

ABSTRACT
Background
KCNQ4 is a common genetic cause of nonsyndromic autosomal dominant hearing loss. We have identified the family in China with a KCNQ4 (c.701A>G; p.His234Arg) missense variation. In this study, a survey and analysis were performed to investigate the audiological and genetic characteristics of the Chinese family.
Methods
The medical history of family members was collected, and the family members underwent pure tone audiometry, acoustic immittance, and physical examination. The proband was additionally examined by ABR (auditory brainstem response) and DPOAE (distortion product otoacoustic emission). DNA samples from family members were collected, and the possible causative gene of the proband was detected by whole‐exome sequencing (WES), which was verified by Sanger sequencing in family members.
Results
The inheritance pattern of the family was an autosomal dominant nonsyndromic type. The hearing loss was characterized by postlingual deafness, high‐frequency hearing loss in the early stage, gradually involving the full frequency. About 32–40 years of age, the hearing gradually became stable, the decline rate slowed down, and the final degree of hearing loss was severe. WES results showed that the KCNQ4 gene had a missense variation (c.701A>G; p.His234Arg).
Conclusion
This family has autosomal dominant nonsyndromic hereditary hearing loss caused by a variation in the KCNQ4 gene, characterized by high‐frequency hearing loss.
Keywords: Chinese family, hereditary hearing loss, KCNQ4 gene, whole‐exome sequencing
A Chinese family with a KCNQ4 (c.701A>G; p.His234Arg) missense variation was identified. The hearing loss was characterized by postlingual deafness, with high‐frequency hearing loss. This finding provides evidence for otolaryngologists to counsel patients, facilitating clinical decisions related to reproductive planning and childcare.

1. Introduction
Deafness, a prevalent hearing and sensory disorder, affects approximately 1.5 billion people worldwide. According to the WHO, this number is projected to increase to 2.5 billion by 2050 (Chadha et al. 2021). Genetic factors are reported to be the most significant contributors, accounting for approximately 50%–60% of all deafness cases (Kvestad et al. 2012; Wolber et al. 2012). Hereditary hearing loss can be categorized into two types based on symptoms: syndromic hearing loss (SHL), which includes other systemic symptoms, and nonsyndromic hearing loss (NSHL), which does not (Sheffield and RJH 2019). To date (December 19, 2024), 156 genes have been reported to be associated with NSHL, and more than 50 genes have been associated with SHL.(https://hereditaryhearingloss.org/). NSHL accounts for about 70% of these cases. Common inheritance patterns of NSHL include autosomal dominant, autosomal recessive, X‐chromosome linked, Y‐chromosome related, and mitochondrial (Del Castillo et al. 2022). Among these, autosomal recessive nonsyndromic hearing loss (ARNSHL) is the most common, typically presenting as prelingual deafness involving all frequencies and ranging from severe to profound hearing loss (Imtiaz 2022). In contrast, autosomal dominant nonsyndromic hearing loss (ADNSHL) accounts for about 20%, often beginning postlingually, is mainly high‐frequency, progressive, and generally milder than ARNSHL (Lezirovitz and Mingroni‐Netto 2022). Other forms of hereditary deafness are rare.
In this study, we identified a c.701A>G (NM_004700) variation in the KCNQ4 (OMIM#603537) gene in a Chinese family with ADNSHL. The clinical and genetic characteristics of the family were investigated in detail. The research process is presented as follows.
2. Methods
2.1. Ethical Compliance
In this study, our survey and analysis were approved by the Ethics Committee of the General Hospital of Central Theater Command.
2.2. Family and Clinical Evaluation
The proband who participated in this study presented at General Hospital of Central Theater Command in 2022 due to binaural hearing loss and had a history of deafness in her family. Therefore, we launched an investigation into her family. The family, from Hubei Province, China, was designated as CHZ‐01. There were 33 members in the CHZ‐01 family across four generations, including five deceased members and 28 living members. All 28 members participated in this investigation. The family pedigree was drawn based on the survey results (Figure 1A).
FIGURE 1.
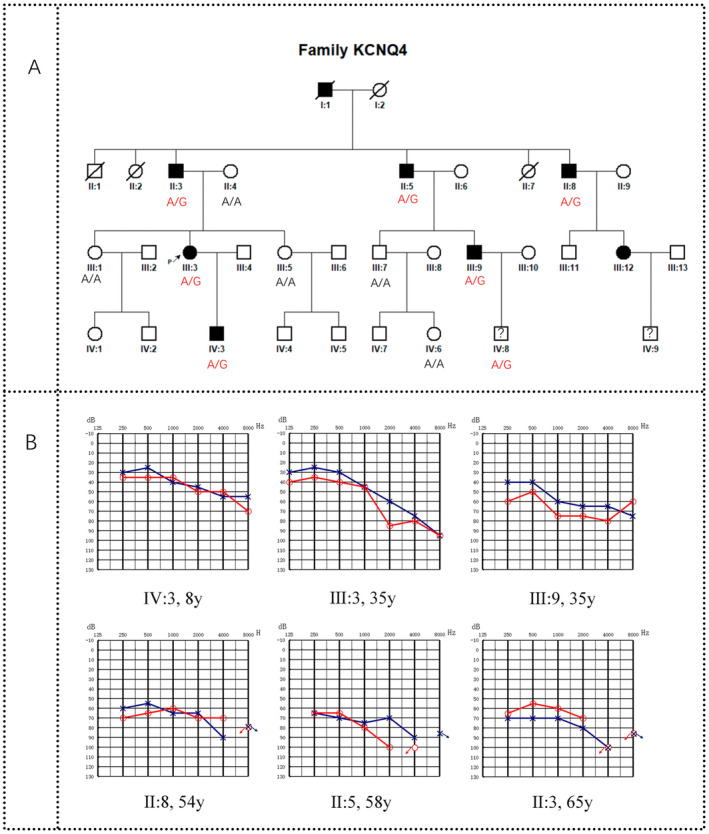
(A) family pedigree of CHZ‐01. This figure shows that there are four generations in this family, included five deceased members and 28 living members, of which seven were deafness and two were clinically uncertain. The blood samples from six individuals with hearing loss and six asymptomatic individuals were used for Sanger sequencing, and the results were indicated as A/A or A/G. (A/A: Wild‐type with no variation; A/G: Heterozygous with variation.) (B) Audiogram of patients in·family·CHZ‐01.
All family members participating in the study signed the informed consent (for minors, the consent was signed by their guardians). After obtaining the consent of the patients and guardians, we took the patients' medical history in detail and conducted a physical examination, audiological examination (including electric otoscopy, pure tone audiometry, acoustic immittance examination), and intelligence assessment. The proband also underwent distortion product otoacoustic emission (DPOAE) and auditory brainstem response (ABR). Peripheral venous blood samples (10 mL each member) were taken from 12 family members (6 patients with hearing loss and 6 asymptomatic individuals) for gene detection.
2.3. Whole‐Exome Sequencing
Five milliliters of peripheral blood was drawn from the proband, and we then extracted the Genomic DNA (gDNA) according to the manufacturer's standard procedure (MagPure Buffy Coat DNA Midi KF Kit, MAGEN). The gDNA was sheared into fragments, and the 200–300 bp fragments were collected by magnetic bead (Vahtstm DNA Clean Beads, VAZYME). The library was enriched for 16 h at 65°C by array hybridization (KAPA Hyper Exome, ROCHE), followed by elution and postcapture amplification. The products were then subjected to Caliper GX_1000 Kit and BMG to estimate the magnitude of enrichment. The qualified products were converted into DNB; finally, sequenced with PE100 + 10 on MGISEQ‐2000.
2.4. Data Analysis
To detect the potential variants in the family, we performed bioinformatics processing and data analysis after receiving the primary sequencing data. We used previously published filtering criteria to generate “clean reads” for further analysis (Wei et al. 2011). The “clean reads” (with a length of 90 bp) derived from targeted sequencing and filtering were then aligned to the human genome reference (hg19) using the BWA (Burrows Wheeler Aligner) Multi‐Vision software package (Li and Durbin 2009). After alignment, the output files were used to perform coverage and depth analysis of the target region, as well as single‐nucleotide variants (SNVs) and INDEL calling. We used GATK software (McKenna et al. 2010) to detect SNVs and indels. All SNVs and indels were filtered and annotated via multiple databases, including NCBI dbSNP and gnomAD. All known deafness genes that have been reported were used for analysis. To predict the effect of missense variants, we used dbNSFP (Lucas et al. 2012) which contains seven well‐established in silico prediction programs (Scale‐Invariant Feature Transform (SIFT), Polyphen2, LRT, Mutation Taster, and PhyloP). Pathogenic variants are assessed under the protocol issued by ACMG (Richards et al. 2015).
2.5. Sanger Sequencing Validation
All mutations and potential pathogenic variants were validated using conventional Sanger sequencing. (Primers for Sanger sequencing, F: CCTTCTGTGTCATCGGTAATG R: ACGAGTGGTCCCTTCAAAC).
3. Results
3.1. Clinical Characteristics
There were four generations in the CHZ‐01 family. All 28 living members of the family participated in the survey, including 17 males and 11 females, aged from 1 to 65 years. Seven patients had hearing loss (II: 3, II: 5, II: 8, III: 3, III: 9, III: 12, IV: 3) (Figure 1B, Patient III: 12 was unable to obtain an audiogram due to refusal of hearing examination), no speech disorder, no other organ and system symptoms, no history of aminoglycoside antibiotic use, no history of head or ear trauma, no history of noise exposure, and no obvious history of ear infection before onset. The age of onset ranged from 8 to 25 years. Two of them (II: 8, III: 9) developed tinnitus but no vestibular symptoms such as vertigo after years of illness (Table 1). No ear abnormalities such as external ear malformation, ear trauma, tympanic membrane perforation, tympanic effusion, or tympanic sclerosis were found in the special examination, and no obvious abnormalities were found in the physical examination. Among the five deceased family members, one family member (I: 1) was considered by his family members to have hearing loss when he was young, and the specific age of onset was unknown. The other four members (I: 2, II: 1, II: 2, II: 7) had no hearing loss. The clinical characteristics of this family were summarized as follows: nonsyndromic deafness, autosomal dominant inheritance, postlingual deafness, onset age between 8 and 25 years old, and hearing loss was gradual. The degree of hearing loss in young people is mild and gradually becomes severe with age.
TABLE 1.
Clinical data of patients in the CHZ‐01 family.
| Subject | Gender | Age of test (years) | Age of onset (years) | Better ear | ||
|---|---|---|---|---|---|---|
| PTA (dB HL) | Hearing loss | Associated symptoms | ||||
| II:3 | Male | 63 | 25 | 71.25 | Severe | Not |
| II:5 | Male | 58 | 25 | 72.5 | Severe | Not |
| II:8 | Male | 54 | 11 | 66.25 | Severe | Tinnitus |
| III:3 | Female | 35 | 11 | 57.5 | Moderately severe | Not |
| III:9 | Male | 35 | 17 | 52.5 | Moderately severe | Tinnitus |
| IV:3 | Male | 11 | 8 | 50 | Moderate | Not |
Note: PTA, pure‐tone air‐conduction averages (0.5, 1, 2, and 4 kHz) for the better hearing ear of affected subjects in family CHZ‐01.
3.2. Causative Gene Analysis
To explore the genetic variation in the CHZ‐01 family, we used the WES method to detect the blood samples of the proband. The WES had a mean depth of 334X, with coverage over 20X at 99.79%. By filtering variants in known deafness genes, a candidate variant in this family was found: a missense variant occurring in exon 4 of the KCNQ4 gene (c.701A>G, p. His234Arg). Subsequently, Sanger sequencing was performed. This variant demonstrated phenotype–genotype co‐segregation in the family (PP1_strong) (Figure 2A,B). The variant was not detected in gnomAD (PM2). The gene variation led to the substitution of histidine at position 234 of the amino acid sequence by arginine, while histidine at position 234 was conserved among species (Figure 2C). Based on prediction, this is a damaging variant for protein function (CADD:27.4, REVEL:0.944) (PP3). His234Arg completely abrogates the potassium channel function of KCNQ4 (PS4_supporting) (Zheng et al. 2022). A similar pathogenic variant (c.701A>T, p.His234Leu) in KCNQ4 was identified by Uehara et al. in a Brazilian family (PM5) (Uehara et al. 2015). According to the ACMG guidelines, this variant was classified as pathogenic.
FIGURE 2.

Sanger sequencing detected a heterozygous variation c.701 A>G in the KCNQ4 gene of the patients(A), and normal members did not detect the variation(B). It is indicated by the red arrow. Conservation analysis of residues 194–238 of the KCNQ4 protein. The 234 position is highlighted by an open‐red rectangle. Histidine at position 234 was conserved among species (C).
4. Discussion
We found that a variation in the KCNQ4 gene, located at position 1p34.2, may be responsible for the hereditary hearing loss in the CHZ‐01 pedigree. The KCNQ4 gene, which contains 14 exons, encodes the Kv7.4 subunit. This includes the N‐ and C‐termini, six transmembrane regions (S1–S6), and a P‐loop (located at S5–S6 linker area), forming a voltage‐gated potassium ion channel on the cell membrane (Zheng et al. 2022). The Kv7.4 potassium channel, primarily situated on the basolateral side of OHCs (outer hair cells), is responsible for releasing potassium ions from the hair cells into the perilymph; therefore, maintaining ion circulation and homeostasis in the inner ear (Maamrah et al. 2023). In our study, we identified the c.701A>G variation in the KCNQ4 gene. The variation, located on exon 4, affects the S4–S5 linker of the Kv7.4 subunit (Zhang et al. 2023). The specific amino acid change involves the replacement of histidine (His) at position 234 with arginine (Arg).
KCNQ4 gene mutations can lead to hereditary deafness, clinically classified as DFNA2A (OMIM#600101) type, characterized by autosomal dominant inheritance, nonsyndromic type, postlingual deafness, and mainly high‐frequency hearing loss (Aldè et al. 2023). So far, 47 KCNQ4 gene mutations have been reported in the literature, with exons 5 and 6, which encode the S5 and S6 transmembrane domains and the P‐loop between them, having the most mutations (24 in total). These are followed by exons 7–14 (11 in total), encoding the C‐terminus of the Kv7.4 potassium channel. The number of mutations in exons 1 and 4 is tied for third; exon 1 (5 in total) encodes the N‐terminus and part of the S1 transmembrane domain of the Kv7.4 potassium channel, while exon 4 (5 in total) encodes the S4 transmembrane domain, S3–S4 linker, and S4–S5 linker. The fewest mutations have been found in exon 3 (2 in total), which encodes the S2 and S3 transmembrane domains and the S2–S3 linker of the Kv7.4 potassium channel. No mutations in exon 2 have been reported (Zhang et al. 2023) (Figure 3).
FIGURE 3.

A total of 47 KCNQ4 mutations have been reported.
The different regions of the Kv7.4 potassium channel protein serve different functions. The P‐loop located in the S5–S6 linker is crucial, primarily facilitating the ion selection function of the potassium channel. Other areas surround the P‐loop and assist in its functions (Peixoto Pinheiro et al. 2022). The variation discovered in our study, KCNQ4 gene (c.701A>G, p. His234Arg), located at the S4–S5 linker of the Kv7.4 potassium channel protein, a key region for sensing voltage and gating ion channels, potentially influences the formation of the core region (P‐loop) structure. According to the pathogenicity calculation method based on the ACMG guideline, the evidence of this variant is PP1_strong + PM2 + PM5 + PP3 + PS4_supporting, and it is classified as pathogenic. Zheng et al. used the whole‐cell patch clamp technique to explore the functional changes of potassium channels caused by 4085 missense mutations in the KCNQ4 gene in CHO‐K1 cells and found that the H234R completely abrogates the potassium channel function of KCNQ4 but does not exert a strong dominant‐negative inhibitory effect on wild‐type KCNQ4 (when co‐expressed); these mutations in the S4–S5 linker are most likely to lead to decreased function of Kv7.4 channels (Zheng et al. 2022). Similarly, the variant found by Uehara et al. in a Brazilian family, KCNQ4 (c.701A>T, p.His234Leu), located at the same position but with a different base change, caused mild to severe high‐frequency hearing loss in the proband, her mother, and her sister. These two similar mutations exhibit similar clinical hearing characteristics, suggesting that mutations at this position are indeed pathogenic, but the mechanism still requires further study (Uehara et al. 2015). In addition, three other mutations have been found in the S4–S5 linker. Among them, Baek et al. discovered a KCNQ4 deletion mutation (c.664_681del, p.T223_ G228del) in 2011 in a Korean family. The hearing of the family was characterized by moderate to severe slow drop‐type high‐frequency hearing loss (Baek et al. 2011). Subsequently, Naito T et al. found a KCNQ4 missense mutation (c.689 T>A, p. Val230Glu) in a Japanese family in 2013. The hearing of this family was characterized by moderate to severe valley‐type mid‐frequency hearing loss (Naito et al. 2013). The latest discovery was a KCNQ4 missense mutation (c.683G>A, p. Gly228Asp) discovered by Cui et al. in 2022 in a Chinese family with mild to moderate slow‐drop high‐frequency hearing loss (Cui et al. 2022). Our findings in the CHZ‐01 family, along with the above mutations in the S4–S5 linker, all led to hearing loss, indicating that the stability of the S4–S5 linker structure is very important for the function of the Kv7.4 potassium channel. On the contrary, mutations in the S4–S5 linker may lead to hearing loss. Quaio et al. also reported the His234Arg variant, but it was found in an individual with normal hearing (the patient's age and other details were not mentioned in the article) (Quaio et al. 2022). The variant carrier might not have reached the age of onset. It is possible that the variant carrier will still experience hearing loss in the future. This may be the same situation as the clinically uncertain cases we found; in our study, IV:8 was an 11‐year‐old male child with no symptoms of hearing loss at the time of investigation, and Sanger sequencing detected a KCNQ4 variant (c.701A>G; p.His234Arg) in him. This highlights how genetic diagnosis helps with early detection and intervention.
Research on the treatment of hereditary deafness caused by KCNQ4 gene mutations is ongoing and includes drug treatments and gene therapy. Some drugs that activate the Kv7.4 channel have been identified, but their clinical use is limited due to side effects. The main challenges include improving the drug's lipophilicity and optimizing the drug according to mechanism studies (Li et al. 2021; Osuma et al. 2019; Wang et al. 2018). Since the advent of gene editing technologies, gene therapy holds the potential to improve patients' hearing by targeting specific genetic mutations (Noh et al. 2022).
In conclusion, we have identified a mutation in the KCNQ4 gene(c.701A>G, p. His234Arg) that leads to structural changes in the S4–S5 linker of the Kv7.4 potassium channel protein. This provides evidence for otolaryngologists to offer consultations to patients, contributing to the implementation of clinical decisions regarding eugenics and childcare. However, the specific treatment plan still depends on further research on the pathogenesis.
Author Contributions
Yi Sun and Yu Lu contributed to study design, genetic counseling and revision of the manuscript; Guo‐Qing Gong and Cheng‐Cheng Huang and Hui‐Yu Jin contributed to family clinical data collection and draft of the manuscript; Chang‐Liang Yang and Guang Yang contributed to protein conservation analysis; Hui‐Fang Lu, Yue‐Bin Yang and Zhao Zhang contributed to WES data analysis; Jing‐Yuan Cao, Li‐Wang and Rui‐Yao Chen contributed to audiological examination; Yi‐Ming Ji contributed to polish the manuscript. All authors have read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding: This work was supported by the Natural Science Foundation of Hubei Province (2018CFB719), the Fund Project of Health Commission of Hubei Province (WJ2023M089), and the Hubei Key Laboratory of Central Nervous System Tumor and Intervention (ZZYKF202206).
Guo‐Qing Gong, Cheng‐Cheng Huang, and Hui‐Yu Jin had equal contributions to this study.
Contributor Information
Yi Sun, Email: sunyi_wuhan@126.com.
Yu Lu, Email: samuelluyu@163.com.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Aldè, M. , Cantarella G., Zanetti D., et al. 2023. “Autosomal Dominant Non‐Syndromic Hearing Loss (DFNA): A Comprehensive Narrative Review.” Biomedicine 11, no. 6: 1616. Published 2023 Jun 1. 10.3390/biomedicines11061616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek, J. I. , Park H. J., Park K., et al. 2011. “Pathogenic Effects of a Novel Mutation (c.664_681del) in KCNQ4 Channels Associated With Auditory Pathology.” Biochimica et Biophysica Acta 1812, no. 4: 536–543. 10.1016/j.bbadis.2010.09.001 Epub 2010 Sep 9. [DOI] [PubMed] [Google Scholar]
- Chadha, S. , Kamenov K., and Cieza A.. 2021. “The World Report on Hearing.” Bulletin of the World Health Organization 99, no. 4: 242. 10.2471/BLT.21.285643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, C. , Zhang L., Qian F., et al. 2022. “A Humanized Murine Model, Demonstrating Dominant Progressive Hearing Loss Caused by a Novel KCNQ4 Mutation (p.G228D) From a Large Chinese Family.” Clinical Genetics 102, no. 2: 149–154. 10.1111/cge.14164 Epub 2022 May 28. [DOI] [PubMed] [Google Scholar]
- Del Castillo, I. , Morín M., Domínguez‐Ruiz M., and Moreno‐Pelayo M. A.. 2022. “Genetic Etiology of Non‐syndromic Hearing Loss in Europe.” Human Genetics 141, no. 3–4: 683–696. 10.1007/s00439-021-02425-6. [DOI] [PubMed] [Google Scholar]
- Imtiaz, A. 2022. “ARNSHL Gene Identification: Past, Present and Future.” Molecular Genetics and Genomics 297, no. 5: 1185–1193. 10.1007/s00438-022-01926-x. [DOI] [PubMed] [Google Scholar]
- Kvestad, E. , Czajkowski N., Krog N. H., Engdahl B., and Tambs K.. 2012. “Heritability of Hearing Loss.” Epidemiology 23, no. 2: 328–331. 10.1097/EDE.0b013e318245996e. [DOI] [PubMed] [Google Scholar]
- Lezirovitz, K. , and Mingroni‐Netto R. C.. 2022. “Genetic Etiology of Non‐syndromic Hearing Loss in Latin America.” Human Genetics 141, no. 3–4: 539–581. 10.1007/s00439-021-02354-4. [DOI] [PubMed] [Google Scholar]
- Li, H. , and Durbin R.. 2009. “Fast and Accurate Short Read Alignment With Burrows‐Wheeler Transform.” Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, T. , Wu K., Yue Z., Wang Y., Zhang F., and Shen H.. 2021. “Structural Basis for the Modulation of Human KCNQ4 by Small‐Molecule Drugs.” Molecular Cell 81, no. 1: 25–37. 10.1016/j.molcel.2020.10.037 Epub 2020 Nov 24. [DOI] [PubMed] [Google Scholar]
- Lucas, F. A. S. , Wang G., Scheet P., and Peng B.. 2012. “Integrated Annotation and Analysis of Genetic Variants From Next‐Generation Sequencing Studies With Variant Tools.” Bioinformatics 28, no. 3: 421–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maamrah, B. , Pocsai K., Bayasgalan T., Csemer A., and Pál B.. 2023. “KCNQ4 Potassium Channel Subunit Deletion Leads to Exaggerated Acoustic Startle Reflex in Mice.” Neuroreport 34, no. 4: 232–237. 10.1097/WNR.0000000000001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna M., Banks E., et al. 2010. “The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next‐Generation DNA Sequencing Data.” Genome Research 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, T. , Nishio S. Y., Iwasa Y., et al. 2013. “Comprehensive Genetic Screening of KCNQ4 in a Large Autosomal Dominant Nonsyndromic Hearing Loss Cohort: Genotype‐Phenotype Correlations and a Founder Mutation.” PLoS One 8, no. 5: e63231. 10.1371/journal.pone.0063231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh, B. , Rim J. H., Gopalappa R., et al. 2022. “In Vivo Outer Hair Cell Gene Editing Ameliorates Progressive Hearing Loss in Dominant‐Negative Kcnq4 Murine Model.” Theranostics 12, no. 5: 2465–2482. 10.7150/thno.67781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuma, A. T. , Xu X., Wang Z., Van Camp J. A., and Freiberg G. M.. 2019. “Design and Evaluation of Pyrazolopyrimidines as KCNQ Channel Modulators.” Bioorganic & Medicinal Chemistry Letters 29, no. 19: 126603. 10.1016/j.bmcl.2019.08.007 Epub 2019 Aug 6. [DOI] [PubMed] [Google Scholar]
- Peixoto Pinheiro, B. , Müller M., Bös M., et al. 2022. “A Potassium Channel Agonist Protects Hearing Function and Promotes Outer Hair Cell Survival in a Mouse Model for Age‐Related Hearing Loss.” Cell Death & Disease 13, no. 7: 595. Published 2022 Jul 11. 10.1038/s41419-022-04915-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaio, C. R. A. C. , Coelho A. V. C., Moura L. M. S., et al. 2022. “Genomic Study of Nonsyndromic Hearing Loss in Unaffected Individuals: Frequency of Pathogenic and Likely Pathogenic Variants in a Brazilian Cohort of 2,097 Genomes.” Frontiers in Genetics 30, no. 13: 921324. 10.3389/fgene.2022.921324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz N., Bale S., et al. 2015. “Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.” Genetics in Medicine 17, no. 5: 405–424. 10.1038/gim.2015.30 Epub 2015 Mar 5, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield, A. M. , and RJH S.. 2019. “The Epidemiology of Deafness.” Cold Spring Harbor Perspectives in Medicine 9, no. 9: a033258. 10.1101/cshperspect.a033258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara, D. T. , Freitas É. L., Alves L. U., et al. 2015. “A Novel KCNQ4 Mutation and a Private IMMP2L‐DOCK4 Duplication Segregating With Nonsyndromic Hearing Loss in a Brazilian Family.” Human Genome Variation 29, no. 2: 15038. 10.1038/hgv.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Qiao G. H., Hu H. N., Gao Z. B., and Nan F. J.. 2018. “Discovery of Novel Retigabine Derivatives as Potent KCNQ4 and KCNQ5 Channel Agonists With Improved Specificity.” ACS Medicinal Chemistry Letters 10, no. 1: 27–33. 10.1021/acsmedchemlett.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, X. , Ju X., Yi X., et al. 2011. “Identification of Sequence Variants in Genetic Diseasecausing Genes Using Targeted Next‐Generation Sequencing.” PLoS One 6: e29500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber, L. E. , Steves C. J., Spector T. D., and Williams F. M.. 2012. “Hearing Ability With Age in Northern European Women: A New Web‐Based Approach to Genetic Studies.” PLoS One 7, no. 4: e35500. 10.1371/journal.pone.0035500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Wang H., Li J., et al. 2023. “The genotype‐phenotype correlation analysis and genetic counseling of hearing loss patients with novel KCNQ4 mutations.” Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 37, no. 1: 25–35. 10.13201/j.issn.2096-7993.2023.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, H. , Yan X., Li G., et al. 2022. “Proactive Functional Classification of all Possible Missense Single‐Nucleotide Variants in KCNQ4.” Genome Research 32, no. 8: 1573–1584. 10.1101/gr.276562.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Y. , Liu H., Chen Y., et al. 2022. “Structural Insights Into the Lipid and Ligand Regulation of a Human Neuronal KCNQ Channel.” Neuron 110, no. 2: 237–247. 10.1016/j.neuron.2021.10.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.