

Abstract
The translocator protein (TSPO) is an 18 kDa protein on the outer mitochondrial membrane. It has gained significant interest in recent years for its potential as a therapeutic target and imaging biomarker, particularly in neuroinflammation, cancer, and central nervous system disorders (CNS). Clinical translation of ligands has been complicated by the presence of a common single nucleotide polymorphism (A147T TSPO), at which many disclosed TSPO ligands lose affinity. One exception is the first-generation TSPO ligand PK 11195, however, this ligand possesses unfavourable pharmacokinetic properties limiting translation in CNS applications. We aimed to investigate which motifs of this ligand contribute to high binding at the wild type (WT) and A147T TSPO isoforms with the aim of identifying elements that may tolerate lipophilicity-reducing substitutions. Affinities for a small library of isoquinoline, quinazoline, indole and azaindole carboxamides with varying aliphatic and aromatic substituents were measured using radioligand binding at both TSPO isoforms, with computational studies performed to rationalise the experimentally measured binding. The heterocycle and acetamide substituents of PK 11195 were found to play a role in its non-discriminating nature. In addition, the study yielded 2a, a high affinity, non-discriminating TSPO ligand. Modelling suggests its high affinity and lack of discrimination results from additional π–π interactions introduced at the binding site of both TSPO isoforms. These findings provide a foundation for developing TSPO ligands with improved clinical properties and insensitivity to A147T polymorphism.
This work investigates which motifs of the TSPO ligand PK 11195 contribute to high binding at the wild type and clinically relevant A147T TSPO isoform, with the aim of identifying elements that may tolerate lipophilicity-reducing substitutions.
Introduction
The 18 kDa translocator protein (TSPO), has five transmembrane domains, and is localised to the outer mitochondrial membrane of many cell types.1 Baseline TSPO expression levels in the brain are low but are upregulated on reactive glia in areas damaged by neurodegenerative diseases and in cancers, such as glioma.1 As such, TSPO has become a target for the development of therapeutics and diagnostic imaging agents for use in cancer and neurodegenerative diseases. To date, the most widely used TSPO PET ligand to assess neuroinflammation has been the isoquinoline carboxamide, [11C]PK 11195, a first-generation TSPO ligand.2 Despite its utility, [11C]PK 11195 is characterised by significant limitations, including poor bioavailability in brain tissue, high non-specific binding, and plasma protein binding due to its high lipophilicity. These issues have led to difficulties in quantifying its binding potential in human PET studies.2
Second-generation TSPO radioligands were developed to address the drawbacks of [11C]PK 11195. Examples include PBR28, and XBD-173, which exhibit improved binding affinity, higher selectivity, and better signal-to-noise ratios. However, clinical translation of these ligands has been complicated by the rs6971 single nucleotide polymorphism (SNP), which results in an alanine-to-threonine substitution at residue 147 (A147T).3 The presence of the polymorphism complicates quantitative assessment of PET data using second generation TSPO ligands, as many of these ligands exhibit a drop-off in affinity at TSPO A147T.4,5 Consequently, binding affinity varies among individuals classified as high-affinity binders (HABs, homozygous for the wild-type TSPO), mixed-affinity binders (MABs, heterozygous for A147T), and low-affinity binders (LABs, homozygous for A147T). The distribution of these genotypes within the population significantly limits the clinical utility of second-generation TSPO ligands without genotype-specific considerations. Whilst extensive structure-affinity relationship studies have been undertaken to determine structural features that govern binding to the WT TSPO,6,7 systematic investigations into ligand sensitivity to TSPO A147T are rare.
[11C]PK 11195 exhibits TSPO A147T insensitivity both in vitro8 and in vivo.4,9 Exploring the contribution of PK 11195's phenyl tail group to its TSPO A147T binding sensitivity revealed the sensitivity is impacted by a complex interplay between the nature (e.g. F, CF3, Cl) and position of the substituents on the ring.10–12 Third generation TSPO radioligands such as ER-176, while not derived from PK 11195 directly, represent an important milestone in developing ligands with improved profiles. ER-176 is a quinazoline-based compound that has shown TSPO A147T insensitivity in vitro and demonstrated adequate pharmacokinetics for brain imaging in vivo.10 However, in human PET studies, ER-176 exhibited a three-fold loss in whole brain non-displaceable binding potential for LABs compared to HABs and a 2.4-fold decrease when compared to MABs.13 These challenges underscore the need for further investigation into new ligands that can efficiently target TSPO across different genotypes, particularly for patients with the A147T polymorphism. In addition, an imidazopyridine analogue of DPA-714 that has recently been described shows TSPO A147T binding insensitivity in vitro but is yet to be explored in human studies.14
Given the absence of a TSPO PET ligand with suitable pharmacokinetics for effective brain penetration and the continued lack of a ligand insensitive to the TSPO A147T variant in vivo, further systematic investigation into the structural features of the PK 11195 scaffold is justified. Whilst these ligands may share the lipophilic characteristics of PK 11195, insights from previous studies could inform design strategies for developing clinically relevant ligands. Such unexplored regions of the PK 11195 scaffold include the heterocyclic ring region and the nature of substitutions to the carboxamide moiety. Our previous work has shown the nature of substituents on the carboxamide moiety of carbazole scaffolds can impact sensitivity to TSPO A147T. For example para-methoxyphenyl substitutions can reduce binding sensitivity on carbazole scaffolds and N-benzyl-N-methyl substituents reduce discrimination across a range of heterocycles, potentially by introducing interactions with Phe 99 or 100 in the binding pocket according to docking studies.5,15,16 In addition, the nature of the heterocycle impacted discrimination, with the degree of influence dependent on substituents on other parts of the molecule.16,17 These findings suggest that the heterocyclic ring and the nature of the carboxamide substituents may be crucial for determining PK 11195's binding sensitivity. Our study aimed to dissect which structural components of PK 11195 contribute to its consistent binding profile. Specifically, we focused on modifications to the heterocyclic core and carboxamide substituents to identify designs that retain binding across TSPO variants. The structure–activity relationships reported herein explore a focused library of isoquinoline carboxamides (represented by 1a–1e, Fig. 1) primarily investigating aliphatic and aromatic substituents on the carboxamide nitrogen, given their ability to impart reduced sensitivity to A147T TSPO in other scaffolds.5,15,16 Furthermore, we used a ‘scaffold hopping’ approach18 for the heterocyclic core to investigate WT and A147T TSPO binding. Illustrated in Fig. 1 are the heterocyclic scaffolds chosen to replace the isoquinoline core. The choice of heterocycles arise from a reduction in their predicted lipophilicity and include the quinazolines (represented by 2a–2f),19 indoles (represented by 3a–3f)20,21 and 7-azaindoles (represented by 4a–4f).22,23
Fig. 1. Proposed TSPO ligands: isoquinoline carboxamides (1a–1e), quinazoline carboxamides (2a–2f), indole carboxamides (3a–3f), and azaindole carboxamides (4a–4f).

Results and discussion
Chemistry
Isoquinoline carboxamide analogues
Synthesis of the isoquinoline carboxamide ligands followed the same synthetic route reported by Stevenson and co-workers (Scheme 1).24 The coupling of methyl 2-iodobenzoate (5) with amidoacrylate (6) afforded oxoisoquinoline 7 in good yield (77%). Bromination of 7 using phosphorus(v) oxybromide afforded brominated intermediate 8 in high yield (82%). A Suzuki–Miyaura cross-coupling reaction of 8 with 2-chlorophenylboronic acid gave methyl ester 9 in good yield (76%). Base-catalysed hydrolysis of the methyl ester 9 to the carboxylic acid 10 was achieved in excellent yield (93%). HBTU-mediated coupling of 10 with the appropriate amine furnished target ligands 1a–1e in good-to-excellent yields (63–84%).
Scheme 1. Synthesis of isoquinoline carboxamide ligands: a) Pd(OAc)2, tetrabutylammonium chloride, DMF, 90 °C, 18 h, 77%; b) POBr3, K2CO3, MeCN, Δ, 24 h, 82%; c) Cs2CO3, Pd(dppf)Cl2, 2-chlorophenylboronic acid, toluene/H2O, 80 °C, 16 h, 76%; d) LiOH, MeOH/H2O, Δ, 6 h, 93%; e) HBTU, DIPEA, appropriate amine, DMF, rt, 16 h, 63–84%.

Quinazoline carboxamide analogues
Synthesis of the quinazoline carboxamide ligands followed a similar synthetic route reported by Castellano and coworkers (Scheme 2).7 Intermediate 13 was obtained in low yield (26%) using 1-bromo-2-chlorobenzene (11) and 2-aminobenzonitrile (12) via a Grignard reaction. The condensation of 13 with glyoxylic acid and ammonium acetate afforded intermediate 14, which was subsequently oxidised, in the presence of light, to afford 15. The HBTU-mediated coupling of carboxylic acid 15 with the appropriate amine, furnished target ligands 2a–2e and intermediate 16 in good yields (66–83%). Target ligands 2a, 2b and 2d have been described previously in the literature.7 Methylation of 16 using methyl iodide, in the presence of potassium tert-butoxide furnished target ligand 2f in high yield (79%). Ligand 2f (ER176) has been described previously in the literature.10,13
Scheme 2. Synthesis of quinazoline carboxamide ligands: a) i) Mg, Et2O, 0 °C–Δ, 24 h; ii) HCl, H2O, rt, 2 h, 26%; b) NH4OAc, glyoxylic acid, EtOH, rt, 1 h, 76%; c) external light irradiation with 20 W halogen tungsten lamp, DMF, rt, 12 h, 71%; d) HBTU, DIPEA, appropriate amine, DMF, rt, 16 h, 66–83%; e) MeI, KOtBu, THF, 0 °C–rt, 17 h, 79%.

Indole carboxamide analogues
The indole carboxamide ligands were synthesised according to Scheme 3. ortho-Nitrophenyl intermediate 19 was obtained in excellent yield (85%) using methyl 1H-indole-2-carboxylate (17) and 1-fluoro-2-nitrobenzene (18), in the presence of sodium hydroxide. Subsequent catalytic hydrogenation of 19 with palladium on carbon (Pd/C) afforded amino intermediate 20 in excellent yield (86%). The transformation of 20 to intermediate 21 was achieved in a good yield (66%) using Sandmeyer conditions. Hydrolysis of 21 under basic conditions gave carboxylic acid 22 in an excellent yield (94%). HBTU-mediated coupling of 22 with the appropriate amine furnished target ligands 3a–3e and intermediate 23 in good yields (73–85%). Methylation of 23 using methyl iodide, in the presence of potassium tert-butoxide furnished target ligand 3f in excellent yield (81%).
Scheme 3. Synthesis of indole carboxamide ligands: a) NaOH, DMSO, 0 °C–rt, 2 h, 85%; b) Pd/C (10% w/w), H2 atmosphere (1 atm), EtOAc, rt, 5 h, 86%; c) CuCl2, tBuONO, MeCN, rt, 1.5 h, 66%; d) LiOH, THF/H2O, Δ, 6 h, 94%; e) HBTU, DIPEA, appropriate amine, DMF, rt, 16 h, 73–85%; f) MeI, KOtBu, THF, 0 °C–rt, 17 h, 81%.

7-Azaindole carboxamide analogues
The 7-azaindole carboxamide ligands were synthesised according to Scheme 4. Intermediate 25 was prepared in an excellent yield (92%) by the acylation of 7-azaindole (24) using trichloroacetyl chloride, in the presence of aluminium trichloride. The transformation of 25, to methyl ester 26, was achieved in an excellent yield (89%) under basic conditions. Nitro intermediate 27 was obtained in an excellent yield (92%) using 26 and 1-fluoro-2-nitrobenzene (18), in the presence of sodium hydride. Subsequent catalytic hydrogenation of 27 with palladium on carbon (Pd/C) afforded amino intermediate 28 in excellent yield (92%). Chlorination of 28 to intermediate 29 was achieved in a moderate yield (56%) via the Sandmeyer reaction. Hydrolysis of 29 under basic conditions gave the carboxylic acid 30. HBTU-mediated coupling of 30 with the appropriate amine furnished target ligands 4a–4e and intermediate 31 in good yields (55–87%). Methylation of 31 using methyl iodide, in the presence of potassium tert-butoxide furnished target ligand 4f in excellent yield (86%).
Scheme 4. Synthesis of azaindole carboxamide ligands: a) trichloroacetyl chloride, AlCl3, CH2Cl2, 0 °C–rt, 3 h, 92%; b) NaHCO3, MeOH, Δ, 4 h, 89%; c) i) NaH, THF, 0 °C, 30 min; ii) 1-fluoro-2-nitrobenzene (18), THF, 0 °C–rt, 8 h, 92%; d) Pd/C (10% w/w), H2 atmosphere (1 atm), EtOAc, rt, 8 h, 92%; e) CuCl2, tBuONO, MeCN, rt, 2 h, 56%; f) LiOH, THF/H2O, Δ, 6 h, 89%; g) HBTU, DIPEA, appropriate amine, DMF, rt, 16 h, 55–87%; h) MeI, KOtBu, THF, 0 °C–rt, 17 h, 86%.

It should be noted that in this study 2f, 3f and 4f were prepared and screened as the (R)-sec-butyl enantiomers only. This was based on the well-established enantiomer-dependent affinity observed for PK 11195, with the (R)-sec-butyl enantiomer displaying potent binding affinity and the (S)-sec-butyl enantiomer lacking affinity for TSPO.25
Biological evaluation
The binding affinities, expressed as Ki (nM), for all newly synthesised ligands were characterised by competition radioligand binding assays using [3H]PK 11195 on membranes derived from HEK-293 cells overexpressing either A147T or WT TSPO.5 The binding affinity between TSPO WT and A147T for each ligand is expressed as a ratio (A147T : WT), where a ratio between A147T : WT closer to 1 indicates no preference between the two isoforms (Table 1).
Table 1. TSPO binding affinities (Ki) of the heterocyclic ligands using membranes from human TSPO WT and A147T over-expressing HEK-293 cells. Values represent the mean ± SD from at least three independent experiments performed in duplicate.
| Ligand | R1 | R2 | K i (nM) | A147T : WT | ||
|---|---|---|---|---|---|---|
| A147T | WT | |||||
| PK11195 | CH3 | (R)-sec-Butyl | 29.8 ± 2.9 | 25.1 ± 6.1 | 1.2 | |

|
1a | CH3 | Bn | 9.9 ± 1.7 | 28.4 ± 4.9 | 0.35 |
| 1b | CH2CH3 | Bn | 50.5 ± 3.6 | 91.4 ± 29.3 | 0.55 | |
| 1c | CH2CH3 | CH2CH3 | 291.1 ± 72.1 | 58.3 ± 13.9 | 5.0 | |
| 1d | CH3 | 3-OCH3Bn | 119.2 ± 34.4 | 330.2 ± 92.1 | 0.36 | |
| 1e | CH2CH3 | 3-OCH3Bn | 72.1 ± 7.3 | 147.3 ± 48.4 | 0.41 | |

|
2a | CH3 | Bn | 8.4 ± 2.5 | 11.1 ± 2.5 | 0.8 |
| 2b | CH2CH3 | Bn | 19.0 ± 5.2 | 10.4 ± 3.1 | 1.8 | |
| 2c | CH2CH3 | CH2CH3 | >10 000 | >10 000 | N/A | |
| 2d | CH3 | 3-OCH3Bn | 229.0 ± 43.1 | 127.9 ± 40.1 | 1.8 | |
| 2e | CH2CH3 | 3-OCH3Bn | 67.5 ± 19.4 | 21.9 ± 2.9 | 3.1 | |
| 2f | CH3 | (R)-sec-Butyl | 118.7 ± 30.4 | 47.8 ± 13.9 | 2.5 | |
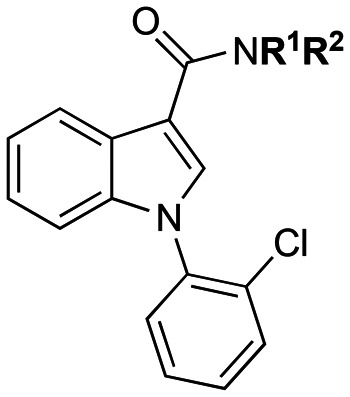
|
3a | CH3 | Bn | 212.0 ± 19.1 | 77.2 ± 19.7 | 2.8 |
| 3b | CH2CH3 | Bn | 304.3 ± 100.2 | 58.5 ± 19.2 | 5.2 | |
| 3c | CH2CH3 | CH2CH3 | 360.8 ± 29.7 | 80.8 ± 25.1 | 4.5 | |
| 3d | CH3 | 3-OCH3Bn | 323.9 ± 100.0 | 84.5 ± 27.2 | 3.8 | |
| 3e | CH2CH3 | 3-OCH3Bn | 266.6 ± 75.4 | 30.1 ± 8.6 | 8.9 | |
| 3f | CH3 | (R)-sec-Butyl | 136.6 ± 37.1 | 40.3 ± 3.3 | 3.4 | |

|
4a | CH3 | Bn | 396.7 ± 72.2 | 182.1 ± 33.8 | 2.2 |
| 4b | CH2CH3 | Bn | >10 000 | 190.0 ± 41.5 | N/A | |
| 4c | CH2CH3 | CH2CH3 | 497.0 ± 70.0 | 252.4 ± 79.1 | 2.0 | |
| 4d | CH3 | 3-OCH3Bn | >10 000 | 468.4 ± 148.9 | N/A | |
| 4e | CH2CH3 | 3-OCH3Bn | 25.9 ± 5.2 | 99.1 ± 24.9 | 0.26 | |
| 4f | CH3 | (R)-sec-Butyl | >10 000 | >10 000 | N/A | |
The isoquinoline carboxamide ligands (represented by ligands 1a–1e) were designed to evaluate the effect of carboxamide substituents for imparting affinity at both TSPO isoforms, with respect to PK 11195. Ligand 1a, featuring a benzyl group at the R2 position and maintaining the methyl group at the R1 position, exhibited similar affinity at the TSPO WT to PK 11195 but a 3-fold improvement in affinity at the TSPO A147T. Replacing the methyl group of 1a with an ethyl group at the R1 position to generate ligand 1b showed a 5.1-fold decrease in affinity at TSPO WT and a 3.2-fold decrease in affinity at A147T. Replacing the R2 benzyl of 1b with an ethyl group to generate 1c produced a 1.6-fold improvement in affinity at TSPO WT, but a 5.8-fold decrease in affinity at A147T compared to 1b. Replacing the R2 group of 1a with a meta-substituted methoxybenzyl group, represented by ligand 1d, produced ∼12-fold decreases in affinity compared to 1a at both TSPO forms. In contrast, replacing the R2 group of 1b with a meta-substituted methoxybenzyl group, represented by ligand 1e, produced a more moderate ∼1.5-fold decrease in affinity compared to 1b at both TSPO forms. All isoquinoline carboxamide ligands, except ligand 1c exhibited preference for TSPO A147T over WT TSPO.
Ligands 2a–2f, 3a–3f and 4a–4f were designed to explore the effects of heterocyclic core and the carboxamide substituents on affinity for both TSPO isoforms. Amongst the quinazoline carboxamide series (represented by ligands 2a–2f), ligand 2a, featuring a methyl group at the R1 position and a benzyl group at the R2 position showed excellent binding affinities at both isoforms (8.4 nM and 11.1 nM for A147T and WT, respectively), with minimal preference between the two isoforms. Replacing the methyl group in 2a with an ethyl group at the R1 position (represented by ligand 2b) resulted in a 2.3-fold decrease in affinity at TSPO A147T, without impacting affinity at TSPO WT. Changing the R2 benzyl group of 2b with an ethyl group (2c) abolished binding at both isoforms, highlighting the importance of the benzyl group. Replacing the R2 group of 2a with a meta-substituted methoxybenzyl group (represented by ligand 2d) produced a marked decrease in affinity, showing an 11.5-fold reduction at WT TSPO and a 27.3-fold decrease at A147T, compared to ligand 2a. In contrast, replacing the R2 group of 2b with a meta-substituted methoxybenzyl group (represented by ligand 2e) produced more moderate 3.6-fold and 2.1-fold reductions in affinity at WT and A147T TSPO, respectively. Lastly, replacing the isoquinoline heterocycle in PK 11195 with a quinazoline scaffold to yield 2f resulted in a 1.9-fold decrease in WT TSPO affinity and a 4-fold decrease in A147T affinity. All quinazoline carboxamide ligands except ligand 2a showed discrimination between the TSPO isoforms. However, the quinazoline scaffold improved A147T TSPO affinity (apart from 2c and 2f) when compared to the corresponding isoquinoline analogues. This replacement was particularly beneficial for ligand 2a, which showed superior affinity to PK 11195 while maintaining a non-discriminatory binding profile.
It should be noted that competition radioligand binding with ligand 2f (ER176) against [3H]PK 11195 in a previous study using human leukocytes and cerebellar homogenates produced only a 1.3-fold loss in affinity at LABs compared to HABs.10 Unexpectedly, though, when examined in a human PET study, [11C]ER176 suffered a 3-fold decrease in whole brain binding potential (BPND) for LABs compared to HABs,13 similar to the 2.5-fold decrease we found in our cellular binding study. The reason for this discrepancy in sensitivity between previous ex vivo radioligand binding studies and our current study is not clear, although there were methodological differences between the two studies (e.g. cell type, temperature). It should also be noted that ligand 2b has been screened previously at WT TSPO, displaying subnanomolar affinity in contrast to the affinity of 10 nM in our hands.7 This previous study was conducted on rat kidney mitochondrial membranes, so this could be explained by species differences. This is supported by the use of 0.6 nM [3H]PK 11195 for the competing radioligand in the previous study,7 presumably the Kd of this tritiated ligand in this system, which is less than the 10 nM Kd measured in our system.5
The indole carboxamide ligands (represented by ligands 3a–3f) displayed moderate to good binding affinities, ranging from 137–361 nM at TSPO A147T and 30–85 nM for TSPO WT. However, they generally showed greater discrimination at A147T compared to the isoquinoline and quinazoline carboxamides, indicating that the indole core may not be ideal for achieving balanced affinity at both TSPO isoforms.
The azaindole carboxamide ligands (4a–4f) demonstrated the weakest binding to both TSPO forms, with most ligands exhibiting moderate to poor affinity (ranging from 397 to >10 000 nM at A147T and 182 to >10 000 nM at WT). The exception was ligand 4e, which displayed good binding affinities at both TSPO A147T and WT and was also the most A147T TSPO-preferring compound out of the library (26 nM and 99 nM for A147T and WT, respectively). These results suggest that azaindole is not a promising scaffold for achieving high affinity at both TSPO isoforms.
Several trends emerged from these four series of compounds. The nature of PK-11195's acetamide substitutions were influential in its non-discriminating nature, with incorporation of benzyl-containing substituents driving binding in favour of A147T TSPO within series 1. In addition, the size and nature of PK 11195's heterocycle contributes to the non-discriminating nature of PK 11195, with incorporation of an additional nitrogen into PK 11195's isoquinoline heterocycle to produce a quinazoline, or contraction of the heterocycle to an indole or azaindole all reducing affinity at both TSPO forms and increasing sensitivity to A147T (2f, 3f, 4f).
More generally, adding a nitrogen to the heterocycle was better tolerated at both TSPO isoforms when the nitrogen was incorporated into the isoquinoline core, producing the quinazoline carboxamides (series 1vs.2), compared to the incorporation of nitrogen into the indole ring to create the azaindole series (series 3vs.4). The introduction of a nitrogen atom into the isoquinoline core generally improved binding at WT TSPO, while its effects on A147T affinity were more variable depending on the substituents. Although WT TSPO affinity of both series 3 and 4 were generally poor overall, affinity decreased at both isoforms when moving from the indole to the azaindole series (e.g., 3a > 4a, 3b > 4b, 3c > 4c, 3d > 4d, 3f > 4f), although ligands 3e and 4e were exceptions, with ligand 4e showing improved A147T affinity compared to its indole counterpart.
A further overall trend could be seen when contracting the size of the heterocycle (i.e. series 1vs.3 and 2vs.4). Although this had variable effects on affinity at WT TSPO depending on the nature of the R1R2 substituents, in general it reduced binding at A147T TSPO (with the exception of diethyl-substituted derivatives), suggesting contraction of the heterocycle is a deleterious strategy for removing discrimination between TSPO isoforms.
When considering the impact of the R1R2 alkyl substitution, a clear trend emerged with improved affinity at both A147T and WT TSPO when the N-methoxybenzyl-N-methyl R1R2 substitution was changed to an N-methoxybenzyl-N-ethyl substitution. This beneficial effect was stable across all heterocyclic cores (1d < 1e, 2d < 2e, 3d < 3e, 4d < 4e), although it should be noted that most of these derivatives had only moderate to poor WT TSPO affinity. In addition, changing the N-benzyl-N-methyl R1R2 substituent to an N-benzyl-N-ethyl also improved affinity at A147T TSPO, although had a mixed effect at WT (1a > 1b, 2a > 2b, 3a > 3b, 4a > 4b at A147T TSPO).
In silico studies
Docking studies
Docking studies were used to investigate the structural basis for how the variations to the carboxamide substituents and heterocyclic scaffold influence the Ki observed for both WT and A147T TSPO. For these studies the N-benzyl-N-methyl (ligands 1a, 2a, 3a and 4a) and the N-(R)-sec-butyl-N-methyl series (ligands 2f, 3f and 4f) were chosen to: i) understand why changing the (R)-sec-butyl group of PK 11195 to a benzyl group at the R2 position can either improve affinity (1a and 2a) or reduce affinity (3a and 4a); ii) to examine why altering the heterocyclic core (ligands 2f, 3f and 4f) causes a reduction in affinity. Induced fit docking and MM-GBSA ΔG binding scores (obtained by combining molecular mechanics (MM) terms with a generalised Born and surface area (MM-GBSA) solvent model) are summarised in Table S1 (see ESI†).
PK 11195 and ligand 1a had similar values for WT docking and MM-GBSA calculations, while only A147T docking scores reflect the observed stronger affinity of ligand 1a. The docking and MM-GBSA scores also reflect the observed stronger affinity of ligand 2a when compared to PK 11195, apart from the WT docking scores. When comparing the binding conformation of PK 11195 to ligands 1a and 2a there is a similar orientation of the heterocyclic scaffold (π–π interactions with Trp95 and/or Trp53) for all three compounds (Fig. 2). Unlike the binding conformation of PK 11195, 1a and 2a were able to generate additional π–π interactions due to the benzyl carboxamide substituent. These additional interactions were observed in both the WT and A147T TSPO studies and could provide a rationale for the improved affinity observed experimentally for both ligands, when compared to the corresponding N-(R)-sec-butyl-N-methyl analogues. However, this data did not provide any significant insight into the influence that the acetamide substitutions may have on whether a ligand will discriminate between WT and A147T TSPO.
Fig. 2. Three-dimensional and two-dimensional ligand binding diagrams of the TSPO ligands 1a (A), 2a (B) and PK 11195 (C) into the TSPO A147T. The crystal structure of the TSPO protein from Bacillus cereus (PDB ID: 4RYI) was used as the template for homology modelling.

The docking scores and the MM-GBSA ΔG values of PK 1195, 1a and 2a were comparable to those of the indole (3a/f) and 7-azaindole (4a/f) analogues and reflected the reduced affinity of these ligands. When comparing the binding of PK 11195 to ligands 3f and 4f, a slight variation in heterocyclic scaffold position can be observed for the two ligands. This shift in the heterocyclic core binding position could be the reason for the reduced binding affinity for these two compounds. The need for an optimal heterocyclic core position is further demonstrated when comparing the binding modes of 1a and 2a to 3a and 4a, respectively. The network of π–π interactions (Trp95 and/or Trp53) with the heterocyclic scaffold of 1a and 2a appears to be crucial in orienting the ligands within the predicted binding site, particularly the benzyl group which is potentially the driving force behind the improved affinity of these compounds. For ligands 3a and 4a the heterocyclic core is in a suboptimal binding pose impacting the interactions of the pendant groups, particularly the benzyl group. This observation could provide a rationale for the reduced affinity observed for these ligands. However, further experimental data would be required to support this hypothesis.
Electrostatic potential surface studies
To further rationalise the effect of the carboxamide substituents and heterocyclic scaffold that were observed experimentally, electrostatic potential surfaces were generated for PK 11195, 1a–4a and 1f–4f and are shown in Fig. S1 and S2 (see ESI†). Comparing PK 11195 to 2f shows a similar distribution of charge for the isoquinoline and quinazoline scaffolds, with a slight increase in partial negative charge around the additional nitrogen at the 4-position. This increase in electron density around this position of the scaffold correlates to a 4-fold and 2-fold decrease in affinity to the A147T and WT TSPO respectively for 2f. Interestingly, the increase in electron density of the carboxamide substituent by the substitution of the (R)-sec-butyl group to a benzyl group for 1a and 2a saw an improvement in activity for both heterocyclic scaffolds. As such, maintaining similar heterocyclic core electronics to PK 11195, as well as introducing more electron rich carboxamide substituents is important for improving the binding affinity to WT and A147T TSPO.
The importance of the heterocyclic core electronics for binding affinity is illustrated when comparing the electrostatic potential surface of PK 11195 to 3f/4f, and 1a/2a to 3a/4a. A significant difference can be seen in the electrostatic potential surfaces of indole and 7-azaindole compared to isoquinoline and quinazoline scaffolds. This is reflected by a decrease in electron density adjacent to the 2-position of both the indole and 7-azaindole scaffold. Additionally for 7-azaindole, there is an increase in electron density imparted by the nitrogen at the 7-position. Both changes resulted in a reduced affinity being observed experimentally and correlates well with the observation in the docking studies around the importance of interactions of the heterocyclic scaffold for positioning the compounds within the binding site.
Conclusion
A series of novel PK 11195 derivates were synthesised and evaluated as TSPO WT and A147T ligands. This study was undertaken with the aim of increasing our understanding about how aspects of the pharmacophore influence discrimination between the two TSPO forms and what structural moieties of PK 11195 contribute to its non-discriminating nature. We demonstrated that the size and nature of PK 11195's heterocycle is one determining factor, with incorporation of an additional nitrogen into PK 11195's isoquinoline heterocycle to produce a quinazoline, or contraction of the heterocycle to an indole or azaindole generally reducing affinity at both TSPO forms and increasing sensitivity to A147T (2f, 3f, 4f). In addition, the nature of PK 11195's acetamide substitutions were demonstrated to be influential, with incorporation of benzyl-containing substituents driving binding in favour of A147T TSPO within series 1.
Although some lipophilicity-reducing strategies like contraction of the heterocycle or shortening alkyl chain length reduced affinity, addition of a nitrogen into the isoquinoline scaffold produced ligand 2a, which displayed greater affinity at TSPO A147T and WT than PK 11195 without discriminating between the two TSPO forms. It may therefore be worthwhile to further explore the quinazoline carboxamide series in an effort to identify pharmacophoric elements that may tolerate lipophilicity-reducing substitutions.
Experimental section
General experimental
Unless otherwise stated, all solvents and reagents were purchased and used from commercial sources. Anhydrous solvents were obtained from an Innovative Technology PureSolv7 purification system. Tetrahydrofuran and dichloromethane were dried through an alumina column. N,N-Dimethylformamide was dried through a 5 Å molecular sieve column. All reagents were weighed out under ambient conditions. All reactions were performed under nitrogen or argon, unless otherwise stated.
Melting points using an Optimelt Automated melting point apparatus from Stanford Research Systems were measured in open capillary tubes. IR spectra were recorded neat using a Bruker ALPHA FT-IR spectrometer and peaks are expressed in wavenumbers (cm−1).
Nuclear magnetic resonance spectra were recorded on Bruker Advance DRX 300 or Bruker Advance DRX 400 Ascend spectrometers at 300 or 400 MHz 1H NMR frequency, 75 or 101 MHz 13C NMR frequency and 282 MHz 19F NMR frequency. Proton (H), carbon (C) and fluorine (F) chemical shifts are expressed as parts per million (ppm). All resonances are reported as chemical shift (δ) and are referenced to the solvent residual peak. Multiplicities are reported as follows: s (singlet), br (broad), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), sextet (sext), pentet (p), and m (multiplet). Coupling constants (J) are reported in Hz. The 1H NMR and 13C NMR spectra of all final compounds show the presence of two different rotamers in a 1 : 1 ratio. For the sake of simplicity, the NMR data for only one rotamer are reported.
Elemental microanalysis was obtained either from the Chemical Analysis Facility in the Department of Chemistry and Biomolecular Sciences, Macquarie University, Australia or the School of Human Sciences at London Metropolitan University, England. Low- and high-resolution mass spectra were obtained through electron ionisation (ESI). Low-resolution mass spectra were performed on a Finnigan LCQ mass spectrometer. High-resolution mass spectra were performed on a Bruker 7T Apex Qe Fourier Transform Ion Cyclotron resonance mass spectrometer equipped with an Apollo II ESI/APCI/MALDI dual source.
High performance liquid chromatography (HPLC) was performed on the Waters Alliance 2695 apparatus equipped with Waters 2996 photodiode array detector, set at 254 nm. Separation using a SunFireTM C18 column (5 μm, 2.1 × 150 mm) was achieved using water (solvent A) and acetonitrile (solvent B) at a flow rate of 0.2 mL min−1. The method consisted of 0% B to 100% B over 30 minutes. Data acquisition and processing was performed with the Waters Empower 2 software. HPLC data is recorded as percentage purity and retention time (τR) in minutes.
Detailed synthetic procedures are listed in the ESI.†
General biological methods
Cell culture and membrane preparation
Human embryonic kidney 293 cells (HEK-293 cells) stably transfected with the human WT or A147T TSPO were cultured in Dulbecco's modified Eagle medium (DMEM) with the addition of 10% fetal bovine serum (FBS; Gibco), 2 mM l-glutamine and 100 U penicillin/streptomycin at 37 °C. The establishment and characterisation of these cells are described in Sokias et al.5 WT and A147T TSPO cells were harvested for membrane preparation using 0.04% EDTA in PBS for 10 min at 37 °C and were then centrifuged at 1000g for 5 min. Supernatants were pooled in to 5 mM Tris HCl (pH 7.4) and homogenised with a hand-held Ultra-Turrax homogeniser (IKA Werke; 3 × 10 s pulses). Cells were centrifuged three times with an Avanti J-E ultracentrifuge (48 000g, 15 min, 4 °C) and resuspended in 50 mM Tris HCl after each spin. A bicinchoninic acid (BCA) protein assay (Life Technologies) was conducted according to the manufacturer's protocol, and plates were read on a POLARstar Omega plate reader (BMG Labtech) to quantify the total protein concentration in the membrane preparations. Values from the BCA protein analysis were plotted with Graphpad Prism 7 to interpolate total protein concentrations of unknown samples from a non-linear regression curve with a 4-parameter logistic fit.
Competition radioligand binding assay
[3H]PK 11195 at ∼Kd concentration (10 nM; Perkin Elmer) and logarithmically-spaced concentrations of the tested compounds (0.1–10 000 nM) in binding buffer (50 mM Tris HCl, pH 7.4) were added to 96-well U-well microplates. Binding buffer containing 0.1% DMSO served as a vehicle control for calculation of total binding. A negative control containing a saturating concentration (1 μM) of unlabelled PK 11195 was included for calculation of non-specific binding. WT (20 μg per well) and A147T (5 μg per well) TSPO membranes were then added to the tritiated ligand and compounds and incubated for 90 min at 4 °C. The mixture was vacuum harvested onto a glass-fibre filter plate (Millipore) before being washed with binding buffer (4 °C) using a vacuum cell harvester (Brandel). The filters were dried overnight before adding Microscint-0 scintillation liquid (Perkin Elmer) and counts per minute were collected from a MicroBeta2 scintillation counter (Perkin Elmer). Non-specific binding values were subtracted from total counts per minute to calculate specific binding, then specific binding in wells with compounds was normalised to the specific binding in wells containing 0.1% DMSO control. These values were then analysed with a one-site competitive binding analysis in GraphPad to determine the inhibition constant (Ki). Data are expressed as mean ± standard deviation, representing at least three biological replicates, each performed in duplicate.
In silico studies
Protein and ligand preparation
The WT and A147T TSPO homology models generated in our previous study,15 were prepared using preparation and refinement protocols, directed by the Protein Preparation Wizard26 embedded in Maestro v12.8 (Schrödinger, LLC, New York, USA). This process includes assigning bond orders, adding hydrogen atoms, and creating zero order bonds to metals and disulphide bonds. The hydrogen bond network within the protein was also optimised with all het groups within the receptor grid bounding box previously removed and the protein structure minimised to a root mean square deviation (RMSD) of 0.3 Å using the OPLS4 force field.26,27 All ligands were prepared using the LigPrep module to generate all potential ionisation states at pH 7 ± 2.
Induced fit docking studies
Selected ligands were docked into the receptor with Induced Fit Docking28 with default settings. Standard protocol and OPLS4 force field27 was used for the calculations. Box centre was set to encompass the identified PK 1195 binding site of TSPO.29 Receptor van der Waals scaling and Ligand van der Waals scaling was set to 0.50. Residues within 20 Å of ligand poses were refined, with optimisation of side chains. Ligands were redocked with Extra Precision (XP)30 to refine binding energy estimates. All ligands were docked with flexible states to allow sampling of the effect of nitrogen inversion, changing ring conformations and non-planar amide functional groups were penalised.
Prime MM-GBSA calculations,31 which combines molecular mechanics (MM) terms, and a generalised Born and surface area (GBSA) solvent model,32 were utilised to calculate the free energy of binding for the ligands. The output poses from Induced Fit docking were used as the basis for these calculations. The calculations were performed using the variable-dielectric generalised Born (VSGB)33 solvation model and the OPLS4 force field. The docked ligand and protein residues within 20 Å of the ligand were allowed to be flexible, with all other atoms remaining rigid.
Electrostatic potential surfaces
Electrostatic potential surfaces were generated for ligands of interest in Maestro v12.8 (Schrödinger, LLC, New York, USA) and Spark™ v10.5.6 (Cresset®, Litlington, Cambridgeshire, UK).
Data availability
Data supporting this article have been included as part of the ESI.†
Author contributions
Conceptualisation was conducted by M. K., R. S., E. L. W., D. E. H. and A. P. M. Funding acquisition was conducted by M. K. Investigation was conducted by R. S., A. Z., E. L. W., A. P. M., M. A. S., A. D. J. M., and G. A. C. Formal analysis was conducted by R. S., A. Z., E. L. W., A. P. M., M. A. S., A. D. J. M., and G. A. C. Project administration was conducted by E. L. W. and A. P. M. Resources were contributed by D. E. H. Supervision was conducted by E. L. W., A. P. M., J. J. D., D. E. H. and M. K. Writing the original draft was conducted by R. S., E. L. W. and A. P. M. Writing (reviewing and editing) was conducted by A. P. M., E. L. W., B. M. A., J. J. D., and M. K.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work is supported by the National Health and Medical Research Council of Australia (NHMRC) M. K. is an NHMRC Principal Research Fellow (APP1154692). J. J. D. is an NHMRC Emerging Leadership Fellow (GNT2008066).
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00798k
References
- Lacapere J. J. Duma L. Finet S. Kassiou M. Papadopoulos V. Trends Pharmacol. Sci. 2020;41:110–122. doi: 10.1016/j.tips.2019.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno S. Viviano M. Baglini E. Poggetti V. Giorgini D. Castagnoli J. Barresi E. Castellano S. Da Settimo F. Taliani S. Molecules. 2024;29:4212. doi: 10.3390/molecules29174212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumbers G. A. Harvey-Latham E. D. Kassiou M. Werry E. L. Danon J. J. Semin. Nucl. Med. 2024;54:856–874. doi: 10.1053/j.semnuclmed.2024.09.007. [DOI] [PubMed] [Google Scholar]
- Owen D. R. Guo Q. Rabiner E. A. Gunn R. N. Clin. Transl. Imaging. 2015;3:417–422. [Google Scholar]
- Sokias R. Werry E. L. Chua S. W. Reekie T. A. Munoz L. Wong E. C. N. Ittner L. M. Kassiou M. MedChemComm. 2017;8:202–210. doi: 10.1039/c6md00523c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano S. Taliani S. Milite C. Pugliesi I. Da Pozzo E. Rizzetto E. Bendinelli S. Costa B. Cosconati S. Greco G. Novellino E. Sbardella G. Stefancich G. Martini C. Da Settimo F. J. Med. Chem. 2012;55:4506–4510. doi: 10.1021/jm201703k. [DOI] [PubMed] [Google Scholar]
- Castellano S. Taliani S. Viviano M. Milite C. Da Pozzo E. Costa B. Barresi E. Bruno A. Cosconati S. Marinelli L. Greco G. Novellino E. Sbardella G. Da Settimo F. Martini C. J. Med. Chem. 2014;57:2413–2428. doi: 10.1021/jm401721h. [DOI] [PubMed] [Google Scholar]
- Owen D. R. Gunn R. N. Rabiner E. A. Bennacef I. Fujita M. Kreisl W. C. Innis R. B. Pike V. W. Reynolds R. Matthews P. M. Parker C. A. J. Nucl. Med. 2011;52:24–32. doi: 10.2967/jnumed.110.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werry E. L. Bright F. M. Piguet O. Ittner L. M. Halliday G. M. Hodges J. R. Kiernan M. C. Loy C. T. Kril J. J. Kassiou M. Int. J. Mol. Sci. 2019;20:3161. doi: 10.3390/ijms20133161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti-Fregonara P. Zhang Y. Jenko K. J. Gladding R. L. Zoghbi S. S. Fujita M. Sbardella G. Castellano S. Taliani S. Martini C. Innis R. B. Da Settimo F. Pike V. W. ACS Chem. Neurosci. 2014;5:963–971. doi: 10.1021/cn500138n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeon F. G. Lee J. H. Morse C. L. Stukes I. Zoghbi S. S. Manly L. S. Liow J. S. Gladding R. L. Dick R. M. Yan X. Taliani S. Costa B. Martini C. Da Settimo F. Castellano S. Innis R. B. Pike V. W. J. Med. Chem. 2021;64:16731–16745. doi: 10.1021/acs.jmedchem.1c01562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H. Simeon F. G. Liow J. S. Morse C. L. Gladding R. L. Santamaria J. A. M. Henter I. D. Zoghbi S. S. Pike V. W. Innis R. B. J. Nucl. Med. 2022;63:1252–1258. doi: 10.2967/jnumed.121.263168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa M. Lohith T. G. Shrestha S. Telu S. Zoghbi S. S. Castellano S. Taliani S. Da Settimo F. Fujita M. Pike V. W. Innis R. B. T. Biomarkers Consortium Radioligand Project J. Nucl. Med. 2017;58:320–325. doi: 10.2967/jnumed.116.178996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H. Denora N. Laquintana V. Mangiatordi G. F. Lopedota A. Lopalco A. Cutrignelli A. Franco M. Delre P. Song I. H. Kim H. W. Kim S. B. Park H. S. Kim K. Lee S. Y. Youn H. Lee B. C. Kim S. E. Eur. J. Nucl. Med. Mol. Imaging. 2021;49:110–124. doi: 10.1007/s00259-021-05617-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H. W. A. Sokias R. Werry E. L. Ittner L. M. Reekie T. A. Du J. Gao Q. Hibbs D. E. Kassiou M. J. Med. Chem. 2019;62:8235–8248. doi: 10.1021/acs.jmedchem.9b00980. [DOI] [PubMed] [Google Scholar]
- Sokias R. Werry E. L. Alison Cheng H. W. Lloyd J. H. Sohler G. Danon J. J. Montgomery A. P. Du J. J. Gao Q. Hibbs D. E. Ittner L. M. Reekie T. A. Kassiou M. Eur. J. Med. Chem. 2020;207:112725. doi: 10.1016/j.ejmech.2020.112725. [DOI] [PubMed] [Google Scholar]
- Vo S. V. Banister S. D. Freelander I. Werry E. L. Reekie T. A. Ittner L. M. Kassiou M. RSC Med. Chem. 2020;11:511–517. doi: 10.1039/c9md00580c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callis T. B. Garrett T. R. Montgomery A. P. Danon J. J. Kassiou M. J. Med. Chem. 2022;65:13483–13504. doi: 10.1021/acs.jmedchem.2c00969. [DOI] [PubMed] [Google Scholar]
- Jafari E. Khajouei M. R. Hassanzadeh F. Hakimelahi G. H. Khodarahmi G. A. Res. Pharm. Sci. 2016;11:1–14. [PMC free article] [PubMed] [Google Scholar]
- Chadha N. Silakari O. Eur. J. Med. Chem. 2017;134:159–184. doi: 10.1016/j.ejmech.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Saini T. Kumar S. Narasimhan B. Cent. Nerv. Syst. Agents Med. Chem. 2015;16:19–28. doi: 10.2174/1871524915666150608103224. [DOI] [PubMed] [Google Scholar]
- McLaughlin M. Palucki M. Davies I. W. Org. Lett. 2006;8:3307–3310. doi: 10.1021/ol061232r. [DOI] [PubMed] [Google Scholar]
- Merour J. Y. Buron F. Ple K. Bonnet P. Routier S. Molecules. 2014;19:19935–19979. doi: 10.3390/molecules191219935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson L. Pimlott S. L. Sutherland A. Tetrahedron Lett. 2007;48:7137–7139. [Google Scholar]
- Shah F. Hume S. P. Pike V. W. Ashworth S. McDermott J. Nucl. Med. Biol. 1994;21:573–581. doi: 10.1016/0969-8051(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Sastry G. M. Adzhigirey M. Day T. Annabhimoju R. Sherman W. J. Comput.-Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Lu C. Wu C. Ghoreishi D. Chen W. Wang L. Damm W. Ross G. A. Dahlgren M. K. Russell E. Von Bargen C. D. Abel R. Friesner R. A. Harder E. D. J. Chem. Theory Comput. 2021;17:4291–4300. doi: 10.1021/acs.jctc.1c00302. [DOI] [PubMed] [Google Scholar]
- Sherman W. Day T. Jacobson M. P. Friesner R. A. Farid R. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Jaremko L. Jaremko M. Giller K. Becker S. Zweckstetter M. Science. 2014;343:1363–1366. doi: 10.1126/science.1248725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A. Murphy R. B. Repasky M. P. Frye L. L. Greenwood J. R. Halgren T. A. Sanschagrin P. C. Mainz D. T. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P. Friesner R. A. Xiang Z. Honig B. J. Mol. Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Lyne P. D. Lamb M. L. Saeh J. C. J. Med. Chem. 2006;49:4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- Li J. Abel R. Zhu K. Cao Y. Zhao S. Friesner R. A. Proteins. 2011;79:2794–2812. doi: 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting this article have been included as part of the ESI.†