. Differential Ca2+ release by physiological agonist from separate compartments in human platelets. J. Physiol. Biochem. 2005;61:257. [Google Scholar]
- 37.Rosado J. A., López J. J., Redondo P. C., Gómez-Arteta E., Pariente J. A., Salido G. M. Agonists regulate Ca2+ signalling by different second messengers in human platelets. J. Physiol. Biochem. 2005;61:251. [Google Scholar]
- 38.Rosado J. A., Sage S. O. Regulation of plasma membrane Ca2+-ATPase by small GTPases and phosphoinositides in human platelets. J. Biol. Chem. 2000;275:19529–19535. doi: 10.1074/jbc.M001319200. [DOI] [PubMed] [Google Scholar]
- 39.Camello C., Pariente J. A., Salido G. M., Camello P. C. Role of proton gradients and vacuolar H+-ATPases in the refilling of intracellular calcium stores in exocrine cells. Curr. Biol. 2000;10:161–164. doi: 10.1016/s0960-9822(00)00313-4. [DOI] [PubMed] [Google Scholar]
- 40.Bowman E. J., Siebers A., Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. U.S.A. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunningham M. L., Soliman M. S., Badr M. Z., Matthews H. B. Rotenone, an anticarcinogen, inhibits cellular proliferation but not prexisome proliferation in mouse liver. Cancer Lett. 1995;95:93–97. doi: 10.1016/0304-3835(95)03869-x. [DOI] [PubMed] [Google Scholar]
- 42.Dunn T., Gable K., Beeler T. Regulation of cellular Ca2+ by yeast vacuoles. J. Biol. Chem. 1994;269:7273–7278. [PubMed] [Google Scholar]
- 43.Ruiz F. A., Lea C. R., Oldfield E., Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J. Biol. Chem. 2004;279:44250–44257. doi: 10.1074/jbc.M406261200. [DOI] [PubMed] [Google Scholar]
- 44.Schulz I., Thevenod F., Dehlinger-Kremer M. Modulation of intracellular free Ca2+ concentration by IP3-sensitive and IP3-insensitive nonmitochondrial Ca2+ pools. Cell Calcium. 1989;10:325–336. doi: 10.1016/0143-4160(89)90058-4. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez A., Pariente J. A., Salido G. M., Camello P. C. Intracellular pH and calcium signalling in rat pancreatic acinar cells. Pflugers Arch. 1997;434:609–614. doi: 10.1007/s004240050443. [DOI] [PubMed] [Google Scholar]
- 46.Swallow C. J., Grinstein S., Rotstein O. D. A vacuolar type H+-ATPase regulates cytoplasmic pH in murine macrophages. J. Biol. Chem. 1990;265:7645–7654. [PubMed] [Google Scholar]