

Abstract
Pore-forming toxins (PFT) are the largest class of bacterial toxins, and contribute to virulence by triggering host cell death. Vertebrates also express endogenous pore-forming proteins that induce cell death as part of host defense. To mitigate damage and promote survival, cells mobilize membrane repair mechanisms to neutralize and counteract pores, but how these pathways are activated is poorly understood. Here we use a transposon-based gene activation screen to discover pathways that counteract the cytotoxicity of the archetypal PFT Staphylococcus aureus α-toxin. We identify the endo-lysosomal protein LITAF as a mediator of cellular resistance to PFT-induced cell death that is active against both bacterial toxins and the endogenous pore, Gasdermin D, a terminal effector of pyroptosis. Activation of the ubiquitin ligase NEDD4 by potassium efflux mobilizes LITAF to recruit the endosomal sorting complexes required for transport (ESCRT) machinery to repair damaged membrane. Cells lacking LITAF, or carrying naturally occurring disease-associated mutations of LITAF, are highly susceptible to pore-induced death. Notably, LITAF-mediated repair occurs at endosomal membranes, resulting in expulsion of damaged membranes as exosomes, rather than through direct excision of pores from the surface plasma membrane. These results identify LITAF as a key effector that links sensing of cellular damage to repair.
One sentence summary:
The endosomal protein LITAF links membrane damage caused by toxins and Gasdermin D to ESCRT-mediated repair, promoting cell survival.
INTRODUCTION
Pore formation in cellular membranes is utilized by many microbes to exert cytotoxic effects, and pore-forming toxins (PFT) make up the largest class of bacterial toxins (1). During infection, disruption of the plasma membrane by PFTs triggers cell death, breaching epithelial and endothelial barriers. PFTs also target and kill immune cells, subverting the immune response to allow pathogen persistence and growth, and can function at intracellular membranes, disrupting phagosomes and lysosomes, allowing internalized bacteria to persist or escape (2). Recently, it was identified that activation of cellular stress pathways or inflammasomes triggers the assembly of endogenous membrane pores, such as Gasdermin D (GSDMD) (3–5). These endogenous pores also cause cell death and allow the release of cytokines and alarmins such as IL-1β (6) to promote inflammation and immune responses. Despite our increasing knowledge of the critical role that membrane pores play in bacterial pathogenesis and triggering inflammation, exactly how host cells defend themselves against these toxins, and why certain cells and tissues show differential sensitivity to their effects, remain poorly understood.
Although pore-forming proteins can cause direct lysis of cells by osmotic shock when present at high concentrations, pore formation more commonly induces cell death indirectly, via activation of programmed cell death pathways. As an example, K+ efflux induced by the archetypal small PFT Staphylococcus aureus α-toxin (also known as α-hemolysin or Hla) causes activation of caspases, resulting in apoptosis or pyroptosis (7–9). Inflammatory cell death pathways, such as pyroptosis and necroptosis, involve activation of endogenous membrane pores including Gasdermin D and MLKL, which act to amplify ion flux and facilitate cell death (3, 10). To counteract the effects of membrane pores, cells can sense membrane disruption by flux of Ca2+ and K+ ions, which trigger membrane repair mechanisms including blocking of pores, shedding or internalization of damaged membrane, and delivery of vesicles to patch larger membrane breaches (11, 12). Loss of membrane repair mechanisms increases sensitivity to cell death induced by membrane damage and endogenous pores (13–19). Conversely, activation of repair mechanisms can prevent cell death and induce resistance to PFTs (20). Hence cell death appears to be a ‘backup’ pathway activated when membrane repair fails, and the opposing outcomes of membrane damage, repair, and survival, versus death and inflammation, exist in equilibrium.
Here we carried out a forward genetic screen to identify mechanisms of host cell resistance to PFTs. We used a transposon mutagenesis approach designed to activate gene expression (21) to selectively screen for genes that confer resistance to S. aureus α-toxin, and identified the gene LITAF as a mediator of protection from α-toxin-induced cell death. We show that LITAF promotes membrane repair and secretion of α-toxin-containing membranes through the activation of ESCRT machinery. We find that LITAF-mediated membrane repair proceeds through sequestration of α-toxin in intracellular multivesicular vesicles, followed by secretion as exosomes, and is distinct from recently described ESCRT-mediated repair mechanisms which act directly at the plasma membrane (13–18). This mechanism is activated by membrane K+ flux, via the ubiquitin ligase NEDD4, and is effective against other PFTs and the endogenous pore Gasdermin D. Hence, we identify LITAF as a mediator of membrane repair, linking damage sensing to membrane reorganization and turnover.
RESULTS
S. aureus alpha-toxin induces K+ efflux and programmed cell death in U2OS cells
We focused initially on cellular responses to the archetypal PFT, Staphylococcus aureus α-toxin (also known as α-hemolysin or Hla). α-toxin forms heptameric membrane pores of 2–3 nm that allow diffusion of K+ and other monovalent ions (fig. S1A), and at low doses, triggers cell death through apoptosis or pyroptosis, depending on the cell type targeted (7–9). For these studies we selected the osteosarcoma cell line, U2OS, which is highly susceptible to α-toxin. α-toxin caused rapid and complete cell death, with loss of membrane integrity beginning 2 hours after exposure (Fig 1A), and death was prevented if cells were cultured in high KCl medium to buffer intracellular [K+] and prevent K+ efflux (fig. S1B). α-toxin-induced death was associated with activation of caspase 3/7 (Fig 1B), which preceded loss of membrane integrity, and cell death could be blocked by addition of the pan-caspase inhibitors QVD and ZVAD, but not the Capsase 1 inhibitor YVAD (Fig 1A). Hence, α-toxin-induced cell death in U2OS cells occurs through a caspase-dependent apoptosis mechanism in response to K+ efflux, in agreement with previous studies (22, 23), and not through Caspase 1-mediated pyroptosis as has been reported for immune cells (8).
Figure 1: Genome-wide identification of genes involved in resistance to α-toxin.
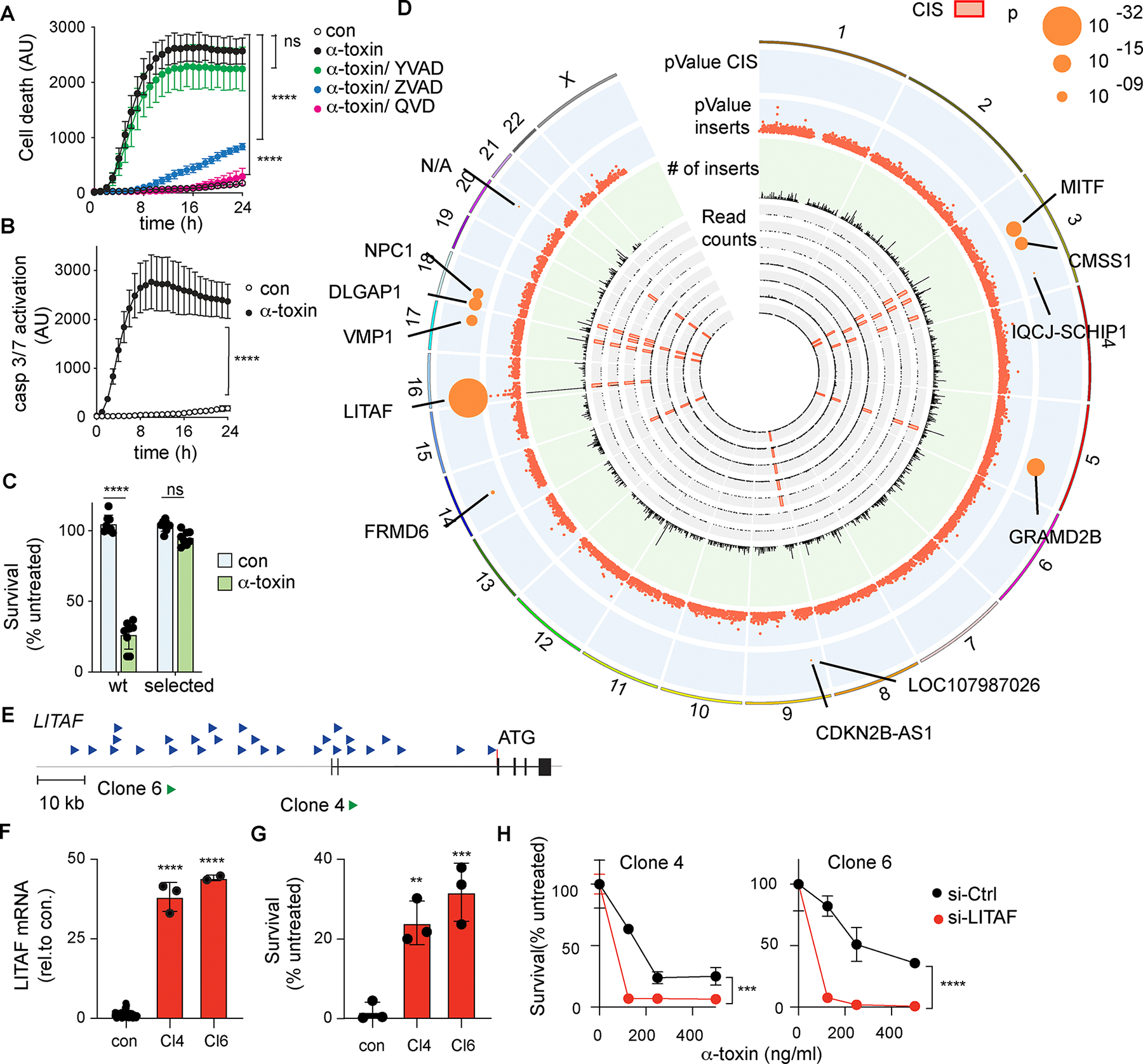
(A) Time course of cell death (yoyo3 incorporation), in control U2OS cells (con), cells treated with α-toxin alone, or α-toxin with the caspase 1 inhibitor YVAD, or the pan-caspase inhibitors QVD and ZVAD (n=3 replicate wells/condition), representative of three independent experiments. (B) Time course of Caspase3/7 activation in control U2OS cells (con) or after treatment with α-toxin (500ng/ml) (n=3 replicate wells/condition), representative of three independent experiments. (C) Survival of non-mutagenized parental U2OS cells and pools of puromycin-selected cells with and without α-toxin (n = 8 replicate wells/condition), combined from two independent experiments. (D) Genome distribution of transposon insertion sites in resistant pools from 7 independent screens. Inner circles (grey) show cumulative transposon insertions in 50 Mb windows from each independent library and locations of common insertion sites (CIS). The middle circle (green) shows numbers of cumulative transposon insertions in 50 Mb windows from all libraries. Outer circles (blue) show −log10 p-values and sites of high-confidence CIS, with predicted target genes. p-values are calculated as described in materials and methods. (E) Location and orientation of transposon insertions close to LITAF in all screens (upper, blue arrows), and in 2 isolated clones selected for further analysis (lower, green arrows). (F) Expression of LITAF mRNA in clones 4 (Cl4) and 6 (Cl6), compared with non-mutagenized U2OS cells (con) (n ≥ 2 replicate cultures). (G) Survival of clones 4 and 6, or non-mutagenized U2OS cells (con), after treatment with α-toxin (500 ng/ml, 24h) (n=3 replicate wells/condition), representative of three independent experiments. (H) Survival of clones 4 and 6 transfected with siRNA against LITAF (si-LITAF) or control si-RNA (si-Ctrl) and treated with α-toxin for 24h (n=2 replicate wells/condition), representative of three independent experiments. Data expressed as mean ± SD. ** p < 0.01, *** p < 0.001, **** p <0.0001, Unpaired Student’s t test (A,B,F), one-way ANOVA (G) or two-way ANOVA (C,H).
A transposon mutagenesis screen identifies genes involved in resistance to α-toxin-induced death
To identify cell intrinsic pathways of resistance to pore-induced cell death, we used a transposon mutagenesis forward genetic screen. This approach leverages a PiggyBac transposon to stimulate or disrupt the expression of neighboring genes, allowing interrogation of both gene activation and inactivation in a single screen (21, 24) (fig. S1C–F). We generated libraries of transposon mutagenized U2OS cells consisting of approximately 107 independently targeted cells. To screen for mutations that induce resistance to α-toxin, transposon-mutagenized libraries were treated with α-toxin at doses that kill non-mutagenized cells, and cells that survived α-toxin treatment were expanded and re-challenged to confirm that they had acquired stable resistance (Fig 1C). Transposon insertion sites from resistant cells were sequenced, mapped to the genome and genomic regions were identified with high numbers of transposon insertions (‘Common Insertion Sites’ or CIS), which represent likely sites of mutations that confer resistance (25). Combining analysis from 7 independent screens identified 17 high confidence CIS, and a further 24 CIS that passed the p<0.05 significance cut off. (Fig. 1D, Table 1).
The highest confidence CIS identified (p < 10−32) was located close to the gene LITAF. All insertions were in the same orientation as the gene, and directly upstream of the first coding exon (Fig 1E), consistent with increased expression of the gene. To confirm the effects of the transposon insertions, 24 independent cell clones from an α-toxin-resistant pool were isolated, sequenced to identify transposon insertions and rechallenged with α-toxin to confirm resistance (fig. S2A,B).
Many clones contained insertions within identified CIS, including multiple clones with insertions upstream of the LITAF ATG (fig. S2B). We analyzed expression of target genes in two selected clones with insertions in LITAF. Both showed increased expression of LITAF (around 40-fold; Fig 1F), as predicted by the insertion position, and increased resistance to toxin-induced cell death (Fig 1G). Furthermore, knockdown of LITAF expression in these clones restored sensitivity to α-toxin, demonstrating that increased expression by transposon insertion was responsible for resistance (Fig 1H). Similarly, clones with insertions in two other CIS (fig. S2C) also showed increased expression of genes downstream of their respective transposon insertions, GRAMD2B and its homolog GRAMD1B (figs. S2, D and F), corresponding with increased resistance to α-toxin (figs. S2, E and G). As we observed for LITAF, knockdown of these transposon target genes in their respective clones restored sensitivity to α-toxin (figs. S2, H and I). Thus, transposon mutagenesis could identify multiple high confidence genes or genomic loci able to confer resistance to α-toxin through gene activation.
LITAF is necessary and sufficient to promote survival against α-toxin.
We focused on determining the mechanism of action of LITAF, which had the most independent insertions and the most significant CIS p-value. We first tested whether increased expression of LITAF was sufficient to induce resistance to α-toxin. Overexpression of LITAF increased survival after treatment with α-toxin and increased the effective LD50 2.5-fold (Fig 2A,B). Similar protective effects were seen if LITAF was expressed stably, or if expression was induced just prior to toxin treatment (Fig 2C,D). Conversely, knockdown of endogenous LITAF in wild-type U2OS cells further increased their sensitivity to α-toxin (Fig 2E,F). To determine whether this property of LITAF was limited to U2OS cells, we tested additional human cell lines for their expression of LITAF and sensitivity to α-toxin. LITAF expression was inversely correlated with sensitivity to α-toxin (R2=0.79) in a range of cell lines (Fig 2G). Over-expression of LITAF in the α-toxin-sensitive cell lines HepG2, HEC1B, HEK293T and HT29 also increased cell survival (Fig 2H). Furthermore, CRISPR knockout of LITAF in the α-toxin-resistant cell line A549 increased sensitivity to α-toxin, demonstrating that expression of high levels of endogenous LITAF was associated with increased resistance (Fig 2I). Hence, LITAF is both necessary and sufficient for resistance to α-toxin-induced programmed cell death in multiple cell types.
Figure 2: LITAF is necessary and sufficient to protect cells against cell death induced by alpha-toxin.
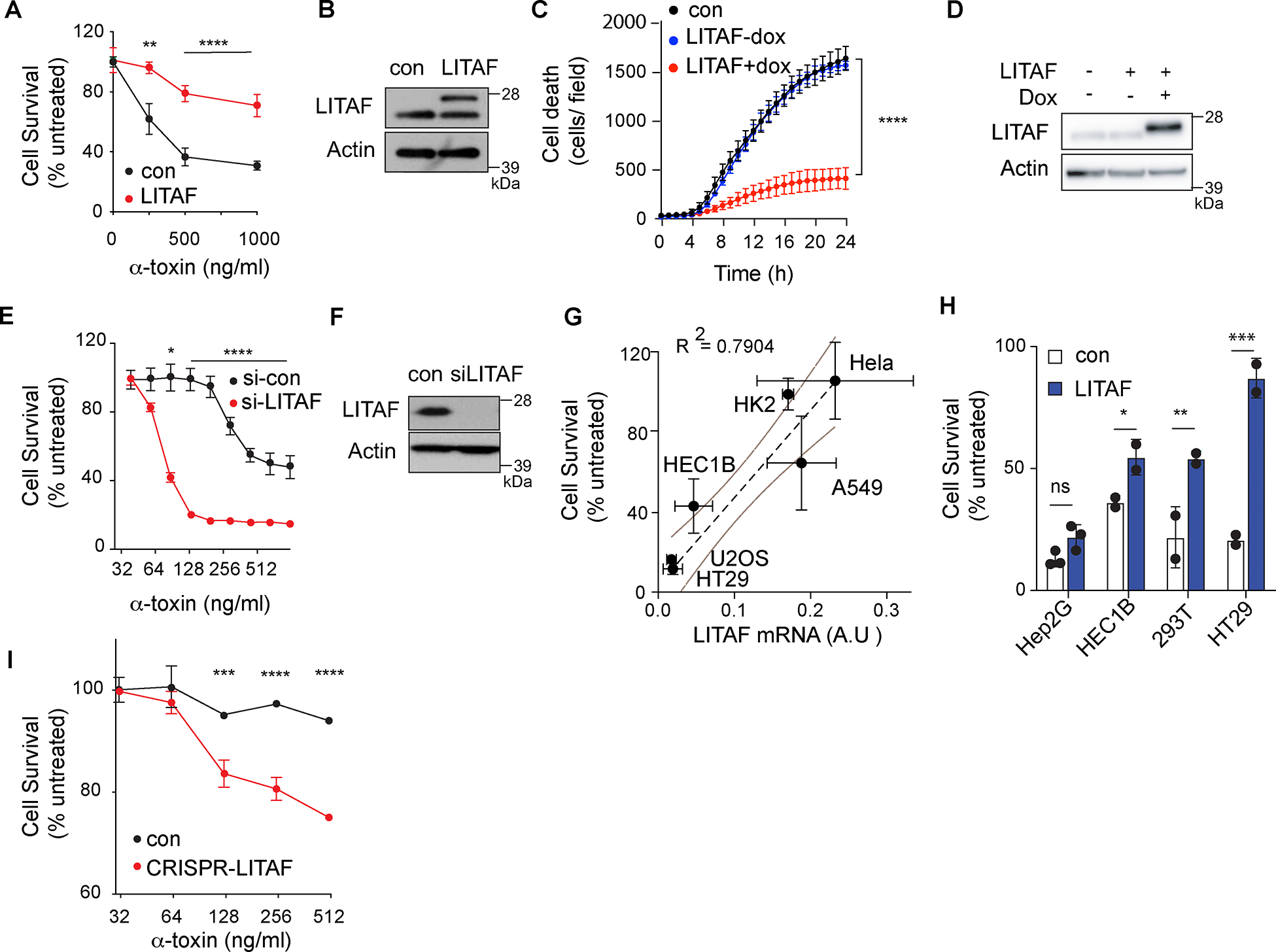
(A,B) Survival 24h after α-toxin treatment (A) and LITAF expression (B) in U2OS cells expressing LITAF-MycFlag (LITAF) or a control plasmid (con). Blot shows endogenous LITAF (lower band) and LITAF-MycFlag (upper band). (n≥6 replicate wells/condition), combined from three independent experiments. (C) Cell death (Sytox Green) following treatment of U2OS cells expressing doxycycline (dox)-inducible LITAF-MycFlag with 500 ng/ml α-toxin with or without doxycycline. (n=8 replicate wells/condition), representative of three independent experiments. (D) LITAF expression after doxycycline treatment, representative of three independent experiments. (E,F) Survival 24h after α-toxin treatment (E) and LITAF expression (F) in U2OS cells with siRNA targeting LITAF (si-LITAF) or a non-targeting control (si-Ctrl). (n=3 replicate wells/condition), representative of three independent experiments. (G) Cell survival after treatment with 500ng/ml of α-toxin plotted against expression of LITAF mRNA (expressed in arbitrary units) for indicated cell lines (n ≥ 2 replicates) representative of three experiments. Dashed line shows linear correlation with 95% confidence intervals (Simple linear regression). (H) Survival 24h after treatment with 500ng/ml α-toxin in indicated cell lines expressing LITAF or control plasmid. (n≥2 replicate wells/condition), representative of three independent experiments. (I) Survival 24h after α-toxin treatment of LITAF-knockout A549 cells (CRISPR-LITAF) or controls with non-targeting gRNA (control). (n=2 replicate wells/condition), representative of three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p <0.0001, two-way ANOVA (A,C,E,H,I).
LITAF promotes resistance to bacterial and endogenous pores through membrane repair
To understand how LITAF increased resistance, we first set out to establish whether LITAF affected α-toxin assembly or function. LITAF did not affect expression of the α-toxin receptor ADAM10 (fig. S3A), which promotes pore formation in the plasma membrane (26). Furthermore, we observed no difference in induction of p38 MAPK phosphorylation following α-toxin treatment between control and LITAF-expressing cells (fig. S3B). MAPK activation is characteristic of the rapid stress response triggered by α-toxin pores(27), and we therefore concluded that overexpression of LITAF does not prevent the assembly of α-toxin pores in the cell membrane and initial K+ efflux.
LITAF, also known as Small Integral Protein of Lysosome/Late Endosome (SIMPLE), is a membrane-associated protein found in endosomes, lysosomes and the cell surface membrane (28, 29). LITAF associates tightly with the cytoplasmic face of cell membranes through an amphipathic helix structure (‘LITAF domain’) (30), and is known to interact with the ESCRT machinery to promote extracellular vesicle (EV) formation (31). ESCRT-mediated removal of damaged membrane as EVs has been shown to promote repair of membranes containing pore-forming toxins, suggesting that LITAF may promote ESCRT-mediated membrane repair (15). To test this, we analyzed EV fractions from the conditioned media of LITAF-overexpressing and control cells after treatment with α-toxin. LITAF over-expression increased production of EV after α-toxin treatment (Fig. 3, A and B) and EVs contained α-toxin (Fig. 3C), supporting a role for LITAF in promoting EV production for membrane repair. Furthermore, EV contained LITAF, and the ESCRT components TSG101 and ALIX, but did not contain the endosomal protein EEA1 which is selectively excluded from EV, confirming that these were not simply cellular fragments or debris due to cell death (Fig. 3C). Notably, LITAF over-expression did not affect basal levels of EV (Fig. 3B), and ESCRT components and LITAF were only detected in EV after α-toxin treatment (Fig. 3C), demonstrating that LITAF-mediated EV production was triggered in response to α-toxin. These data were therefore consistent with LITAF promoting membrane repair through EV production.
Figure 3: LITAF promotes Extracellular Vesicle production and α-toxin trafficking to multivesicular bodies.

(A-B) Quantification of exosomes secreted by wild type and LITAF-expressing U2OS cells with and without treatment with α-toxin (500 ng/ml, 2 hrs). (A) Representative histogram of CD9 fluorescence. (B) Exosome production expressed as percentage of α-toxin-treated wild-type U2OS cells. (n = 5 replicate / condition), combined from five independent experiments. **, p<0.01; one-way ANOVA. (C) Representative immunoblot of exosome fractions stained for α-toxin, Tsg101, Alix, LITAF and EEA1. Bottom panel shows immunoblot of equivalent cell lysates stained for EEA1 and actin. (D) Representative confocal images of GFP-LITAF (green) and nucleus (white) in U2OS cells, untreated or following α-toxin treatment (500 ng/ml, 1h). (E) Relative numbers of U2OS cells untreated or α-toxin treated as in (D) with LITAF localized predominantly to the plasma membrane or predominantly in internal vesicles from analysis of ≥ 100 cells per condition. Mean ± SD of n≥3 replicate wells/condition, representative of three independent experiments (F) Representative confocal images of U2OS cells expressing GFP-LITAF and RFP-Rab5, untreated or treated with α-toxin (500 ng/ml, 1h) and stained for CD63. Regions within dotted boxes are enlarged in adjacent images. (G) Pixel intensity of indicated stains along the dotted line in (F) (H) Co-localization (Pearson’s correlation) of LITAF and Rab5, or LITAF and CD63, in GFP-LITAF-expressing U2OS cells untreated or treated with α-toxin (500 ng/ml, 1h). Each point represents a single image of n≥6 replicate images/condition, combined from four independent experiments. (I) Representative confocal images of U2OS cells treated with α-toxin (500 ng/ml, 1h) and stained for α-toxin, CD63 and LBPA. Regions within dotted boxes are enlarged in adjacent images. (J) Pixel intensity of indicated stains along the dotted lines in enlarged panels in (I). (K) Co-localization (Pearson’s correlation) of α-toxin and CD63, or α-toxin and LBPA in control and LITAF-expressing U2OS cells following treatment with α-toxin as in (E). Each point represents a single image of n≥6 replicate images/condition, representative of three independent experiments. (L) Representative confocal images of α-toxin-treated U2OS cells stained for LITAF and α-toxin. Region in dotted box is enlarged in adjacent images (M) Pixel intensity of indicated stains along the dotted line in (L) Scale bar in all images is 10 μm. * p < 0.05, ** p < 0.01, **** p <0.0001, Unpaired Student’s t test (H,K) or two-way ANOVA (E).
LITAF promotes intracellular traffic of α-toxin to multivesicular bodies.
ESCRT-mediated repair of membranes damaged by toxins or GSDMD has been reported to occur by direct shedding of EV from the plasma membrane (14, 15, 18). In unchallenged cells, LITAF localized at the plasma membrane, as well as in small and intermediate-sized Rab5+ vacuoles, consistent with previous reports (28, 29, 32) (Fig. 3D–H). However, treatment with α-toxin did not increase localization of LITAF to the plasma membrane as would be expected if it was acting to promote EV shedding. Instead, LITAF re-localized away from the plasma membrane within 30 minutes of α-toxin treatment, and accumulated in large CD63+ intracellular vacuoles (Fig. 3D–H). α-toxin was also rapidly internalized by U2OS cells and accumulated in CD63+ vacuoles (Fig. 3I–K) in agreement with previous studies in other cell types (33). In addition to its role in producing EV that are shed directly from the cell surface (termed ‘ectosomes’), ESCRT can be recruited to intracellular CD63+ vacuoles, where it generates multivesicular bodies (MVB) containing intraluminal vesicles that are ultimately secreted as exosomes. We therefore speculated that LITAF may promote membrane repair through MVBs, rather than at the plasma membrane as has been described in other settings. Visualization of ESCRT by expression of CHMP4B-GFP confirmed that ESCRT accumulated in large multivesicular vacuoles after α-toxin treatment (movie S1, Fig S3C). Furthermore, consistent with this model, α-toxin was often positioned inside CD63+ vacuoles, rather than on the vacuole membrane (Fig. 3, I and J) and closely co-localized with lysobisphosphatidic acid (LBPA), an unconventional phospholipid that is enriched in intraluminal vesicles. LITAF expression significantly increased the accumulation of α-toxin within these LBPA+ intraluminal vesicles (Fig. 3L), and LITAF could be detected co-localized with α-toxin in intracellular puncta (Fig. 3M). These imaging data suggested that LITAF activated ESCRT-mediated membrane repair, but that this may not occur through direct ESCRT activation at the plasma membrane.
LITAF promotes resistance by endosomal activation of ESCRT.
To test whether LITAF-mediated repair was distinct from described mechanisms of ectosome shedding from the plasma membrane, we tested specific components of the ESCRT activation and vesicle formation machinery. ESCRT assembly and function occurs through the sequential action of various complexes and accessory proteins, which mediate recognition of target proteins and membranes (termed ESCRT-0 and ESCRT-I components), initiate ESCRT assembly (ESCRT-II) and finally execute membrane remodeling and scission (ESCRT-III)(13). Treatment with siRNA against the ESCRT-III component CHMP4B, which is required for all ESCRT activity, completely reversed resistance to α-toxin in LITAF-expressing cells (Fig. 4A), confirming that LITAF acted through ESCRT. The requirement for ESCRT-0 and ESCRT-I components varies depending on the mechanisms and sub-cellular location of ESCRT activation. Of note, membrane repair at the cell surface occurs through direct activation of the ESCRT-I component TSG101 independently of ESCRT-0 components. However, we found that siRNA against the ESCRT-0 component STAM1 inhibited LITAF-mediated repair and resistance to α-toxin (Fig 4B), consistent with previous reports that LITAF activation of ESCRT at endosomal membranes requires STAM1 and the ESCRT-0 complex (34). To further confirm that LITAF-mediated repair occurs at MVBs followed by release of exosomes, we tested the effects of inhibiting exosome secretion by pharmacological blockade of neutral sphingomyelinase, which is required for the fusion of MVB to the plasma membrane, and by knockdown of Rab27a, which mediates traffic and secretion of exosomes from MVBs. Both treatments blocked LITAF-mediated resistance to α-toxin (Fig. 4A and 4C). Finally we tested the requirement for membrane ion flux in triggering LITAF-mediated repair. Repair mechanisms at the plasma membrane require Ca2+ influx, but we found that LITAF re-localization occurred independently of intracellular Ca2+ signaling, but was instead dependent on K+ efflux (Fig 4D). Furthermore, intracellular Ca2+ chelation did not significantly inhibit LITAF-mediated cell survival after α-toxin treatment (Fig 4E). These data therefore demonstrate that LITAF-mediated repair is distinct from described mechanisms of direct membrane shedding from the plasma membrane, and confirm that this occurs through secretion of damaged membrane as exosomes.
Figure 4: LITAF-mediated repair occurs through endosomal action of ESCRT and requires interaction with NEDD4, Tsg101 and endosomes.
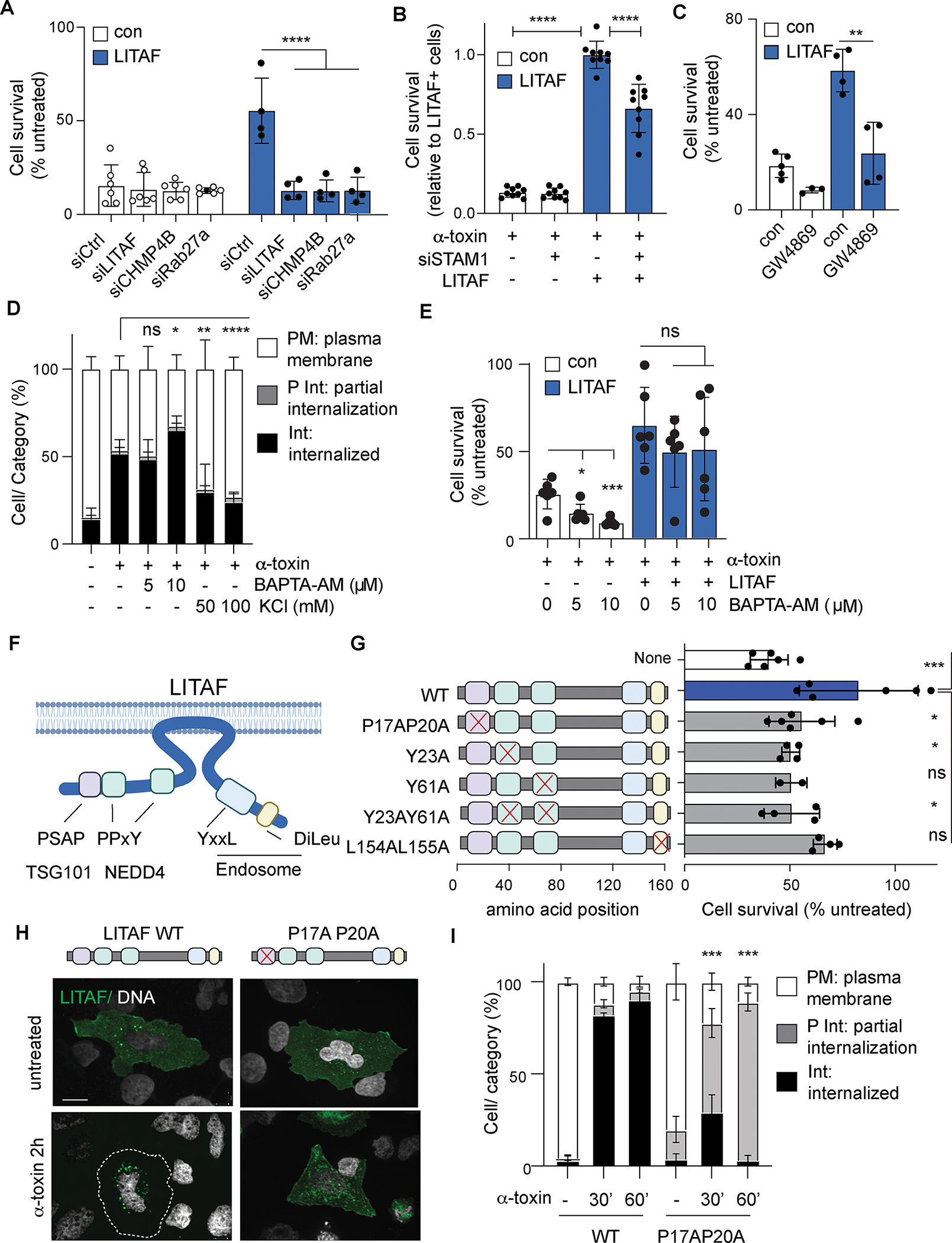
(A-C) Survival 24h after α-toxin treatment of control (con) and LITAF-expressing (LITAF) U2OS cells treated with indicated siRNA or exosome inhibitor GW4869. Data are mean ± SD of n ≥ 3 replicate wells/condition, combined from three independent experiments. (D) Distribution of LITAF after 2h treatment with α-toxin, BAPTA-AM and KCl as indicated. Data expressed as proportions of cells with LITAF localized predominantly to the plasma membrane (PM), internal vesicles (Int), or distributed between both (P Int). Mean ± SD of percentages of cells, n=6 replicate wells/condition, combined from two independent experiments (≥ 100 cells were analyzed per condition in each experiment). (E) Survival 24h after α-toxin treatment of control (con) and LITAF-expressing (LITAF) U2OS cells treated with BAPTA-AM. Data are mean ± SD of n=6 replicate wells/condition, combined from two independent experiments. (F) Schematic of LITAF orientation on the cytoplasmic face of the plasma membrane and location of motifs that mediate interactìon with NEDD4, Tsg101 and endosomes. (G) Survival 24h after α-toxin of U2OS cells expressing LITAF with point mutations in interaction motifs, wild type LITAF (WT), or no LITAF (none). (n≥2 replicate well/condition), representative of three independent experiments. (H-I) Representative confocal images (H) and distribution (I) of wild type GFP-LITAF or LITAF P17AP20A mutant in U2OS cells treated with α-toxin. Mean ± SD of n=3 replicate wells/condition, representative of three independent experiments (≥ 100 cells were analyzed per condition in each experiment). * p < 0.05, ** p < 0.01, *** p < 0.001, **** p <0.0001, unpaired Student’s t test (E), one-way ANOVA (A-C, G) or two-way ANOVA (D,I). Scale bar in microscopy images is 10 μm.
LITAF activates ESCRT at endosomes through TSG101 binding
LITAF associates tightly with the cytoplasmic face of cell membranes through an unusual amphipathic helix structure (referred to as a ‘LITAF domain’) (30), and interacts with several endosomal and cytoplasmic proteins through well characterized protein binding domains (Fig 4F). To determine the molecular mechanism of LITAF activity, mutations were introduced into LITAF to disrupt these interaction and binding domains. Naturally occurring autosomal dominant mutations in LITAF in or around the membrane-binding LITAF domain prevent membrane association or endosomal localization and are responsible for Charcot Marie Tooth disease type 1C (CMT1C), a rare demyelinating peripheral neuropathy (35). None of these disease-associated mutant forms of LITAF promoted cell survival when overexpressed in U2OS cells (fig. S4A). Mutations of the endosome membrane-targeting LL motif similarly impaired LITAF membrane and endosome localization, and promoted only a small increase in cell survival that was not statistically significantly different from non-transfected controls (figs. S4B and 4G). Thus, the association of LITAF with endosomal membranes is required for resistance to α-toxin.
LITAF recruits ESCRT to the endosome and MVB by binding to the ESCRT-I component TSG101 through a PSAP motif (36). Mutation of the TSG101 binding site blocked the ability of LITAF to increase survival in response to α-toxin (Fig 4G), consistent with our observations that LITAF-mediated resistance was dependent on ESCRT. Furthermore, these mutations also prevented LITAF accumulation in MVBs after α-toxin treatment; TSG101-binding mutants translocated normally from the plasma membrane in response to α-toxin, but accumulated in smaller endosomes rather than MVB (Fig 4H–I). These data are consistent with a failure of LITAF to activate ESCRT at endosomes to form MVBs, and provide further evidence that LITAF promotes resistance by ESCRT-mediated sequestration of internalized α-toxin in MVBs.
NEDD4 triggers re-localization of LITAF to endosomes in response to K+ efflux
To understand how LITAF was engaged in response to membrane pores, we focused on its interactions with the NEDD4 family of E3-ubiquitin ligases which are involved in protein internalization and recycling (37). LITAF mutants that lack either of the two PPxY NEDD4 interaction motifs failed to confer increased resistance to α-toxin, implicating the LITAF-NEDD4 interactions in membrane repair (Fig. 4G). Consistent with this possibility, knockdown of the major NEDD4 family member, NEDD4.1, also prevented LITAF-mediated resistance to α-toxin (Fig. 5A). Furthermore, ubiquitylation of NEDD4.1, a proxy for NEDD4 activation (38, 39), was induced by α-toxin treatment (Fig. 5B), and NEDD4 ubiquitin-ligase activity was required for resistance as overexpression of a ubiquitin-ligase deficient NEDD4 mutant (NEDD4 H1284A/C1286A) inhibited the ability of LITAF to protect cells (Fig 5C). This NEDD4.1 ubiquitylation in response to α-toxin was blocked by the addition of extracellular K+ to buffer ion flux (fig. S5A) but was unaffected by LITAF overexpression (Fig. 5B). Taken together, these data place NEDD4 downstream of pore formation and K+ efflux, but upstream of LITAF, in the membrane repair pathway.
Figure 5: NEDD4 connects membrane damage sensing to LITAF activation and trafficking.
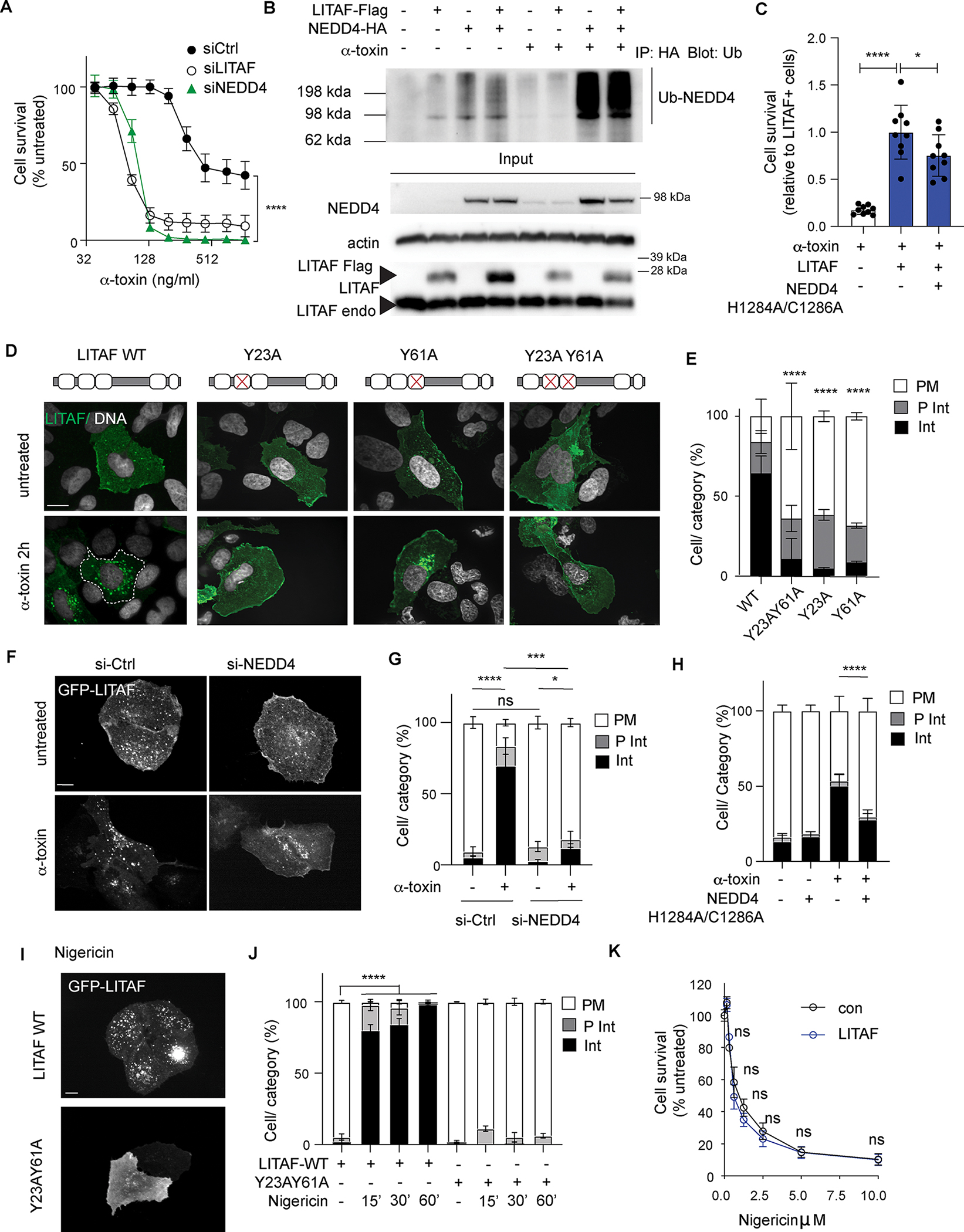
(A) Survival 24h after α-toxin treatment of U2OS cells transfected with LITAF alone (no siRNA) or LITAF and siRNA targeting LITAF or NEDD4. Mean ± S.D of n=6 replicate wells/condition, combined from two independent experiments. (B) Immunoblot of anti-HA immunoprecipitations from α-toxin treated and untreated U2OS cells, expressing NEDD4-HA and LITAF-Flag, stained for Ubiquitin (Ub). Lower panels show immunoblots of corresponding total cell lysates. Similar results were seen in at least 3 independent experiments. (C) Survival 24h after α-toxin of U2OS cells transfected with LITAF and catalytically inactive H1284A/C1286A NEDD4. Mean ± S.D of n=9 replicate wells/condition, combined from three independent experiments. (D,E) Representative confocal images (D) and quantification of LITAF localization (E) of U2OS cells expressing wild type GFP-LITAF or NEDD4-binding mutants. Localization recorded as predominantly plasma membrane (PM), internal vesicles (Int), or distributed between both (P Int). mean ± S.D or range of n≥2 replicate wells/condition, representative of three independent experiments. (F-G) Representative confocal images (F) and quantification (G) of GFP-LITAF localization in U2OS cells treated with α-toxin and siRNA against NEDD4. Mean ± S.D of n=3 replicate wells/condition, representative of three independent experiments. (H) LITAF re-localization in U2OS cells treated wexpressing wild-type NEDD4 or catalytically inactive H1284A/C1286A mutant. Mean ± S.D. of n=9 replicate wells/condition, combined from three independent experiments. (I,J) Representative confocal images (I) and quantification (J) of LITAF localization in U2OS cells expressing wild-type LITAF or Y23AY60A mutant LITAF treated with Nigericin for indicated time (minutes). Mean ± S.D. of n=3 replicate wells/condition, representative of three independent experiments. (K) Survival (relative to untreated cells) of wild type (con) and LITAF-overexpressing U2OS cells 24h after treatment with Nigericin. Mean ± S.D. of n≥6 replicate wells/condition) combined from two independent experiments. * p < 0.05, *** p < 0.001, **** p <0.0001, one-way ANOVA (C), or two-way ANOVA (A,E,G,H,J,K). ≥ 100 cells were analyzed per condition for LITA localization. Scale bars 10 μm.
To understand how LITAF and NEDD4 cooperate in repair we further analyzed their interaction and trafficking. Although LITAF bound to NEDD4 and was ubiquitylated through a NEDD4-dependent mechanism (figs. S5, B to D), α-toxin treatment did not increase LITAF ubiquitylation (fig. S5C) suggesting that NEDD4-mediated ubiquitylation of LITAF was unlikely to regulate this process. α-toxin did not increase the interaction between LITAF and NEDD4, but uncoupling of LITAF from NEDD4 by mutation of either of the NEDD4-binding PPxY motifs prevented LITAF re-localization in response to α-toxin. Similarly, LITAF relocalization did not occur in NEDD4.1 knockdown cells (Fig. 5F and G), or in cells expressing the ubiquitin-ligase deficient NEDD4 mutant (NEDD4 H1284A/C1286A) (Fig. 5H). Notably, in contrast with TSG101-interaction mutants of LITAF, which arrest at endosomal compartments after α-toxin treatment (Fig. 4H and I), NEDD4 interaction mutants completely failed to relocate from the plasma membrane (Fig. 5D and E). We therefore concluded that LITAF trafficking occurs in 2 stages, with interactions with NEDD4 and TSG101 playing distinct and sequential roles. First, LITAF re-localizes from the plasma membrane to endosomes, through a NEDD4-dependent mechanism. Second, TSG101 promotes ESCRT recruitment and LITAF accumulation in MVBs and exosomes.
Membrane repair in response to PFTs has previously been shown to be activated by ion flux through pores, rather than by direct recognition of pore complexes. α-toxin causes efflux of K+ from cells, and we have shown that this is necessary for the activation of NEDD4 (Fig S5A) and mobilization of LITAF (Fig 4D). To determine whether K+ efflux is sufficient for LITAF relocalization, U2OS cells were treated with Nigericin, an ionophore that induces K+ and Ca2+ efflux but does not cause membrane damage, thus uncoupling ion flux from pore-formation. Similar to the relocalization seen with α-toxin, treatment with Nigericin induced rapid internalization of LITAF to large vacuoles, and this required interaction between LITAF and NEDD4 (Fig. 5I,5J). However, LITAF did not impact survival of U2OS cells in response to Nigericin (Fig. 5K), which mediates U2OS death by caspase-9 dependent apoptosis, confirming that LITAF does not confer resistance by blocking programmed cell death but by enhancing membrane repair (40). Together, these data support a model in which de-polarization of membrane K+ triggers NEDD4 activation and mobilization of LITAF to endosomes, where the NEDD4/LITAF complex is poised to initiate membrane repair.
LITAF promotes resistance to additional bacterial toxins and to Gasdermin D
The role of LITAF in activating ESCRT at internal membranes rather than the plasma membrane may reflect the use of α-toxin in our study, which is predominantly removed via internalization and sequestration (20, 33, 41). To test whether LITAF could protect against other membrane pores, we tested the sensitivity of LITAF-expressing cells to death induced by the PFT, Streptolysin O (SLO). SLO binds to the plasma membrane though a receptor-independent mechanism (20), forming much larger pores than α-toxin (25–30 nm), and can be countered by plasma-membrane repair mechanisms (15). We found that LITAF overexpression prevented SLO-induced cell death in U2OS cells (Fig 6A) and this occurred through the same endosomal membrane repair pathway as for α-toxin. Specifically, SLO triggered NEDD4-dependent re-localization from the plasma membrane (Fig 6B and 6C, Fig S6A) and survival required NEDD4 activity (Fig S6B). In addition, SLO treatment of LITAF-expressing cells caused accumulation of the ESCRT component CHMP4 at intracellular vesicles (movie S2, Fig S6C), co-localized with LITAF (movie S3, Fig S6D), rather than at the plasma membrane. Although SLO pores are large enough to allow Ca2+ influx, which is responsible for initiating repair at the plasma membrane (42), activation of LITAF by SLO and protection from cell death occurred through sensing of K+ efflux independently of Ca2+ (Fig S6E and S6F) as we had observed for α-toxin. Furthermore, LITAF-mediated survival was dependent on STAM1 and Rab27a (Fig S6G), further confirming that repair occurs through the same intracellular pathway. We, therefore, concluded that LITAF-mediated repair may represent a general mechanism of cell response to PFTs.
Figure 6: LITAF protects against membrane damage by SLO or Gasdermin D.

(A-D) Survival 24h after SLO treatment of control (con) and LITAF-expressing (LITAF) U2OS cells. Mean ± S./D or range of n≥2 replicate wells/condition, representative of three independent experiments. (B-C) Representative confocal images (B) and quantification of LITAF localization (C) of U2OS cells expressing wild type GFP-LITAF or NEDD4-binding mutant and treated with SLO (500 ng/ml, 30 min). Localization recorded as predominantly plasma membrane (PM), internal vesicles (Int), or distributed between both (P Int). Mean ± S.D of n=3 replicate wells/condition, representative of three independent experiments (D) Cell death (yoyo3 incorporation) in control (con) and LITAF-expressing (LITAF-Flag, GFP-LITAF) U2OS cells, transfected with an activated GSDMD construct, or treated with α-toxin. Mean ± S.D. of n=3 replicate wells/condition, representative of three independent experiments. (E-F) Representative confocal images (E) and quantification of LITAF localization (F) of U2OS cells expressing wild type GFP-LITAF or NEDD4-binding mutant, transfected with activated GSDMD construct Mean ± S.D. of n=3 replicate wells/condition, combined from three independent experiments. (G) Immunoblot of mouse LITAF and GAPDH in BMDM derived from wild-type (WT) or LITAF-knockout (ko) mice. (H) Cell death in wild-type and LITAF-ko BMDMs primed with LPS or Pam3CSK4 (4h) and treated with Nigericin for indicated time. Mean ± S.D. of n≥4 mice/condition, combined from three independent experiments. ** p < 0.01, *** p < 0.001, **** p <0.0001, one-way ANOVA (D), or two-way ANOVA (A,C,F,H). ≥ 100 cells were analyzed per condition for LITAF localization. Scale bars 10 μm.
Endogenous pore-forming proteins are increasingly implicated as effectors of inflammation and cell death. Gasdermin D (GSDMD) is a pore-forming protein that is activated by Caspase 1 and 11, and is required to promote cell death and release of IL-1β and IL-18 during pyroptosis (4). Recently ESCRT-mediated membrane repair was shown to counteract GSDMD-induced cell death (18). To test whether LITAF promotes repair of GSDMD pores, U2OS cells expressing wild-type and mutant forms of LITAF were transfected with constructs expressing active GSDMD. Active GSDMD induced rapid cell death in wild type cells U2OS cells (Fig 6D). However, expression of LITAF completely blocked cell death caused by active GSDMD (Fig 6D). Expression of active GSDMD caused re-localization of LITAF to intracellular vacuoles (Fig 6E, F), and this was dependent on LITAF interaction with NEDD4, similar to our findings with α-toxin and SLO treatment. These experiments relied on overexpression of truncated constitutively active GSDMD. To test whether LITAF could protect cells against GSDMD activation in a physiological setting, we prepared macrophages from wild type and LITAF-knockout mice (Fig 6G, Fig S6H and S6I). LITAF-knockout macrophages showed increased cell death after priming and inflammasome activation than wild-type macrophages (Fig 6H).
DISCUSSION
Using an unbiased forward genetic screen we have identified a key role for the endosomal protein LITAF in sensing disrupted plasma membrane integrity and linking it with membrane repair. We find that LITAF promotes sequestration of membrane pores intracellularly in the MVB, and subsequent expulsion of the damaged membrane in exosomes. This pathway is activated by K+ efflux, which triggers NEDD4 activation and LITAF mobilization to α-toxin-containing endosomes, where it activates the ESCRT machinery. LITAF-mediated membrane repair protects cells against cell death induced by both bacterial toxins and endogenous pore-forming proteins and occurs through a distinct pathway from previously reported examples of ESCRT-dependent repair of the plasma membrane in response to bacterial toxins and endogenous pores(14–19). We therefore propose that LITAF represents a key link between sensing of pores and directed repair, and provides an additional pathway of cellular resilience to membrane damage and cell death.
Based on our data, we propose a model (fig. S7) in which K+ efflux through pores causes activation of NEDD4, which mediates the targeting of LITAF to endosomes containing α-toxin. At the endosome, LITAF initiates assembly of the ESCRT machinery, causing internalization of α-toxin into intraluminal vesicles (ILVs) in MVBs. These are then degraded or secreted from the cell, expelling damaged membranes. Notably, several additional lines of evidence show that this pathway of LITAF-mediated repair is distinct from the ESCRT-mediated budding and fission of damaged membrane as ‘ectosomes’ at the plasma membrane previously implicated in repair of SLO and listeriolysin O toxin pores and the endogenous pores MLKL and GSDMD (14, 15, 18, 19). We show that LITAF-mediated ESCRT activation requires the ESCRT-0 component STAM1, but LITAF-mobilization occurs independently of Ca2+ influx, in contrast with plasma membrane repair, which occurs independently of STAM1 but is triggered by intracellular Ca2+ changes. We therefore propose that LITAF provides a repair mechanism that acts in parallel with plasma membrane shedding. LITAF is selectively expressed in myeloid cells, particularly macrophages, and plasmacytoid DCs, and in lung epithelial cells, and is upregulated in response to inflammatory signals (43). LITAF may therefore provide an additional protective mechanism for cells involved in immune defense that are exposed to potentially high levels of membrane damage from pathogen effectors or endogenous pores. Recently, LITAF was reported to promote sensitivity to the Bacillus cereus Hemolysin BL toxin (44), in contrast with our findings that LITAF reduces sensitivity to S. aureus α-toxin and SLO. The role of LITAF in Hemolysin BL sensitivity was independent of interactions with ESCRT, and LITAF is proposed to act as a receptor for the toxin, although it remains unclear how this occurs given the localization of LITAF to the inner leaflet of the membrane. The differences between our work and this previous study therefore appear to reflect different mechanisms of LITAF action.
The mechanism we propose is consistent with many other studies of LITAF function. LITAF is known to regulate generation of MVBs and exosome formation through recruitment to internal membranes and directed activation of ESCRT (29, 31, 34). LITAF is implicated in the sorting of cell surface proteins, including EGFR (34) and ion channels (45), to MVB for degradation. However, LITAF has not previously been linked with membrane repair, and its cellular function is unknown. Mutations in LITAF are responsible for a form of the inherited neurological disorder Charcot Marie Tooth disease (CMT), known as CMT1C (35). LITAF mutations found in CMT1C are mostly around the membrane-binding LITAF domain and endosomal localization sites, and disrupt LITAF trafficking to the endosome and lysosome and exosome formation (31, 32). Demyelinating forms of CMT, such as CMT1C, arise from defects in Schwann cells, which are responsible for generation and maintenance of the myelin sheath that protects peripheral nerves. Mutations in additional endosomal-lysosomal proteins, including Rab7, SH3TC2, MTMR2, MTMR13/SBF2 and FIG 4 also cause demyelinating CMT (46–52), indicating that defects in vesicular trafficking have an important role in this disease, although the exact role of LITAF and other genes in Schwann cells remains unclear. Schwann cells produce very large amounts of myelin to maintain the myelin sheath resulting in significant levels of endoplasmic reticulum stress, and disruption of the unfolded protein response in these cells results in demyelination (53, 54). It has been therefore proposed that LITAF contributes to this process by targeting misfolded proteins for degradation (29), and this is consistent with our data, supporting a general role for LITAF in targeting removal of aberrant protein or membrane to counteract cellular stress. Our data also raise the possibility that demyelination in CMT may arise from compromised membrane repair in Schwann cells.
Membrane repair by LITAF requires binding to NEDD4, an E3-ubiquitin ligase (55) involved in targeting proteins for lysosomal degradation (56). NEDD4 mono-ubiquitylates proteins at the plasma membrane or Golgi, resulting in their traffic to sorting endosomes and then to MVBs (57). It is likely, therefore, that NEDD4 is responsible for labeling α-toxin or neighboring membrane proteins for internalization and removal by LITAF. Our data support this, showing that NEDD4 acts upstream of LITAF in membrane repair, and is required for targeting LITAF to endosomes for activation of ESCRT activity. The exact mechanism of NEDD4 activation by membrane damage remains to be determined. NEDD4 can be activated through a variety of mechanisms, including phosphorylation by PKA, MAPK and Src-family kinases, many of which are active during cellular stress responses. Our observation that LITAF internalization by NEDD4 also occurs in cells treated with the K+ ionophore Nigericin, and is dependent on K+ efflux, implicate ion flux as the critical signal activating NEDD4. Intriguingly, NEDD4 regulates surface expression of a range of ion channels and exchangers, including Na+, K+ and Ca2+ channels (58–61), and has been shown to be activated by increases in local Ca2+, which releases auto-inhibition by the C2 domain (62). Hence, it is possible that NEDD4 activation may be linked to changes in ion flux or local membrane potential, and that NEDD4 functions as a direct sensor of pore formation.
A potential limitation of the present study is that the transposon mutagenesis approach we have used is designed to identify strong ‘gain-of-function’ phenotypes due to gene activation, rather than an exhaustive description of all of the proteins involved in LITAF-mediated repair, or other membrane repair pathways. Interestingly, two additional genes identified in this screen, GRAMD1B and GRAMD2B, contain pleckstrin-homology-like GRAM domains, which mediate protein binding to PIP moieties on lipid bilayers (63), and direct interactions between internal membranes and the cell surface (64, 65). Mutations in 2 GRAM-domain containing proteins, MTMR2 and MTMR13/SBF2 (66), also cause demyelinating forms of CMT (46, 49, 51), and we therefore speculate that GRAMD1B and GRAMD2B may work with LITAF or in the same pathway to traffic α-toxin for removal and facilitate membrane repair.
Our finding that LITAF protects against cell death induced by SLO and GSDMD, indicate that LITAF-mediated repair is not limited to α-toxin and represents a general repair mechanism against membrane damage. LITAF is expressed at mucosal barrier surfaces and in macrophages and other innate immune cells and expression increases during inflammation (43) and in response to TLR stimulation or viral infection (67), leading us to speculate that upregulation of LITAF may be a component of innate immune defense during infection. It will be interesting in future studies to determine whether LITAF expression contributes to resistance or resilience to infection through effects on epithelial, endothelial or immune cells. The relationship between loss of membrane integrity caused by exogenous or endogenous pores, and the induction of cell death, is complex and it is now well established that cell death in response to membrane pores is due to regulated cell death, rather than a direct process of cell swelling and leakage of key cellular components. This complexity is underscored by the recent identification of NINJ1, which forms large pores, as a ‘final’ effector of programmed cell death triggered by the endogenous membrane pore GSDMD, or by exogenous PFTs (68). A number of studies, including our own, demonstrate that loss of repair leads to increased cell death, while increasing membrane repair can allow cells to survive damage that would otherwise lead to death. Induction of cell death and resultant inflammation may therefore be a consequence of insufficient repair, and that promoting membrane repair may offer a novel therapeutic approach to reduce tissue damage and inflammation from infection or sterile injury at barrier surfaces.
MATERIAL AND METHODS
Study Design
The main objectives of this study are to identify and characterize genes that confer resistance to cell death induced by membrane pores. A forward genetic screen based on transposon mutagenesis was used to identify candidate genes. The strongest candidate (LITAF) was validated via gene overexpression, knockout and mutation approaches in assays of cell survival. Confocal microscopy and biochemical assays (western blot, immunoprecipitation) were used to determine the mechanism of action. We generated a LITAF-knockout mouse using CRISPR to validate the role of LITAF in primary macrophages. Unless noted, all experiments were performed independently at least 3 times. Investigators were blinded to experimental groups in all experiments involving image analysis.
Tissue culture
All cell lines were purchased from ATCC (Manassas, VA, USA), and were used at passage below 5. U2OS and HT29 were maintained in McCoy’s 5A media. A549, Hela, HK2, HEC1B, MRC5, HepG2 and 293T cells (ATCC) were maintained in DMEM media. All media were supplemented with 2mM GlutaMAX, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco), and 10% FBS (Seradigm/VWR). All cells were maintained at 37°C with 5% CO2.
Constructs
RFP-Rab5 was a gift from C. Becker. CD63-pEGFP C2 was a gift from Paul Luzio (Addgene plasmid # 62964). pSLIK-LITAF was a gift from A. Oberst. All LITAF mutants were generated using quick-change mutagenesis following vendor protocol on plasmid LITAF-MycDDK (origene) (Table S2). Following protocol from Gray et al 2017 (69), we generated LITAF CRISPR constructs using guide RNA (Table S3) cloned into Cas9-t2a-puro pRRL lentivirus plasmid (gift from D. Stetson). Briefly, we used a lentivirus in which a U6 promoter-driven guide RNA and MND promoter-driven Cas9-T2A-puromycin resistance cassette are constitutively expressed from a single, self-inactivating lentivirus. A549 cells were transduced with lentivirus and selected with 3μg/ml puromycin. Gene targeting was evaluated by western blot for endogenous protein. GFP-LITAF constructs were generated by cloning LITAF sequences (WT and mutants), inside pEGFP-C2 using TOPO cloning. The active truncated form of GSDMD (amino acids 1–275) was amplified from cDNA and cloned (Infusion HD Cloning Kit, TakaBio) into the AAV backbone pAAV.MND.GFP vector (Gift from Dr. David Rawlings, Seattle Children’s Research Institute), directly downstream of the MND promoter, and upstream of an α globin polyadenylation site.
Antibodies and reagents
List of antibodies and reagents used are available is Table S4 and Table S5 respectively.
Cell treatment
α-toxin and SLO were used at 1 μg/ml or 500 ng/ml or in dose response as indicated in figure legends. Unless otherwise stated, Nigericin was used at 5 μM. Caspases inhibitors YVAD, ZVAD and QVD were used at 2 μM, 2 μM and 5 μM respectively. The exosome inhibitor GW4869 was used at 20 μM. Cells were treated with BAPTA-AM at 5 or 10 μM at the same time as toxin treatment to inhibit Ca2+-dependent signaling. To inhibit K+ efflux, cells were cultured in medium supplemented with KCl at 50–100 mM unless otherwise indicated. Caspase 3/7 activation was measured following manufacturer guidelines. Activation of inducible LITAF expression was done by pretreatment overnight with doxycycline at 1 μg/ml. For measuring inflammasome activation, BMDMs were treated with LPS at 100 ng/ml or Pam3CSK4 at 1 μg/ml for 4h, and then Nigericin at 5 μM.
Transposon Mutagenesis and Screening
Transposon mutagenesis and screening were performed following protocol described in Bruchez et al 2020 (24). In brief, U2OS cells were co-transfected with piggyBac plasmid pPB-SB-CMV-puro-SD3 (6 μg DNA/6×107 cells) and p-hyPBASE (6 μg DNA/6×107 cells) using Fugene 6 transfection reagent (Promega, Madison WI, USA). For each library, between 107–108 cells were transfected, cultured with the addition of 3 μg/ml puromycin (Gibco) for one week to select cells that had incorporated the transposon, and then used for screens of α-toxin resistance with minimal further expansion. Each cell carries an average of 5 insertions and libraries are predicted to contain > 5 × 107 independent transposon insertion events, >2-fold higher than the estimated 2 × 107 PB insertion sites in the human genome. Non-mutagenized U2OS cells were treated with α-toxin under the same conditions to confirm that no resistance was seen in the absence of transposon mutagenesis. Cells were treated with α-toxin (500ng/ml) for 48h. Cells were washed in PBS to remove dead cells and were treated again for 48h to ensure the selection of only highly resistant cells, which were pooled and sequenced. Clonal populations of resistant cells were generated by limiting dilution cloning, expanded and sequenced.
Sequencing and analysis
Sequencing and analysis were performed following protocol described in Bruchez et al 2020 (24). In brief, genomic DNA (gDNA) was isolated from 5–10 × 106 cells. 4 mg of gDNA were digested with BfaI or CviQI (New England Biolabs, Ipswich MA, USA) and purified (QIAquick PCR purification kits, Qiagen, Hilden, Germany) then ligated with barcoded P7-side adaptors using T4 DNA Ligase (New England BioLabs). Ligated gDNA fragments were then combined and amplified by nested PCR. The first round of PCR used primers corresponding to transposon sequence (KS15) and the P7-side adaptor (LP1). A second round of PCR (primers P2A and LP22) added the full Illumina P5 and P7 sequences. Sequencing was performed by the BRI Genomics platform using a HiScan or HiSeq platform (Illumina, San Diego CA, USA) with a custom sequencing primer (Rd1SeqP.ipdC) to generate 50 or 100 bp single end reads.
Sequences were pre-processed (Trimmomatic v. 0.33) to remove Illumina adaptor sequences, filter out sequences < 40 bp and crop longer sequences to 50 bp. Sequences were then aligned to human genome hg38 (Bowtie v. 2.1.0). Sequences that did not map to unique sites or showed significant mismatch or error (edit distance >2 over 50bp) were excluded from further analysis. Finally, reads were mapped and filtered to a known PiggyBac consensus insertion site (TTAA) at their start.
Common insertion sites (CIS) were identified using the Poisson Regression Insertion Model (PRIM) published by Bergemann et al 2012(70). The PRIM model was run using a p-value cutoff of 0.01 and Bonferroni correction on both selected, unselected, and in silico-generated libraries based on known TTAA sites in the genome, using window sizes of 10 kb, 25 kb, 50 kb, 75 kb, and 100 kb. CIS in experimental libraries that were also found in unselected or in silico generated libraries were excluded.
Cell survival assay
After toxin treatment cells were washed twice with PBS to remove dead cells. Cell death was assessed by incorporation of non-permeable DNA dyes, YOYO3 (thermo fisher) at 0.2 mM or Sytox Green (thermo fisher) at 1 mM for 20 min. Total number of cells was measured by incorporation of permeable DNA staining Hoechst for 20 min. Cells were imaged and quantified using Cytation 3 imager and Gen5 software v3.04 for end point survival; or using Incucyte plate reader and Incucyte ZOOM 2015A Software for time-course analysis.
Transfections and lentivirus production
For transient overexpression transfection, all cell lines were transfected following the same protocol. Briefly, 2×10ˆ5 cells were plated on 6 wells plates and left overnight. Cells were then transfected with 4.5 ml of Fugene 6 (Promega) and 1.5 mg of DNA following manufacturer instructions. For transient knockdowns, the same number of cells were transfected with 2 ml of RNAiMax (Invitrogen) and 2 ml of siRNA following manufacturer instructions. Details about siRNA used in this study are available in Table S6. To produce lentivirus for transduction, 4.5×10ˆ6 HEK293T cells were first plated and transfected the next day with 42 ml of PEI (1mg/ml) and 4:2:1 ratio of vector : viral packaging (psPAX2) : viral envelope(pVSVg) contained in 500 ml of diluent (5 mM Hepes, 150mM NaCL at pH 7.05). Supernatant containing lentivirus were collected 48h after transfection and filtered 0.22 mm before adding on the cells to transduce.
Exosome preparation
U2OS were cultivated in McCoy media with 10% FBS-exosome free (GIBCO) and treated or left untreated for 2 h with α-toxin. Exosomes from culture supernatants were isolated by differential centrifugation, at 300g for 10 min, 2,000g for 10 min, 10,000g for 30 min and 100,000g for 70 min, followed by one wash with PBS and purification by centrifugation at 100,000g for 70 min. Purified exosomes were then characterized by immunoblot analysis as described in Li et al 2013 (71).
Exosome analysis by flow cytometry
30 ml of purified exosomes were combined with 5 ml of latex beads aldehyde sulfate 4% w/v 4 mm, at 40×10^6 beads/ml, for 15 min at RT. Beads containing exosomes were then washed for 10 min with BCB (PBS, 0.1%BSA, 0.01%NaN3) and centrifuged at 9000 rpm for 2 min. Beads were then incubated for 30 min with 0.5 mg of CD9 antibody. After 10 min wash in BCB, beads were incubated with anti-mouse 488 for 20 min. Finally, beads were washed in BCB and PBS before being analyzed on an Aria FACS Calibur(72).
Immunofluorescence and confocal microscopy
Immunofluorescence studies were performed on cells fixed in 4% paraformaldehyde (Sigma) 15 min. Cells were incubated with blocking buffer (10% FBS, 0.05% saponin in PBS) for 1 h at room temperature, then with primary antibodies in dilution buffer (1% FBS, 0.05% saponin in PBS). All washes between staining were performed in 0.05% saponin in PBS for three times 5 min, to maintain permeabilization. Secondary antibodies were added in dilution buffer for 1 h at room temperature. Cell nuclei were stained with Hoechst 33342, and the cover slips were mounted with Prolong Diamond Antifade (Invitrogen). Cells were imaged using ×60 objective and ×100 oil objective, on Nikon Ti (Eclipse) inverted microscope with Ultraview Spinning Disc (CSU-X1) confocal scanner (Perkin Elmer). Images were captured with an Orca-ER Camera using Volocity (Quorum technologies). Post-acquisition analysis such as contrast adjustment, deconvolution through iterative restoration and colocalization were performed using Volocity software.
Live imaging
Cells were imaged using the ImageXpress HT.ai (Molecular devices) inside a controlled environment with 5% CO2 and at 37c. Imaging was performed using the spinning disk 50um slit, with 60X water immersion objective. ROIs were selected at 10x using SpotID prior to intoxication, and live imaging of untreated or toxin-treated cells was performed on those ROIs at 60X every 5 seconds. Video montage was performed from raw tiff images using ImageJ/Fiji.
Immunoblotting.
Cells were washed in PBS and lysed for 10 min on ice in RIPA buffer supplemented with 1 mM DTT, 1 mM PMSF and protease inhibitor cocktail (Pierce protease inhibitor). Buffer was also supplemented with NEM for ubiquitinylation studies. Lysates were centrifuged for 10 min at 12,000g to pellet the nuclei and supernatant was collected for analysis. Proteins were separated by electrophoresis using NuPage-Bis-Tris gels (Lifesciences) and transferred onto PVDF membranes (Millipore). Non-specific binding was blocked with 5% BSA in TBS Tween (0.1%) or 5% Milk in PBS Tween (0.1%) for 2 hours. Primary and secondary antibodies were used in 2.5% BSA in TBS Tween (0.1%), diluted at 1/5000 or 1/10,000 for actin. HRP signal was revealed using ECL reagents (Millipore), and Biorad imager (Chemidoc touch). Uncropped versions of all immunoblots are provided in Supplementary materials.
Immunoprecipitation.
U2OS cells (107 cells) were transfected using Fugene 6 with pCMV6-LITAF-Flag. Cells were lysed in 1mL of immunoprecipitation buffer (IPB) (RIPA, 1 mM PMSF, 1 mM DTT, 10mM NEM and protease inhibitor cocktail). Cleared lysates were incubated with 4 μg of mouse Flag-M2 antibody or 4 μg of rabbit-HA antibody for 1h at 4°C. Immunoprecipitates were immobilized on Dynabeads® Protein G (Lifescience) for 1h at 4°C. Beads were washed three times with 1 mL of IPB and resuspended in 50 μL of IPB, LDS and sample reducing agent (NuPage). Protocol adapted from Visvikis et al 2011(73).
Circos Plot
Genomic data were visualized using Circos v0.67 human karyotype hg38(74).
Quantitative RT-PCR
Reverse transcription was performed on 1ug of purified RNA by using the high-capacity cDNA reverse transcription kit (Applied Biosystems; Thermo-Fisher). Power SYBR Green PCR Master Mix (Applied Biosystems; Thermo-Fisher) was used to perform quantitative PCR on Applied Biosystem 7900 HT QPCR (Table S7).
LITAF knockout mice
CRISPR ko mice from C57Bl6 background were generated by the Genetically Engineered Mouse Modeling Program from the Fred Hutchinson Cancer Research Center (Seattle). Using Benchling and CRISPR guide analysis tools, two gRNA predicting high specificity and efficiency and Low off-targets were selected within exon 2 (Table S8). The selected gRNAs were co-injected with Cas9 mRNA and protein in 0.5 dpc embryos. Mutations and deletions were confirmed by PCR (Table S7) and sequencing of mice’s genomic DNA. Males homozygous for the CRISPR-deleted allele aged 9–18wks, and male C57Bl/6 control mice of the same age were used for BMDM inflammasome experiments. All animal experiments were pre-approved by the Benaroya Research Institute Animal Care and Use Committee and performed under national guidelines for animal care.
BMDM inflammasome experiments
BMDMs were generated from bone marrow from femurs and/or tibias of mice by culture for 7 days in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% heat-inactivated FBS, 10 mM Hepes, NEAA, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco), and with 10% conditioned culture supernatant from MCSF-expressing L929 cells. Macrophages were primed with Salmonella minnesota R595 LPS (100 ng/ml; InvivoGen) or Pam3CSK4 (1 μg/ml, InvivoGen) for 4 hours before nigericin treatment for the indicated time. CytoTox 96 kit (Promega) was used to determine LDH release.
Statistical analysis
The data were analyzed using one-way analysis of variance (ANOVA) with Bonferroni post hoc, or two way student t-test, as indicated in the figure legends. The significance level was set as p ≤ 0.05. All statistical analyses were performed using Prism V6.0b software.
Supplementary Material
Acknowledgments:
We are grateful to K Sha, and other past and present members of the Lacy-Hulbert laboratory for assistance and suggestions. We thank V Gersuk and the BRI genomics core for sequencing, and members of the Bioinformatics Department at BRI for support in data analysis.
Funding:
National Institutes of Health grant R33AI119341 (AL-H)
National Institutes of Health grant R01GM102482 (LMS.)
Footnotes
Competing interests: The authors declare that they have no competing interests.
Supplementary Materials
Fig S1–S7
Table S1–S8
Movie S1–S3
Supplementary data file S1. Raw data excel file
Data and materials availability:
Full analysis of screen results are presented in Supplementary Material. DNA Sequencing data is deposited at GEO (GSE# GSE242913). The PB transposon was obtained under a material transfer agreement with the Wellcome Trust Sanger Institute. All other data are available in the manuscript or the supplementary materials.
References and Notes
- 1.Dal Peraro M, van der Goot FG, Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol 14, 77–92 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Los FC, Randis TM, Aroian RV, Ratner AJ, Role of pore-forming toxins in bacterial infectious diseases. Microbiol Mol Biol Rev 77, 173–207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devant P, Kagan JC, Molecular mechanisms of gasdermin D pore-forming activity. Nat Immunol 24, 1064–1075 (2023). [DOI] [PubMed] [Google Scholar]
- 4.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J, Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25, 1285–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H, Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nygaard TK, Pallister KB, DuMont AL, DeWald M, Watkins RL, Pallister EQ, Malone C, Griffith S, Horswill AR, Torres VJ, Voyich JM, Alpha-toxin induces programmed cell death of human T cells, B cells, and monocytes during USA300 infection. PLoS One 7, e36532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA, Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One 4, e7446 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhakdi S, Tranum-Jensen J, Alpha-toxin of Staphylococcus aureus. Microbiol Rev 55, 733–751 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Xu W, Zhou R, NLRP3 inflammasome activation and cell death. Cell Mol Immunol 18, 2114–2127 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brito C, Cabanes D, Sarmento Mesquita F, Sousa S, Mechanisms protecting host cells against bacterial pore-forming toxins. Cell Mol Life Sci 76, 1319–1339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ammendolia DA, Bement WM, Brumell JH, Plasma membrane integrity: implications for health and disease. BMC Biol 19, 71 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatta AT, Carlton JG, The ESCRT-machinery: closing holes and expanding roles. Curr Opin Cell Biol 59, 121–132 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR, ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300 e216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jimenez AJ, Maiuri P, Lafaurie-Janvore J, Divoux S, Piel M, Perez F, ESCRT machinery is required for plasma membrane repair. Science 343, 1247136 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Mo F, Wang Y, Li W, Chen Y, Liu J, Chen-Mayfield TJ, Hu Q, Enhancing Gasdermin-induced tumor pyroptosis through preventing ESCRT-dependent cell membrane repair augments antitumor immune response. Nat Commun 13, 6321 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ritter AT, Shtengel G, Xu CS, Weigel A, Hoffman DP, Freeman M, Iyer N, Alivodej N, Ackerman D, Voskoboinik I, Trapani J, Hess HF, Mellman I, ESCRT-mediated membrane repair protects tumor-derived cells against T cell attack. Science 376, 377–382 (2022). [DOI] [PubMed] [Google Scholar]
- 18.Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P, ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Yoon S, Kovalenko A, Bogdanov K, Wallach D, MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 47, 51–65 e57 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Corrotte M, Fernandes MC, Tam C, Andrews NW, Toxin pores endocytosed during plasma membrane repair traffic into the lumen of MVBs for degradation. Traffic 13, 483–494 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Stuart L, Ohsumi TK, Burgess S, Varshney GK, Dastur A, Borowsky M, Benes C, Lacy-Hulbert A, Schmidt EV, Transposon activation mutagenesis as a screening tool for identifying resistance to cancer therapeutics. BMC Cancer 13, 93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haslinger B, Strangfeld K, Peters G, Schulze-Osthoff K, Sinha B, Staphylococcus aureus alpha-toxin induces apoptosis in peripheral blood mononuclear cells: role of endogenous tumour necrosis factor-alpha and the mitochondrial death pathway. Cell Microbiol 5, 729–741 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Menzies BE, Kourteva I, Staphylococcus aureus alpha-toxin induces apoptosis in endothelial cells. FEMS Immunol Med Microbiol 29, 39–45 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Bruchez A, Sha K, Johnson J, Chen L, Stefani C, McConnell H, Gaucherand L, Prins R, Matreyek KA, Hume AJ, Muhlberger E, Schmidt EV, Olinger GG, Stuart LM, Lacy-Hulbert A, MHC class II transactivator CIITA induces cell resistance to Ebola virus and SARS-like coronaviruses. Science 370, 241–247 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarver AL, Erdman J, Starr T, Largaespada DA, Silverstein KA, TAPDANCE: an automated tool to identify and annotate transposon insertion CISs and associations between CISs from next generation sequence data. BMC Bioinformatics 13, 154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J, A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med 17, 1310–1314 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kloft N, Busch T, Neukirch C, Weis S, Boukhallouk F, Bobkiewicz W, Cibis I, Bhakdi S, Husmann M, Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem Biophys Res Commun 385, 503–506 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Ho AK, Wagstaff JL, Manna PT, Wartosch L, Qamar S, Garman EF, Freund SM, Roberts RC, The topology, structure and PE interaction of LITAF underpin a Charcot-Marie-Tooth disease type 1C. BMC Biol 14, 109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SM, Olzmann JA, Chin LS, Li L, Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J Cell Sci 124, 3319–3331 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin W, Wunderley L, Barrett AL, High S, Woodman PG, The Charcot Marie Tooth disease protein LITAF is a zinc-binding monotopic membrane protein. Biochem J 473, 3965–3978 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu H, Guariglia S, Yu RY, Li W, Brancho D, Peinado H, Lyden D, Salzer J, Bennett C, Chow CW, Mutation of SIMPLE in Charcot-Marie-Tooth 1C alters production of exosomes. Mol Biol Cell 24, 1619–1637, S1611–1613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lacerda AF, Hartjes E, Brunetti CR, LITAF mutations associated with Charcot-Marie-Tooth disease 1C show mislocalization from the late endosome/lysosome to the mitochondria. PLoS One 9, e103454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Husmann M, Beckmann E, Boller K, Kloft N, Tenzer S, Bobkiewicz W, Neukirch C, Bayley H, Bhakdi S, Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett 583, 337–344 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Lee SM, Chin LS, Li L, Charcot-Marie-Tooth disease-linked protein SIMPLE functions with the ESCRT machinery in endosomal trafficking. J Cell Biol 199, 799–816 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Street VA, Bennett CL, Goldy JD, Shirk AJ, Kleopa KA, Tempel BL, Lipe HP, Scherer SS, Bird TD, Chance PF, Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology 60, 22–26 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Shirk AJ, Anderson SK, Hashemi SH, Chance PF, Bennett CL, SIMPLE interacts with NEDD4 and TSG101: evidence for a role in lysosomal sorting and implications for Charcot-Marie-Tooth disease. J Neurosci Res 82, 43–50 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Marmor MD, Yarden Y, Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 23, 2057–2070 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Grimsey NJ, Narala R, Rada CC, Mehta S, Stephens BS, Kufareva I, Lapek J, Gonzalez DJ, Handel TM, Zhang J, Trejo J, A Tyrosine Switch on NEDD4–2 E3 Ligase Transmits GPCR Inflammatory Signaling. Cell Rep 24, 3312–3323 e3315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie W, Jin S, Wu Y, Xian H, Tian S, Liu DA, Guo Z, Cui J, Auto-ubiquitination of NEDD4-1 Recruits USP13 to Facilitate Autophagy through Deubiquitinating VPS34. Cell Rep 30, 2807–2819 e2804 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Xie J, Fang J, Lv M, Yang M, Deng Z, Xie Y, Cai L, Nigericin exerts anticancer effects through inhibition of the SRC/STAT3/BCL-2 in osteosarcoma. Biochem Pharmacol 198, 114938 (2022). [DOI] [PubMed] [Google Scholar]
- 41.Corrotte M, Almeida PE, Tam C, Castro-Gomes T, Fernandes MC, Millis BA, Cortez M, Miller H, Song W, Maugel TK, Andrews NW, Caveolae internalization repairs wounded cells and muscle fibers. Elife 2, e00926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romero M, Keyel M, Shi G, Bhattacharjee P, Roth R, Heuser JE, Keyel PA, Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ 24, 798–808 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bushell KN, Leeman SE, Gillespie E, Gower AC, Reed KL, Stucchi AF, Becker JM, Amar S, LITAF mediation of increased TNF-alpha secretion from inflamed colonic lamina propria macrophages. PLoS One 6, e25849 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Zuo Z, Sastalla I, Liu C, Jang JY, Sekine Y, Li Y, Pirooznia M, Leppla SH, Finkel T, Liu S, Sequential CRISPR-Based Screens Identify LITAF and CDIP1 as the Bacillus cereus Hemolysin BL Toxin Host Receptors. Cell Host Microbe 28, 402–410 e405 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moshal KS, Roder K, Kabakov AY, Werdich AA, Chiang DY, Turan NN, Xie A, Kim TY, Cooper LL, Lu Y, Zhong M, Li W, Terentyev D, Choi BR, Karma A, MacRae CA, Koren G, LITAF (Lipopolysaccharide-Induced Tumor Necrosis Factor) Regulates Cardiac L-Type Calcium Channels by Modulating NEDD (Neural Precursor Cell Expressed Developmentally Downregulated Protein) 4–1 Ubiquitin Ligase. Circ Genom Precis Med 12, 407–420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolino A, Bolis A, Previtali SC, Dina G, Bussini S, Dati G, Amadio S, Del Carro U, Mruk DD, Feltri ML, Cheng CY, Quattrini A, Wrabetz L, Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol 167, 711–721 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, Lupski JR, Weisman LS, Meisler MH, Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448, 68–72 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lupo V, Galindo MI, Martinez-Rubio D, Sevilla T, Vilchez JJ, Palau F, Espinos C, Missense mutations in the SH3TC2 protein causing Charcot-Marie-Tooth disease type 4C affect its localization in the plasma membrane and endocytic pathway. Hum Mol Genet 18, 4603–4614 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Previtali SC, Quattrini A, Bolino A, Charcot-Marie-Tooth type 4B demyelinating neuropathy: deciphering the role of MTMR phosphatases. Expert Rev Mol Med 9, 1–16 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Senderek J, Bergmann C, Weber S, Ketelsen UP, Schorle H, Rudnik-Schoneborn S, Buttner R, Buchheim E, Zerres K, Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet 12, 349–356 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Tersar K, Boentert M, Berger P, Bonneick S, Wessig C, Toyka KV, Young P, Suter U, Mtmr13/Sbf2-deficient mice: an animal model for CMT4B2. Hum Mol Genet 16, 2991–3001 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, FitzPatrick D, Schmedding E, De Vriendt E, Jacobs A, Van Gerwen V, Wagner K, Hartung HP, Timmerman V, Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 72, 722–727 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hussien Y, Podojil JR, Robinson AP, Lee AS, Miller SD, Popko B, ER Chaperone BiP/GRP78 Is Required for Myelinating Cell Survival and Provides Protection during Experimental Autoimmune Encephalomyelitis. J Neurosci 35, 15921–15933 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin W, Stone S, Unfolded protein response in myelin disorders. Neural Regen Res 15, 636–645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ingham RJ, Gish G, Pawson T, The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23, 1972–1984 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Rotin D, Staub O, Haguenauer-Tsapis R, Ubiquitination and endocytosis of plasma membrane proteins: role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J Membr Biol 176, 1–17 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Horák J, The role of ubiquitin in down-regulation and intracellular sorting of membrane proteins: insights from yeast. Biochimica et Biophysica Acta (BBA) - Biomembranes 1614, 139–155 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D, Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J 16, 6325–6336 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simonin A, Fuster D, Nedd4–1 and beta-arrestin-1 are key regulators of Na+/H+ exchanger 1 ubiquitylation, endocytosis, and function. J Biol Chem 285, 38293–38303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rougier JS, Albesa M, Abriel H, Viard P, Neuronal precursor cell-expressed developmentally down-regulated 4–1 (NEDD4-1) controls the sorting of newly synthesized Ca(V)1.2 calcium channels. J Biol Chem 286, 8829–8838 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fotia AB, Ekberg J, Adams DJ, Cook DI, Poronnik P, Kumar S, Regulation of neuronal voltage-gated sodium channels by the ubiquitin-protein ligases Nedd4 and Nedd4-2. J Biol Chem 279, 28930–28935 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Wang J, Peng Q, Lin Q, Childress C, Carey D, Yang W, Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J Biol Chem 285, 12279–12288 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, Nunnari J, Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J Cell Biol 209, 539–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Besprozvannaya M, Dickson E, Li H, Ginburg KS, Bers DM, Auwerx J, Nunnari J, GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanada K, Lipid transfer proteins rectify inter-organelle flux and accurately deliver lipids at membrane contact sites. J Lipid Res 59, 1341–1366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson FL, Dixon JE, Myotubularin phosphatases: policing 3-phosphoinositides. Trends Cell Biol 16, 403–412 (2006). [DOI] [PubMed] [Google Scholar]
- 67.Kang Y, Feng M, Zhao X, Dai X, Xiang B, Gao P, Li Y, Li Y, Ren T, Newcastle disease virus infection in chicken embryonic fibroblasts but not duck embryonic fibroblasts is associated with elevated host innate immune response. Virol J 13, 41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, Sandoval W, Yan D, Kang J, Xu M, Zhang J, Lee WP, McKenzie BS, Ulas G, Payandeh J, Roose-Girma M, Modrusan Z, Reja R, Sagolla M, Webster JD, Cho V, Andrews TD, Morris LX, Miosge LA, Goodnow CC, Bertram EM, Dixit VM, NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM, Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8(+) T Cell Terminal Differentiation and Loss of Multipotency. Immunity 46, 596–608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bergemann TL, Starr TK, Yu H, Steinbach M, Erdmann J, Chen Y, Cormier RT, Largaespada DA, Silverstein KA, New methods for finding common insertion sites and co-occurring common insertion sites in transposon- and virus-based genetic screens. Nucleic Acids Res 40, 3822–3833 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H, Liu J, Pan T, Chen J, Wu M, Zhou X, Yuan Z, Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat Immunol 14, 793–803 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF, Thery C, Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12, 19–30; sup pp 11–13 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Visvikis O, Boyer L, Torrino S, Doye A, Lemonnier M, Lores P, Rolando M, Flatau G, Mettouchi A, Bouvard D, Veiga E, Gacon G, Cossart P, Lemichez E, Escherichia coli producing CNF1 toxin hijacks Tollip to trigger Rac1-dependent cell invasion. Traffic 12, 579–590 (2011). [DOI] [PubMed] [Google Scholar]
- 74.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA, Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full analysis of screen results are presented in Supplementary Material. DNA Sequencing data is deposited at GEO (GSE# GSE242913). The PB transposon was obtained under a material transfer agreement with the Wellcome Trust Sanger Institute. All other data are available in the manuscript or the supplementary materials.