

Abstract
A terminator is a DNA region that ends the transcription process. Currently, multiple computational tools are available for predicting bacterial terminators. However, these methods are specialized for certain bacteria or terminator type (i.e. intrinsic or factor-dependent). In this work, we developed BacTermFinder using an ensemble of convolutional neural networks (CNNs) receiving as input four different representations of terminator sequences. To develop BacTermFinder, we collected roughly 41 000 bacterial terminators (intrinsic and factor-dependent) of 22 species with varying GC-content (from 28% to 71%) from published studies that used RNA-seq technologies. We evaluated BacTermFinder’s performance on terminators of five bacterial species (not used for training BacTermFinder) and two archaeal species. BacTermFinder’s performance was compared with that of four other bacterial terminator prediction tools. Based on our results, BacTermFinder outperforms all other four approaches in terms of average recall without increasing the number of false positives. Moreover, BacTermFinder identifies both types of terminators (intrinsic and factor-dependent) and generalizes to archaeal terminators. Additionally, we visualized the saliency map of the CNNs to gain insights on terminator motif per species. BacTermFinder is publicly available at https://github.com/BioinformaticsLabAtMUN/BacTermFinder.
Introduction
Transcription starts at the promoter region and ends at the terminator region. A terminator is a DNA segment that indicates the end of a gene or operon [1]. The termination of transcription is a process crucial for the accurate synthesis of RNA. In prokaryotes, termination can occur through either factor-dependent or factor-independent mechanisms, with the latter known as intrinsic termination. Intrinsic terminators are a DNA region rich in cytosine (C) and guanine (G) nucleotides followed by a poly-thymine (T) sequence. In the canonical terminator structure, the RNA created from the CG-rich region binds with itself, forming a hairpin structure that causes the RNA polymerase to stall. The weak base pairing between the adenine (A) nucleotides of the DNA template and the uridines of the RNA transcript let the transcript to detach from the template thus, terminating the transcription [2]. However, alternative intrinsic terminator structures have been identified in many bacteria [3]. While intrinsic termination requires only cis-acting elements on nascent RNAs, factor-dependent termination depends on both cis-acting RNA elements and a protein such as Rho (ρ), which mediates termination via three distinct mechanisms [4].
Identifying terminators is crucial for understanding operon structure and transcriptional regulation. As both terminator types rely, at least partially, on cis-acting elements on the DNA template, they can be predicted from genomic sequence. This can be done via computational identification. Genome-wide terminator prediction via computational approaches is more affordable and efficient than via wet lab approaches. Although terminator predictions should be considered putative terminators or terminator-like regions requiring further validation. Since the development of TransTermHP [5], which is arguably the most cited terminator prediction tool, there have been several computational tools developed for bacterial terminator prediction (summarized in Table 1). However, as one can see in Table 1, these tools have either (i) focused on few (one to three) bacterial species (mostly Escherichia coli and Bacillus subtilis), (ii) used a relatively small number of terminators for generating their model, (iii) predicted a single terminator type (factor-dependent or intrinsic), or (iv) a combination of the above. Thus, there is still the need for a species- and terminator type-independent tool for predicting bacterial terminators. The availability of genome-wide transcription termination sites (TTSs, also called in other studies 3′ termini or 3′ ends) identified by RNA-seq technologies such as Term-seq [6], SEnd-seq [7], SMRT-cappable [8], RendSeq [9], RNATag-seq [10], and dRNA-seq [11] in several bacterial species opens the door to generate a species-agnostic machine learning-based model using a large number (i.e. thousands) of terminator sequences of a wide range of bacterial species. Here, we generated such a method by (i) gathering a large collection of TTSs from published studies, (ii) exploring thousands of features to represent (encode) terminator sequences, (iii) generating and assessing 11 different machine learning models to identify bacterial terminators, and (iv) comparatively assessing the performance of our best model (BacTermFinder) with the performance of four other bacterial terminator prediction methods (namely, TermNN [12], iTerm-PseKNC [13], RhoTermPredict [14], and TransTermHP [5]). Our results show that BacTermFinder can detect intrinsic and factor-dependent terminators and even archaeal terminators at a higher recall rate than current tools.
Table 1.
Summary of tools for predicting bacterial terminators
| Methods | Year | Terminator type | Method | Software available? | # of terminators | # of species |
|---|---|---|---|---|---|---|
| InterPin [42] | 2023 | Intrinsic | SM | * | N/A | N/A |
| TermNN [12] | 2022 | Intrinsic | DL | Yes | 1175 | 2 |
| ITT prediction [49] | 2021 | Intrinsic | Statistical | No | 137 | 1 |
| iterb-PPse [50] | 2020 | Both | ML | No | 928 | 2 |
| iTerm-PseKNC [13] | 2019 | Both | ML | Yes | 852 | 1 |
| RhoTermPredict [14] | 2019 | Factor-dep. | DP | Yes | 1298 | 3 |
| OPLS-DA [51] | 2018 | Factor-dep. | DA | Yes | 104 | 2 |
| PASIFIC [52] | 2017 | Intrinsic | SM | Yes | 330 | 89 |
| RNIE [53] | 2011 | Intrinsic | CM | Yes | 1062 | 2 |
| TransTermHP [5] | 2007 | Intrinsic | DP | Yes | N/A | N/A |
# of terminators indicates how many terminator sequences were used in the study. The # of species indicates how many bacterial species were considered. CM = covariance model, DA = discriminant analysis, DL = deep learning, DP = Dynamic programming, ML = machine learning, and SM = Structure matching. * Database with predictions available.
Materials and methods
Terminator sequences collection
We searched the National Center for Biotechnology Information (NCBI) PubMed [15] and Gene Expression Omnibus database [16] using as keywords Term-seq, SEnd-seq, SMRT-cappable, Rend-seq, RNATag-seq, and dRNA-seq to find published studies, which experimentally identified bacterial TTSs. For each identified study, using an IPython Notebook [17] and pandas data manipulation library [18], we stored in a BED file the genomic location of identified TTSs (provided in the supplementary material of the corresponding publication). With BEDTools’ [19] slopeBed and FastaFromBed commands, we extracted the genomic sequences corresponding to 100 nt flanking the TTSs (50 nt on either side) into a FASTA file. In each operation, strandedness was taken into account. We chose 50 nt on either side of the TTSs as a previous study [20] found intrinsic terminator regions in E. coli as long as 65 nt. Thus, a sequence of 100 nt long would be enough to contain this region and allow for some variation. Terminators present in plasmids were disregarded. We also collected archaeal TTSs during this process and kept these for the comparative assessment. Additionally, we collected TTSs available in the following databases: RegulonDB [21], DBTBS [22], and BSGatlasDB [23]. As we collected multiple data sets for some of the bacterial species, we needed to remove duplicated sequences from our data. To achieve this, we merged genomic locations if they had at least a 60% genomic location overlap. When the merged sequence was >100 nt, we symmetrically removed the extra nucleotides from the beginning and the end of the sequence. Additionally, sequences containing ambiguous characters (such as N, Y, etc) were removed. As some of the studies we collected data from have used different genome assemblies for the same bacterium, after getting the terminator sequences, we removed duplicated sequences (i.e. sequences 100% identical). The bacterial species with their number of non-redundant terminators used for training and comparative assessment are listed in Tables 2 and 3, respectively. Details about each study are listed in Supplementary Table S1.
Table 2.
Number of non-redundant terminators (No. Term.) per genome accession used for training
| Data source | Species | Phylum | Genome | No. | GC | Avg. |
|---|---|---|---|---|---|---|
| Accession | Term. | (%) | Pre. | |||
| [6, 54] | Bacillus subtilis 168 | Bacillota | AL009126.3 | 1800 | 42.9 | 0.88 |
| [55–57], | ||||||
| [23, 58, 59] | Bacillus subtilis 168 | Bacillota | NC_000964.3 | 4872 | 42.9 | 0.72 |
| [57] | Caulobacter vibrioides NA1000 | Pseudomonadota | CP001340.1 | 341 | 66.2 | 0.93 |
| [60] | Clostridioides difficile 630 | Bacillota | CP010905.2 | 1646 | 28.6 | 0.84 |
| [61] | Dickeya dadantii 3937 | Pseudomonadota | NC_014500.1 | 1786 | 55.5 | 0.49 |
| [62] | Escherichia coli BW25113 | Pseudomonadota | CP009273.1 | 1095 | 50.1 | 0.81 |
| [7, 57, 58, 63], | ||||||
| [8, 37, 64] | E. coli str. K-12 substr. MG1655 | Pseudomonadota | NC_000913.3 | 4139 | 50.1 | 0.62 |
| [65] | Pseudomonas aeruginosa PAO1 | Pseudomonadota | NC_002516.2 | 805 | 65.6 | 0.89 |
| [66] | Staphylococcus aureus JKD6009 | Bacillota | LR027876.1 | 978 | 32.4 | 0.81 |
| [67] | Staphylococcus aureus NCTC 8325 | Bacillota | NC_007795.1 | 566 | 32.4 | 0.90 |
| [68] | Streptococcus pneumoniae D39V | Bacillota | CP027540.1 | 747 | 39.1 | 0.94 |
| [69] | Streptococcus pneumoniae TIGR4 | Bacillota | NC_003028.3 | 1810 | 39.1 | 0.67 |
| [70, 71] | Streptomyces avermitilis MA-4680 | Actinomycetota | BA000030.4 | 2006 | 69.7 | 0.63 |
| [71, 38] | Streptomyces clavuligerus ATCC27064 | Actinomycetota | CP027858.1 | 1583 | 71.6 | 0.62 |
| [71] | Streptomyces coelicolor M145 | Actinomycetota | NC_003888.3 | 1308 | 71.1 | 0.73 |
| [71, 72] | Streptomyces griseus NBRC13350 | Actinomycetota | NC_010572.1 | 2724 | 71.2 | 0.76 |
| [71, 73] | Streptomyces lividans TK24 | Actinomycetota | CP009124.1 | 1999 | 71.2 | 0.59 |
| [71] | Streptomyces tsukubaensis NBRC108819 | Actinomycetota | CP020700.1 | 1283 | 70.8 | 0.55 |
| [57] | Vibrio natriegens ATCC 14048 | Pseudomonadota | CP009977.1 | 905 | 44.7 | 0.97 |
| [57] | Vibrio natriegens ATCC 14048 | Pseudomonadota | CP009978.1 | 257 | 44.1 | 0.90 |
| [74] | Vibrio parahaemolyticus RIMD 2210633 | Pseudomonadota | NC_004603.1 | 1852 | 44.7 | 0.67 |
| [75] | Zymomonas mobilis ZM4 = ATCC 31821 | Pseudomonadota | CP023715.1 | 2040 | 45.7 | 0.54 |
There is a total of 36 542 terminator sequences. Avg. Pre. shows the average precision obtained during cross-validation as in Fig. 5.
Table 3.
Number of non-redundant terminators (No. Term.) per species used for comparative assessment for a total of 4626 and 3581 bacterial and archaeal terminators, respectively
| Data source | Species | Phylum | Genome | No. | GC | Seq. |
|---|---|---|---|---|---|---|
| Accession | Term. | (%) | Tech. | |||
| Bacteria | ||||||
| [34] | Mycobacterium tuberculosis H37Rv | Actinomycetota | AL123456.3 | 2202 | 64.7 | Term-seq |
| [76] | Streptococcus agalactiae NEM316 | Bacillota | NC_004368.1 | 655 | 35.1 | dRNA-seq |
| [71] | Streptomyces gardneri ATCC 15439 | Actinomycetota | CP059991.1 | 870 | 70.7 | Term-seq |
| [77] | Synechocystis PCC 6803 | Cyanobacteriota | NC_000911.1 | 553 | 47.0 | Term-seq |
| [78] | Synechocystis PCC 7338 | Cyanobacteriota | CP054306.1 | 346 | 47.1 | Term-seq |
| Archaea | ||||||
| [79] | Haloferax volcanii DS2 | Methanobacteriota | NC_013967 | 1227 | 65.7 | Term-seq |
| [80] | Methanococcus maripaludis S2 | Methanobacteriota | NC_005791 | 2354 | 32.6 | Term-seq |
Confirming the sequence length for terminator identification
To confirm that a sequence pattern was within the 100 nt extracted, we used relative nucleotide frequency graphs to visualize whether a distinct pattern was present within this region of interest (ROI). The relative log2 nucleotide frequency Fi, j for a specific position j and nucleotide i (i ∈ {A, T, C, G}) was calculated by  where Ni,j is the total count of nucleotide i at position j and M is the total number of sequences. The log2 ratio of the relative nucleotide frequency in the ROI for each position j and nucleotide i was obtained by subtracting from Fi, j the log2 of the nucleotide i-content in the corresponding genome.
where Ni,j is the total count of nucleotide i at position j and M is the total number of sequences. The log2 ratio of the relative nucleotide frequency in the ROI for each position j and nucleotide i was obtained by subtracting from Fi, j the log2 of the nucleotide i-content in the corresponding genome.
Non-terminator sequences generation
To train machine learning-based models, we needed instances of non-terminators of the same length and from the same genome as the terminator-containing sequences. To generate these negative examples, we used BEDTools’ shuffle command to randomly generate genomic coordinates different from the terminator regions. These random genomic regions are taken from anywhere in the genome and, thus, include intergenic and intragenic sequences. We allowed a maximum sequence overlap between positive and negative sequences of 20 nt. A ratio of 1–10 positive to negative was used as it was shown in [24] that there should be more negative than positive instances to lower the false-positive rate during genome scan. Furthermore, the terminator detection problem has a natural imbalance between terminator and non-terminator sequences, as the estimated number of terminators in a bacterial genome is relatively tiny compared to the number of all possible non-terminator sequences of the same length. Henceforth, we refer to our complete data set (including terminator and non-terminator sequences) as BacTermData.
Feature engineering and selection
BacTermData contains DNA sequences consisting of ATCG characters. However, machine learning methods expect to receive a numerical representation of the sequences as input. There are many approaches to numerically representing sequences, such as one-hot (binary) encoding, k-mer frequencies, etc. As it is not feasible to determine a priori which representation would generate the ‘best-performing’ machine learning model, one needs to try out several distinct representations. Here, the best-performing model refers to a model that maximizes a specific performance metric, such as the F1-score or area under the precision-recall curve (AUPRC). There are several libraries or software (e.g. MathFeature [25], iLearnPlus [26], and RepDNA [27]) to generate features from DNA sequences. Here we decided to use iLearnPlus to represent the sequences in BacTermData, as it can generate a wide variety of feature sets. Using iLearnPlus, we computed 6208 features (belonging to 28 unique feature sets) per sequence. A feature set contains features created by one feature generation method (e.g. one-hot encoding). After generating the features, we applied feature selection methods to identify informative features.
To measure the importance of the features, we used two methods: SHAP [28] and Gini measure of light gradient-boosting machine (LGBM) [29]. We used an iterative algorithm to drop features: (i) train an LGBM model with all remaining features and calculate feature importance values using SHAP and Gini measure, (ii) remove features in the bottom 20% after sorting the features based on their importance as per both feature importance methods, (iii) repeat step (i) and (ii) until the average of the AUPRC obtained during 10-fold cross-validation decreases. We observed a decrease in the AUPRC after reaching 1694 features, down from 6208 features (Fig. 1). We called these 1694 features (belonging to 22 feature sets) the 22-sets features. Supplementary Table S2 describes the 1694 selected features.
Figure 1.

Average precision (an estimate of AUPRC) as a function of the number of features included in the training set. Each dot indicates an iteration of the algorithm used to remove non-informative features. The shaded area is the standard deviation of 10 cross-validation folds. The vertical dash line indicates the point where a drop of AUPRC occurred.
Generating the 1694 features with ILearnPlus is computationally intensive [roughly, it processes 30 sequences/second in a high-performance computing (HPC) cluster]. To reduce the computational requirements, we selected the complete six feature sets comprising 60% of the 1694 features (Supplementary Table S2) and called the corresponding features the six-sets features. We also kept two feature sets (ENAC and one-hot/binary encoding) as they preserve the spatial location of patterns in the ROI.
Machine learning approaches
We utilized convolutional neural networks (CNNs) and fully connected neural networks (FCNNs) as they have been shown to perform well in various domains [30]. We also used a boosting method because of boosting methods’ ability to handle tabular data better than other techniques [31]. Table 4 describes the ML approaches considered. Table 5 describes the CNNs architecture.
Table 4.
Machine learning approaches considered
| # | ML method | Features | Description |
|---|---|---|---|
| 1 | CNN | PS2, ENAC, OH, six-sets features | Fusiona |
| 2 | CNN | OH, six-sets features | Fusion |
| 3 | CNN | PS2, ENAC, OH, six-sets features | Appendb |
| 4 | CNN | PS2 | Singlec |
| 5 | CNN | OH | Single |
| 6 | CNN | ENAC | Single |
| 7 | CNN | NCP | Single |
| 8 | CNN | PS2, ENAC, OH, NCP | Ensembled |
| 9 | FCNN | Six-sets features | FCNN trained on six-sets features |
| 10 | LGBM | Six-sets features | A LGBM trained on six-sets features |
| 11 | LGBM | 22-sets features | A LGBM trained on 22-sets features |
Feature sets are described in Supplementary Table S2.
a Train one CNN for each of the listed feature sets followed by fully-connected layer(s).
b Train a CNN receiving as input the concatenated listed feature sets followed by fully-connected layer(s).
c Train a CNN receiving as input the listed feature set followed by fully-connected layer(s).
d Train independent CNNs followed by fully-connected layer(s) on each listed feature set and the combined prediction is their average output.
Table 5.
CNN architecture description
| Layer type | Hyperparameter | Value |
|---|---|---|
| Conv1D | Filters | 64 |
| Kernel_size | 10 | |
| Activation | PReLU | |
| AveragePooling1D | Pool_size | 2 |
| Conv1D | Filters | 64 |
| Kernel_size | 10 | |
| Activation | PReLU | |
| AveragePooling1D | Pool_size | 2 |
| BatchNormalization | – | – |
| Conv1D | Filters | 64 |
| Kernel_size | 10 | |
| Activation | PReLU | |
| Dropout | Rate | 0.1 |
| Flatten | – | – |
| Dense (dropout) | Units & (rate) | 500 (0.3) |
| Activation | PReLU | |
| Dense (dropout) | Units & (rate) | 600 (0.3) |
| Activation | PReLU | |
| Dense (dropout) | Units & (rate) | 600 (0.3) |
| Activation | PReLU | |
| Dense (dropout) | Units & (rate) | 200 (0.4) |
| Activation | PReLU | |
| Dense (dropout) | Units & (rate) | 200 (0.4) |
| Activation | PReLU | |
| Dense | Units | 1 |
| Activation | Sigmoid |
The architecture of the FCNN is a dense layer containing 400 units and, using the ReLU activation function, is applied to the categorical input. This is followed by a dropout layer with a rate of 0.3 to reduce overfitting. This pattern of a dense layer with 400 units and ReLU activation followed by a dropout layer with a rate of 0.3 is repeated six times more. The output from these repeated layers is then fed into a dense layer with 200 units and parametric ReLU activation, followed by a dropout layer with a rate of 0.4. This configuration is repeated once more before the final output layer, which consists of a single unit with a sigmoid activation function to provide a binary classification output.
Model generation and assessment
We used stratified Monte Carlo cross-validation (SMCCV) [32] (also called repeated random subsampling) to select the optimal hyperparameters of the neural network models. Monte Carlo cross-validation is a method that randomly determines the training and validation set in different iterations. The positive-to-negative data ratio is maintained in each iteration as it is stratified. We used SMCCV with 100 folds and 10 iterations to find the optimal hyperparameters for the models. With 100 folds, 99% of the data is for training and 1% for testing on each fold. Neural networks were implemented in TensorFlow version 2.8.0. We used randomized cross-validation with 50 iterations to find the best hyperparameters for the LGBM model (Table 6). We used the Python LGBM implementation version 3.3.3.
Table 6.
Best hyperparameters for LightGBM model
| Hyperparameter | Value |
|---|---|
| Boosting_type | Goss |
| Colsample_bytree | 0.8 |
| Learning_rate | 0.1 |
| Max_depth | 7 |
| Min_child_weight | 1 |
| n_estimators | 3000 |
| Num_leaves | 64 |
| Objective | Cross_entropy_lambda |
| Random_state | 42 |
| Reg_lambda | 30 |
| Unbalance | True |
| n_jobs | −1 |
We used SMCCV to assess the models’ performance, and calculated average precision, recall, and F-score per species. We calculated score thresholds for classification that maximize the F0.5, F1, and F2 scores. Average precision is calculated as AP = ∑n(Rn − Rn − 1)Pn where Rn and Pn are the precision and recall at the nth threshold. Recall is the proportion of actual positive instances correctly identified by a model (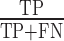 ), while precision measures the proportion of predicted positive instances that are actually true positives (
), while precision measures the proportion of predicted positive instances that are actually true positives ( ) where TP is the number of true positives, FN is the number of false negatives, and FP is the number of false positives. F-score is calculated as
) where TP is the number of true positives, FN is the number of false negatives, and FP is the number of false positives. F-score is calculated as  where β can be 0.5, 1, or 2 for the corresponding F-Score.
where β can be 0.5, 1, or 2 for the corresponding F-Score.
To support the reproducibility of the machine learning method of this study, the machine learning summary table is included in the supplementary material as per DOME recommendations [33].
Results and discussion
Clearly visible pattern in ROI in BacTermData
We visualized the log2 ratio of the relative nucleotide frequency across the ROI for each study data included in BacTermData and confirmed the presence of a clear pattern around the middle of the ROI in all the data. Figure 2 shows the aggregated pattern around the TTS (middle of the sequence) in the terminator sequences in BacTermData. This indicates that there is indeed a pattern in BacTermData, which machine learning approaches could learn to identify terminator sequences. BacTermData consists of 41 168 bacterial terminator sequences (36 542 used for training) of 25 bacterial strains belonging to four phyla: Pseudomonadota (formerly Proteobacteria), Bacillota (formerly Firmicutes), Actinomycetota (formerly Actinobacteria), and Cyanobacteriota. To test the generalization capabilities of our final model, the terminator sequences of five bacterial strains (Table 3), including the only two belonging to the phylum Cyanobacteriota, were left out of training and used only for the comparative assessment.
Figure 2.

Log2 ratio of the relative nucleotide frequency of all terminator sequences in BacTermData.
Selecting a best performing model: BacTermFinder
We assessed various ML approaches and sequence representation combinations (Table 4). This assessment involved training and evaluating these combinations on the data shown in Table 2 using SMCCV. The SMCCV results are presented in Table 7. The LGBM model trained on the 22-sets features achieved performance comparable to that of single CNNs, however, computing the 22-sets features is resource-intensive and time-consuming. During training the models, we noticed that the single CNN models were performing well based on average precision and thus, we decided to build an ensemble with these four models by simply averaging their outputs. This approach consisting of an ensemble of the four single CNNs (Fig. 3) achieved an average precision of 0.7080 ± 0.0248, outperforming all the other approaches considered (Table 7). We selected this as our final model and called it BacTermFinder. Figure 4 shows the precision-recall curve (PRC) and receiver operating characteristic curve (ROC) of BacTermFinder over all SMCCV iterations. Clearly, BacTermFinder’s performance is well above a random classifier’s performance (dashed lines in Fig. 4).
Table 7.
SMCCV average precision ± standard deviation
| # | Approach | Average precision ± S.D. |
|---|---|---|
| 1 | CNN_fusion | 0.5955 ± 0.0232 |
| 2 | CNN_fusion | 0.6553 ± 0.0252 |
| 3 | CNN_append | 0.5949 ± 0.0257 |
| 4 | CNN_single | 0.6738 ± 0.0253 |
| 5 | CNN_single | 0.6775 ± 0.0214 |
| 6 | CNN_single | 0.6544 ± 0.0284 |
| 7 | CNN_single | 0.6730 ± 0.0243 |
| 8 | CNN_ensemble | 0.7080 ± 0.0248 |
| 9 | FCNN | 0.5012 ± 0.0319 |
| 10 | LGBM | 0.5933 ± 0.0269 |
| 11 | LGBM | 0.6476 ± 0.0217 |
The highest SMCCV average precision is highlighted in bold. See Table 4 for a description of the ML approaches.
Figure 3.

Depiction of BacTermFinder architecture.
Figure 4.

PRC in blue [starting at coordinates (0,1)] and ROC curve in orange [starting at coordinates (0,0)] of BacTermFinder's performance on the aggregated results of SMCCV iterations. Dashed lines show the performance of a random classifier.
Effect of GC content and phylum on terminator identification
It has been shown that there is a correlation between GC content and prevalence of certain terminator shapes in bacteria [3]. To explore whether there is an effect of GC content or phylum on terminator identification, we decided to visualize the average precision per bacterium versus their GC content and coloured it by the phylum (Fig. 5). A linear regression line fitted to this data indicates a moderate relationship between GC content and average precision: As the GC content increases, the performance decreases (Spearman correlation value of −0.46, P-value .031). For a 10% increase in GC content, the average precision decreases by 0.038. Thus, BacTermFinder tends to achieve higher average precision in bacteria with lower GC content. Bacteria with high GC content tend to have more factor-dependent terminators [34] and less intrinsic terminators with the canonical shape [3]. Factor-dependent terminators do not always have the hairpin structure of intrinsic terminators [4], and thus, their sequence motif might be weaker. Additionally, the Rho utilization (rut) site is 60–90 nt upstream of the terminator [35], and thus, often outside our ROI. These issues might partially explain why BacTermFinder’s performance is lower on high GC bacteria; however, this pattern is not observed within phyla, and BacTermFinder’s performance is more consistent (with less variation) for Bacillota and Actinomycetota than for Pseudomonadota (Fig. 5). All the bacteria in the phylum Actinomycetota is from the Streptomyces genus (Table 2) and that might explain why BacTermFinder’s performance is similar for these bacteria; however, this is not the case for the phylum Bacillota. Other factors, in addition to GC content, probably affect terminator identification between and within phyla and further investigation is needed.
Figure 5.

Average precision versus GC content of BacTermFinder SMCCV results per bacterial strain. Bacterial strains are coloured based on their phylum with Bacillota in the GC % between 30 and 43%, Pseudomonadota between 44 and 66%, and Actinomycetota above 66%.
Assessing BacTermFinder’s predictions on independent validation data
To confirm that BacTermFinder had learned terminator-like sequence patterns present in prokaryotic organisms not included in the training data, we tested BacTermFinder on an independent set of bacterial and archaeal terminators. For this assessment, we used the experimentally determined terminators described in Table 3 as the reference set of true terminators and compare them against BacTermFinder’s genome-wide terminator predictions for these organisms. We used BacTermFinder’s predictions with a probability threshold of at least 0.30 (this is the probability threshold that maximized the F1 score during cross-validation) and merged those predictions three or less nucleotides apart. BacTermFinder predictions used are available in BacTermFinder’s GitHub repository under the folder ‘data/BacTermFinder predictions validation set’.
First we looked at whether predicted terminators have a log2 ratio of the relative nucleotide frequency similar to that of experimentally identified terminators. Log2 ratios are provided in the Supplementary File 2. To estimate the similarity between these distributions, we calculated Kendall’s Tau between both frequency distributions per nucleotide. As it can be seen from Fig. 6 for Streptococcus agalactiae, the log2 ratio of the relative nucleotide frequency of predicted terminators is similar to that of experimentally identified terminators. The saliency map (Fig. 6C) shows the contribution of each nucleotide per position in BacTermFinder’s predictions. Negative numbers reduce the probability of predicting a terminator, while positive numbers increase the probability of predicting a terminator. Figures 7 and 8 show the same visual analysis for the archaea Haloferax volcanii and the cyanobacterium Synechocystis PCC 6803. Figures for the the other four organisms can be seen in Supplementary Figs S1–S4. The average Kendall’s Tau coefficient value per nucleotide across the seven organisms in the validation data are: A 0.42 ± 0.08, T 0.35 ± 0.17, C 0.29 ± 0.06, and G 0.30 ± 0.18. This suggests that BacTermFinder predicted terminators have similar sequence patterns to those present in experimentally determined terminators. The lowest Kendall’s Tau correlation is observed in Methanococcus maripaludis followed by Mycobacterium tuberculosis; while the highest correlation is observed in Streptococcus agalactiae followed by Synechocystis PCC 6803.
Figure 6.

Visualization of Streptococcus agalactiae experimentally identified terminators (A) versus BacTermFinder genome-wide predictions (B) on the Y-axis is the log2 ratio and on the X-axis is the nucleotide position. Kendall’s Tau coefficient values per nucleotide are A 0.56 (P-value 9.41 × 10−17), T 0.62 (P-value 4.88 × 10−20), C 0.26 (P-value 1.04 × 10−4), and G 0.38 (P-value 1.84 × 10−8). (A) Log2 ratio of the relative nucleotide frequency of experimentally determined terminators. (B) Log2 ratio of the relative nucleotide frequency of genome-wide predicted terminators. (C) Saliency map generated using using [81].
Figure 7.

Visualization of H. volcanii DS2 experimentally identified terminators (A) versus BacTermFinder genome-wide predictions (B) on the Y-axis is the log2 ratio and on the X-axis is the nucleotide position. Kendall’s Tau coefficient values per nucleotide are A 0.43 (P-value 2.57 × 10−10), T 0.26 (P-value 1.2 × 10−4), C 0.39 (P-value 1.04 × 10−8), and G 0.32 (P-value 2.09 × 10−6). (A) Log2 ratio of the relative nucleotide frequency of experimentally determined terminators. (B) Log2 ratio of the relative nucleotide frequency of genome-wide predicted terminators. (C) Saliency map generated using [81].
Figure 8.

Visualization of Synechocystis PCC 6803 experimentally identified terminators (A) versus BacTermFinder genome-wide predictions (B) on the Y-axis is the log2 ratio and on the X-axis is the nucleotide position. Kendall’s Tau coefficient values per nucleotide are A 0.43 (P-value 1.82 × 10−10), T 0.47 (P-value 4.98 × 10−12), C 0.29 (P-value 1.70 × 10−5), and G 0.36 (P-value 7.64 × 10−8). (A) Log2 ratio of the relative nucleotide frequency of experimentally determined terminators. (B) Log2 ratio of the relative nucleotide frequency of genome-wide predicted terminators. (C) Saliency map generated using [81].
As previously done by Bar A et al. [36], we classified BacTermFinder predicted terminators as primary (≤200-nt downstream of an annotated stop codon) and internal (intragenic). On average 16.09 ± 2.16% of BacTermFinder’s predictions are primary and 48.67 ± 5.35% are internal. On average, 23.73 ± 10.60% of TTSs detected by sequencing methods are located within genes [36–38]. Even though, most BacTermFinder predictions are nearby annotated stop codons (Supplementary Fig. S5), the proportion of intragenic predictions among BacTermFinder’s predictions is on the upper range of those experimentally detected by sequencing methods. In addition to false positive predictions, these intragenic predictions might be due to other reasons; for example, intragenic intrinsic terminators have been observed in many bacteria where they might act as attenuators and terminator-like structures may have other functions [39].
We constructed average profiles of structural and physical characteristics of the predicted terminators using the values collected for RNA dinucleotides in the DiProDB database [40] (accessed on 9th December 2024) and compared them against the average profiles of same-length randomly selected sequences from the corresponding genome (control sequences). Predicted terminators showed a clearly distinct average profile from that of the control sequences in all RNA characteristics available in DiProDB. Figure 9 shows the RNA free energy [41] profiles of BacTermFinder predicted terminators for the seven organism in our validation data. This indicates that predicted terminator sequences have a clearly distinct structural characteristics than control sequences.
Figure 9.

RNA free energy average profiles of predicted terminator sequences (orange, jagged line) and control sequences (gray, horizontal line). Dinucleotide free energy values were obtained from the DiProDB database. AL123456.3 = Mycobacterium tuberculosis H37Rv, CP054306.1 = Synechocystis PCC 7338, CP059991.1 = Streptomyces gardneri ATCC 15439, NC_000911.1 = Synechocystis PCC 6803, NC_004368.1 = Streptococcus agalactiae NEM316, NC_005791 = Methanococcus maripaludis S2, NC_013967 = H. volcanii DS2.
Comparative assessment for genome-wide terminator prediction
We compared BacTermFinder’s performance with that of TermNN [12], iTerm-PseKNC [13], RhoTermPredict [14], and TransTermHP [5] on terminators of five bacterial species (Table 3) not used for generating our model. We decided to include TermNN, RhoTermPredict, and iTerm-PseKNC in the comparative assessment because they are the most recently developed tools available for predicting intrinsic, factor-dependent, and both types of terminators, respectively (Table 1). We included TransTermHP because, as mentioned earlier, is the most cited tool for bacterial terminator prediction. All programs were used to obtain genome-wide terminator predictions. Additionally, we downloaded (on 9th December 2024) genome-wide intrinsic terminator predictions from the InterPin database [42] for Synechocystis sp. PCC 6803 and Streptococcus agalactiae NEM316. The InterPin database predictions for Mycobacterium tuberculosis H37Rv and Streptomyces gardneri ATCC 15439 were done in a different genome assembly than the one used in this study. There were not predictions available for Synechocystis sp. PCC 7338 in the InterPin database.
Statistical significance of the difference in recall between methods was estimated using the Friedman test which is a non-parametric test recommended for comparison of more than two classifiers over multiple datasets [43]. To find out which models differ in terms of recall, we used several post-hoc pair-wise tests as recommended in [44], namely Quade, Miller, Nemenyi, and Siegel post-hoc tests. All statistical tests were carried out in R using the packages PMCMRplus [45] and scmamp [46].
As the experimentally identified terminators by sequencing methods in our validation data are not exhaustive (i.e. there are actual terminators missing due to lack of expression of some transcripts in the growth conditions sampled for sequencing), absence of a experimentally-detected terminator in a specific region does not prove that there is not a terminator in that region. Considering all predicted terminators absent in the validation data as false positives would overestimate the false positive rate of the tools. Thus, we evaluated predictive performance using recall (also called sensitivity or true positive rate) at 10 sequence overlapping thresholds between the predicted terminators and the actual terminators (Table 8). That is, we first considered as correct predictions those predictions with at least 10% overlap with an actual terminator, then considered those with at least 20% overlap with an actual terminator as correct, and so on until considering only those with 100% overlap with an actual terminator as correct. In this way, we evaluated how accurately the terminator location is predicted by the tools. To do this, we used BEDtools’ intersect command and calculated recall at each overlap threshold.
Table 8.
Average recall over 10 overlap thresholds ± standard deviation for four other terminator prediction tools and BacTermFinder
| Bacterium | RhoTermPredict | ITerm-PseKNC | TransTermHP | TermNN | BacTermFinder |
|---|---|---|---|---|---|
| Streptomyces gardneri ATCC 15439 | 0.25 ± 0.13 | 0.01 ± 0.004 | 0.17 ± 0.10 | 0.37 ± 0.22 | 0.60 ± 0.22 |
| CP059991.1, GC = 70.7% | |||||
| Mycobacterium tuberculosis H37Rv | 0.24 ± 0.16 | 0.18 ± 0.11 | 0.003 ± 0.002 | 0.14 ± 0.09 | 0.23 ± 0.12 |
| AL123456.3, GC = 64.7% | |||||
| Synechocystis sp. PCC 7338 | 0.10 ± 0.04 | 0.12 ± 0.05 | 0.28 ± 0.15 | 0.51 ± 0.26 | 0.64 ± 0.22 |
| CP054306.1, GC = 47.1% | |||||
| Synechocystis sp. PCC 6803 | 0.12 ± 0.08 | 0.10 ± 0.05 | 0.22 ± 0.10 | 0.48 ± 0.24 | 0.54 ± 0.18 |
| NC_000911.1, GC = 47.0% | |||||
| Streptococcus agalactiae NEM316 | 0.02 ± 0.006 | 0.24 ± 0.13 | 0.56 ± 0.27 | 0.77 ± 0.32 | 0.82 ± 0.30 |
| NC_004368.1, GC = 35.1% | |||||
| Overall mean recall | 0.15 ± 0.10 | 0.13 ± 0.09 | 0.25 ± 0.20 | 0.46 ± 0.23 | 0.57 ± 0.21 |
The corresponding genome accession and GC content (%) is provided below each bacterium name. The highest recall per row is highlighted in bold.
We included in the comparative assessment bacteria with different GC content. Bacteria with high GC content tend to have a larger proportion of factor-dependent terminators [34]. For example, Mycobacterium tuberculosis is reported to have a large proportion (up to 54%) of factor-dependent terminators [34]. As RhoTermPredict was designed to predict factor-dependent terminators, we hypothesized it would perform better on bacteria with high GC content than on bacteria with low GC content. On the other hand, we hypothesized that termNN and TransTermHP would perform better on bacteria with low GC content than on bacteria with high GC content, as these two tools were designed to identify intrinsic terminators. Our results (Table 8) indeed support these hypotheses. RhoTermPredict achieved its highest recall (roughly 24%) on Mycobacterium tuberculosis and Streptomyces gardneri, which are the two organisms with the highest proportion of factor-dependent terminators. BacTermFinder’s recall on these two organisms (i.e. Mycobacterium tuberculosis and Streptomyces gardneri) is comparable to or better than that of RhoTermPredict. This indicates that BacTermFinder can indeed predict some factor-dependent terminators. All tools but RhoTermPredict achieved their highest recall on Streptococcus agalactiae (lowest GC content). InterPin’s recall for Synechocystis sp. PCC 6803 is 0.28 ± 0.19 and for Streptococcus agalactiae NEM316 is 0.49 ± 0.23. InterPin’s recall is comparable to that of TransTermHP (Table 8). This is unsurprising as both tools (InterPin and TransTermHP) predict terminators by seeking sequences that match the canonical hairpin intrinsic terminator structure. Tools’ recall varied between species with an overall recall range of [0.03, 0.82]. BacTermFinder outperformed ITerm-PseKNC, TermNN, and TransTermHP in terms of mean recall across various overlap thresholds in all the species in the validation set, and it achieved the highest mean recall overall (Table 8). This result suggests that BacTermFinder is able to find both types of terminators (intrinsic and factor-dependent) and can generalize to phyla not seen during training (e.g. Cyanobacteriota).
The Friedman test (P-value <2.2 × 10−16) indicated that the average recall rank obtained by some of the classifiers is significantly different from the mean rank expected under the null hypothesis. All post-hoc pair-wise tests indicated that BacTermFinder’s recall rankings were significantly better than those of all other tools, and that termNN’s recall rankings were significantly better than those of iTerm-PseKNC, TransTermHP, and RhoTermPredict (Supplementary Fig. S6 shows the critical difference plot and results per post-hoc test are shown in Supplementary Table S3).
As we have an incomplete annotation of all terminators in any given bacterial genome, it is hard to estimate the genome-wide false positive rate of terminator prediction tools since a prediction might indeed be correct even though a terminator has not yet been determined experimentally in that location. However, the number of predicted terminators per gene can provide a rough estimate of the number of false positives. Figure 10 shows the distribution of number of terminators predicted per gene by TermNN, RhoTermPredict, and BacTermFinder across the five bacteria in our independent validation data. BacTermFinder displays less variation in the number of predicted terminators per gene, and predicts, on average, 6.62 ± 1.18 terminators per gene, while TermNN and RhoTermPredict predict 8.89 ± 3.52 and 4.30 ± 3.20, respectively. RhoTermPredict predicts less terminators per gene; however, its recall is substantially lower than BacTermFinder’s for four out of five bacteria in our independent validation data. The results provided in Fig. 10 and Table 8 indicate that BacTermFinder’s false positive rate is lower than that of TermNN (the second best tool) while achieving a higher recall rate.
Figure 10.

Distribution of the number of predicted terminators per gene across the five bacterial species in our independent validation data. The horizontal line inside each box indicates the median value, and the bottom and top of each box indicate the 25 and 75 percentile, respectively.
We used TermNN and BacTermFinder, which are the two tools with the highest recall as per Table 8, to find archaeal terminators. BacTermFinder outperforms TermNN in terms of recall in predicting archaeal terminators (Table 9). This indicates that BacTermFinder can generalize to identify some archaeal terminators.
Table 9.
Average recall over 10 overlap thresholds on two archaeal species of the two best performing terminator prediction tools (TermNN and BacTermFinder) as per the results shown in Table 8
| Archaea | TermNN | BacTermFinder |
|---|---|---|
| Haloferax volcanii | 0.15 ± 0.11 | 0.32 ± 0.20 |
| NC_013967, GC = 65.7% | ||
| Methanococcus maripaludis | 0.40 ± 0.22 | 0.57 ± 0.25 |
| NC_00579, GC = 32.6% |
The highest recall per Archaea is highlighted in bold.
BacTermFinder can predict the location of terminators accurately
We assessed how closely our predictions aligned with actual terminator locations. To do this, we visualized the recall rate as a function of sequence overlap of the predicted terminators with the actual terminators (Fig. 11). We hypothesized that we would observe a declining trend as we moved towards stricter overlap thresholds, which is indeed the case. We compared our overlap versus recall with that of TermNN. Our results show that (i) on every overlap threshold, BacTermFinder outperforms or is comparable to TermNN, and (ii) BacTermFinder’s recall drops at stricter overlaps than TermNN’s recall. The latter indicates that BacTermFinder can find the location of terminators more accurately than TermNN. However, BacTermFinder’s recall sharply decreases at overlap thresholds >0.8, which suggests the potential need for a nucleotide-wise segmentation approach to achieve accuracy at the nucleotide level.
Figure 11.

Recall as a function of percentage sequence overlap between predicted and actual terminators. All sequences are 100 nt long. The dotted lines are TermNN, and the solid lines are BacTermFinder. Each colour represents a bacterium in our independent validation data. The area under the curves is shown in the legend.
Additionally, we assessed the accuracy of the probability of being a terminator outputted by TermNN and BacTermFinder. To do this, we sorted their predictions based on their estimated probabilities of a sequence being a terminator, and selected the top n% to calculate recall at ten different percentage overlaps with actual terminators. Subsequently, we calculated the mean recall and standard deviation across the percentage overlap levels. We visualized mean recall versus top n predictions for a bacterium with high GC content (Streptomyces gardneri, Fig. 12A), a bacterium with approximately equiprobable nucleotide distribution (Synechocystis PCC 7338, Fig. 12B) and a bacterium with low GC content (Streptomyces gardneri, Fig. 12C). For these three bacteria, BacTermFinder achieves higher recall rates at any top n% predictions than TermNN. BacTermFinder has a wider margin over TermNN as the GC content increases. BacTermFinder has also less variation in recall across different overlap thresholds (shaded area in Fig. 12) than TermNN. For Streptomyces gardneri, the worst recall level of BacTermFinder is similar to or higher than the best recall level of TermNN across all top n% of predictions. With BacTermFinder’s 10% most confident predictions on any of these three bacteria, one is able to identify close to 40% of their known terminators.
Figure 12.

Average recall (solid lines) versus top n% most confident predictions for BacTermFinder (blue, top line) and TernNN (orange, bottom line) for (A) Streptomyces gardneri, (B) Synechocystis PCC 7338, and (C) Streptococcus agalactiae. The shaded area indicates standard deviation.
Low agreement between terminator prediction tools
To see the agreement among RhoTermPredict, TermNN, and BacTermFinder, we generated a Venn diagram for two bacteria in the extremes of the GC content axis in our validation data. That is Mycobacterium tuberculosis with 64.69% GC content and Streptococcus agalactiae with 35.12% GC content. We chose Mycobacterium tuberculosis as it has a large proportion of factor-dependent terminators [34]. For Mycobacterium tuberculosis, the highest agreement (14% of the total predicted terminators) is between BacTermFinder and RhoTermPredict (Fig. 13, intersection areas in the center and top-center). For Streptococcus agalactiae, a low GC bacteria, the highest agreement (92.8% of the total predicted terminators) is between BacTermFinder and TermNN (Fig. 14, intersection areas in the center and left-center). This suggests that there is lower agreement among tools to predict factor-dependent terminators (Fig. 13) than intrinsic terminators (Fig. 14). BacTermFinder predicts a similar number of terminators in Mycobacterium tuberculosis and Streptococcus agalactiae as RhoTermPredict and TermNN, respectively. This suggests that BacTermFinder is as effective in identifying factor-dependent and intrinsic terminators as tools specialized on each terminator type. The number of terminators predicted by all three tools is quite low for both bacteria: 23 (or 1.9%) and 9 (or 1.4%) for Mycobacterium tuberculosis and Streptococcus agalactiae, respectively.
Figure 13.

Agreement among 1220 predicted terminators (with at least 50% sequence overlap with an actual terminator) of BacTermFinder, RhoTermPredict, and TermNN for Mycobacterium tuberculosis.
Figure 14.

Agreement among 642 predicted terminators (with at least 50% sequence overlap with an actual terminator) of BacTermFinder, RhoTermPredict, and TermNN for Streptococcus agalactiae.
Case study: the Rhodobacter capsulatus SB1003 puc operon
As a case study, we looked specifically at terminator predictions made by BacTermFinder, TermNN, RhoTermPredict, and InterPin for the puc operon of Rhodobacter capsulatus SB 1003 (chromosome sequence RefSeq ID: NC_014034.1). R. capsulatus has a GC content of 66.5%, is from the phylum Pseudomonadota and has both types of termination (intrinsic and Rho-dependent). All predictions on the reverse strand were discarded for this analysis. We chose this operon because its transcription initiation, attenuation, and termination have been previously studied using gene fusions, high-resolution RNA 5’ -end mappings by primer extensions, and RNA blot hybridization by LeBlanc et al. [47]. Additionally, R. capsulatus was not included in BacTermFinder training data. LeBlanc et al.’s model of puc RNA regulation indicated that there are two termination sites in this operon: one located immediately downstream of pucE and another between pucA and pucC (circles in Fig. 15). We deemed predictions overlapping the vertical rectangles in Fig. 15 as predicting the corresponding experimentally-determined termination site. Only BacTermFinder and InterPin predicted both termination sites identified by LeBlanc et al. (i.e. their recall is 100%). RhoTermPredict and TermNN only predicted the site directly downstream of pucA (i.e. their recall is 50%). The web version of ITerm-PseKNC failed to predict any termination site in this region. BacTermFinder and RhoTermPredict have a prediction in common in the middle of pucDE. LeBlanc et al. identified several RNA 5’ ends in the middle of pucDE and concluded that there is a promoter internal to pucD. Thus, BacTermFinder and RhoTermPredict are likely both mistakenly identifying this promoter as a terminator. Interestingly, BacTermFinder and RhoTermPredict predicted a pucB-internal terminator, which is immediately downstream of a small RNA (sRNA) predicted with high confidence (probability of being a sRNA of 0.99) by Grüll et al. [48] (rectangle in the third track of Fig. 15). Based on these results, if only the two terminator sites identified by LeBlanc et al. are considered active termination sites, then the precision of the tools is 100%, 40%, 20%, and 11% for InterPin, BacTermFinder, TermNN, and RhoTermPredict, respectively. However, if one assumes that the terminator internal to pucB is bona fide, then the recall of InterPin and BacTermFinder would be 66% and 100%, respectively, while the precision of RhoTermPredict and BacTermFinder would increase to 22% and 60%, respectively. From the perspective of using predictions to guide further research, the predicted pucB-internal termination site downstream of the predicted sRNA is a promising lead. This case study suggests that BacTermFinder might be more sensitive for identifying non-canonical termination sites than InterPin; while, keeping its false positive rate lower than that of other tools.
Figure 15.

R. capsulatus puc operon. Circles in the first track indicate attenuation and termination sites experimentally identified by LeBlanc et al. [47]. NCBI annotated coding sequences and stop codons are shown as rectangles and thin lines in the first and second track, respectively. A sRNA predicted with high-confidence in [48] is shown as a rectangle in the third track. The bottom tracks shown terminator predictions of BacTermFinder (fourth track, dark green rectangles), TermNN (fifth track, magenta rectangles), RhoTermPredict (sixth track, orange rectangles), and InterPin (bottom track, gray rectangles). Predictions within the two large vertical rectangles overlap with the experimentally identified attenuation/termination sites. The number below BacTermFinder’s predictions is the probability of being a terminator calculated by BacTermFinder. Image was partially created using Integrative Genomics Viewer (IGV) [82].
Conclusions
In this work, we have collected, to our knowledge, the largest data set of bacterial terminators identified by sequencing technologies. This comprehensive dataset includes terminators from 25 bacterial strains with a wide range of GC content identified by various sequencing technologies. We expect these data to be valuable for further developments in bacterial terminator prediction. Additionally, we have developed BacTermFinder, a general model for finding bacterial terminators. Future work to improve BacTermFinder includes (i) increasing the ROI to include the rut site; (ii) adding archaeal terminators into the training data; and (iii) determining the terminator type, as BacTermFinder can find both terminator types but does not indicate which type is found.
BacTermFinder outperforms in terms of recall other existing tools in an independent validation data set. BacTermFinder’s average recall over five bacterial species is 0.57 ± 0.21, and TermNN’s (the second-best tool) recall is 0.46 ± 0.23. This increase in recall is achieved by BacTermFinder while predicting, on average, two terminators less per gene than TermNN. BacTermFinder recall in all prokaryotic data (bacterial and archaeal) is 0.53 ± 0.20, and TermNN’s is 0.40 ± 0.22. Furthermore, BacTermFinder can identify both types of terminators as good as or better than tools specialized on that specific terminator type. BacTermFinder and BacTermData are available at:
Supplementary Material
Acknowledgements
This research was partly enabled by computing infrastructure provided by Acenet (ace-net.ca) and the Digital Research Alliance of Canada (alliancecan.ca).
Author contributions: L.P-C. funding acquisition, conceptualization, supervision, formal analysis, visualization and writing. S.M.A.T.G. data curation, investigation, software, formal analysis, visualization and writing.
Contributor Information
Seyed Mohammad Amin Taheri Ghahfarokhi, Department of Computer Science, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3X5, Canada.
Lourdes Peña-Castillo, Department of Computer Science, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3X5, Canada; Department of Biology, Memorial University of Newfoundland, St. John’s, Newfoundland A1B 3X9, Canada.
Supplementary data
Supplementary data is available at NAR Genomics & Bioinformatics online.
Conflict of interest
None declared.
Funding
This work was supported by funds from an Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (2019-05247) to L.P-C. and a graduate fellowship from Memorial University School of Graduate Studies to S.M.A.T.G.
Data availability
Software and data are available at https://github.com/BioinformaticsLabAtMUN/BacTermFinder and https://doi.org/10.5281/zenodo.14498242.
References
- 1. Ray-Soni A, Bellecourt MJ, Landick R Mechanisms of bacterial transcription termination: all good things must end. Annu Rev Biochem. 2016; 85:319–47. 10.1146/ANNUREV-BIOCHEM-060815-014844. [DOI] [PubMed] [Google Scholar]
- 2. Santangelo TJ, Artsimovitch I Termination and antitermination: RNA polymerase runs a stop sign. Nat Rev Microbiol. 2011; 9:319–29. 10.1038/nrmicro2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mitra A, Angamuthu K, Jayashree HV et al. Occurrence, divergence and evolution of intrinsic terminators across eubacteria. Genomics. 2009; 94:110–6. 10.1016/j.ygeno.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 4. Song E, Uhm H, Munasingha PR et al. Rho-dependent transcription termination proceeds via three routes. Nat Commun. 2022; 13:1663. 10.1038/s41467-022-29321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kingsford CL, Ayanbule K, Salzberg SL Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007; 8:R22. 10.1186/GB-2007-8-2-R22/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dar D, Shamir M, Mellin JR et al. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science. 2016; 352:aad9822. 10.1126/science.aad9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ju X, Li D, Liu S Full-length RNA profiling reveals pervasive bidirectional transcription terminators in bacteria. Nat Microbiol. 2019; 4:1907–1918. 10.1038/S41564-019-0500-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yan B, Boitano M, Clark TA et al. SMRT-Cappable-seq reveals complex operon variants in bacteria. Nat Commun. 2018; 9:3676. 10.1038/S41467-018-05997-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lalanne JB, Taggart JC, Guo MS et al. Evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell. 2018; 173:749–61. 10.1016/j.cell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shishkin AA, Giannoukos G, Kucukural A et al. Simultaneous generation of many RNA-seq libraries in a single reaction. Nat Methods. 2015; 12:323. 10.1038/NMETH.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharma CM, Vogel J Differential RNA-seq: the approach behind and the biological insight gained. Curr Opin Microbiol. 2014; 19:97–105. 10.1016/J.MIB.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 12. Brandenburg VB, Narberhaus F, Mosig A Inverse folding based pre-training for the reliable identification of intrinsic transcription terminators. PLoS Comput Biol. 2022; 18:e1010240. 10.1371/JOURNAL.PCBI.1010240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feng CQ, Zhang ZY, Zhu XJ et al. iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics. 2019; 35:1469–77. 10.1093/bioinformatics/bty827. [DOI] [PubMed] [Google Scholar]
- 14. Di Salvo M, Puccio S, Peano C et al. RhoTermPredict: an algorithm for predicting Rho-dependent transcription terminators based on Escherichia coli, Bacillus subtilis and Salmonella enterica databases. BMC Bioinformatics. 2019; 20:117. 10.1186/s12859-019-2704-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. NCBI National Center for Biotechnology Information (NCBI) PubMed 1988–2023. https://pubmed.ncbi.nlm.nih.gov/10 November 2023, date last accessed.
- 16. NCBI National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) 1999–2023. https://www.ncbi.nlm.nih.gov/geo/10 November 2023, date last accessed.
- 17. Kluyver T, Ragan-Kelley B, Pérez F et al.. Loizides F, Schmidt B Jupyter Notebooks—a publishing format for reproducible computational workflows. Positioning and Power in Academic Publishing: Players, Agents and Agendas. 2016; Amsterdam: IOS Press; 87–90. [Google Scholar]
- 18. The pandas development team pandas-dev/pandas: Pandas. 2020; Zenodo; 10.5281/zenodo.3509134. [DOI] [Google Scholar]
- 19. Quinlan AR, Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–2. 10.1093/BIOINFORMATICS/BTQ033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lesnik EA, Sampath R, Levene HB et al. Prediction of rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 2001; 29:3583–94. 10.1093/nar/29.17.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gama-Castro S, Salgado H, Santos-Zavaleta A et al. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016; 44:D133–43. 10.1093/NAR/GKV1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ishii T, Yoshida KI, Terai G et al. DBTBS: a database of Bacillus subtilis promoters and transcription factors. Nucleic Acids Res. 2001; 29:278–80. 10.1093/NAR/29.1.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geissler AS, Anthon C, Alkan F et al. BSGatlas: a unified Bacillus subtilis genome and transcriptome annotation atlas with enhanced information access. Microb Genom. 2021; 7:524. 10.1099/MGEN.0.000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chevez-Guardado R, Peña-Castillo L Promotech: a general tool for bacterial promoter recognition. Genome Biol. 2021; 22:318. 10.1186/S13059-021-02514-9/TABLES/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonidia RP, Domingues DS, Sanches DS et al. MathFeature: feature extraction package for DNA, RNA and protein sequences based on mathematical descriptors. Brief Bioinform. 2022; 23:bbab434. 10.1093/BIB/BBAB434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Z, Zhao P, Li C et al. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021; 49:e60. 10.1093/NAR/GKAB122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu B, Liu F, Fang L et al. repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics. 2015; 31:1307–9. 10.1093/BIOINFORMATICS/BTU820. [DOI] [PubMed] [Google Scholar]
- 28. Lundberg SM, Erion G, Chen H et al. From local explanations to global understanding with explainable AI for trees. Nat Mach Intell. 2020; 2:2522–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ke G, Meng Q, Finley T et al.. Guyon I, Luxburg UV, Bengio S et al. LightGBM: a highly efficient gradient boosting decision tree. Advances in Neural Information Processing Systems. 2017; 30:New York, USA: Curran Associates, Inc. [Google Scholar]
- 30. LeCun Y, Bengio Y, Hinton G Deep Learning. Nature. 2015; 521:436–44. 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 31. Grinsztajn L, Oyallon E, Varoquaux G Why do tree-based models still outperform deep learning on typical tabular data?. arXiv18 July 2022, preprint: not peer reviewedhttps://arxiv.org/abs/2207.08815.
- 32. Xu QS, Liang YZ Monte Carlo cross validation. Chemom Intell Lab Syst. 2001; 56:1–11. 10.1016/S0169-7439(00)00122-2. [DOI] [Google Scholar]
- 33. Walsh I, Fishman D, Garcia-Gasulla D et al. DOME: recommendations for supervised machine learning validation in biology. Nat Methods. 2021; 18:1122–7. 10.1038/s41592-021-01205-4. [DOI] [PubMed] [Google Scholar]
- 34. D’Halluin A, Polgar P, Kipkorir T et al. Premature termination of transcription is shaped by Rho and translated uORFS in Mycobacterium tuberculosis. iScience. 2023; 26:106465. 10.1016/j.isci.2023.106465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Banerjee S, Chalissery J, Bandey I et al. Rho-dependent transcription termination: more questions than answers. J Microbiol. 2006; 44:11–22. [PMC free article] [PubMed] [Google Scholar]
- 36. Bar A, Argaman L, Eldar M et al. TRS: a method for determining transcript termini from RNAtag-seq sequencing data. Nat Commun. 2023; 14:7843. 10.1038/s41467-023-43534-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Adams PP, Baniulyte G, Esnault C et al. Regulatory roles of Escherichia coli 5′ UTR and ORF-internal RNAs detected by 3′ end mapping. eLife. 2021; 10:e62438. 10.7554/ELIFE.62438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hwang S, Lee N, Choe D et al. Elucidating the regulatory elements for transcription termination and posttranscriptional processing in the Streptomyces clavuligerus genome. mSystems. 2021; 6:e01013-20. 10.1128/MSYSTEMS.01013-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peters JM, Vangeloff AD, Landick R Bacterial transcription terminators: the RNA 3′-end chronicles. J Mol Biol. 2011; 412:793–813. 10.1016/j.jmb.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Friedel M, Nikolajewa S, Sühnel J et al. DiProDB: a database for dinucleotide properties. Nucleic Acids Res. 2009; 37:D37–40. 10.1093/nar/gkn597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xia T, SantaLucia J Jr, Burkard ME et al. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson–Crick base pairs. Biochemistry. 1998; 37:14719–35. 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 42. Gupta S, Padmashali N, Pal D INTERPIN: a repository for intrinsic transcription termination hairpins in bacteria. Biochimie. 2023; 214(Pt B):228–36. 10.1016/j.biochi.2023.07.018. [DOI] [PubMed] [Google Scholar]
- 43. Demšar J Statistical comparisons of classifiers over multiple data sets. J Mach Learn Res. 2006; 7:1–30. [Google Scholar]
- 44. García S, Fernández A, Luengo J et al. Advanced nonparametric tests for multiple comparisons in the design of experiments in computational intelligence and data mining: experimental analysis of power. Inform Sci. 2010; 180:2044–64. 10.1016/j.ins.2009.12.010. [DOI] [Google Scholar]
- 45. Pohlert T PMCMRplus: calculate pairwise multiple comparisons of mean rank sums extended. 2024; R package version 1.9.12https://cran.r-project.org/package=PMCMRplus.
- 46. Calvo B, Santafe G scmamp: statistical comparison of multiple algorithms in multiple problems. The R Journal. 2016; 8:248–56. [Google Scholar]
- 47. LeBlanc H, Lang AS, Beatty JT Transcript cleavage, attenuation, and an internal promoter in the Rhodobacter capsulatus puc operon. J Bacteriol. 1999; 181:4955–60. 10.1128/JB.181.16.4955-4960.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grüll MP, Peña-Castillo L, Mulligan ME et al. Genome-wide identification and characterization of small RNAs in Rhodobacter capsulatus and identification of small RNAs affected by loss of the response regulator CtrA. RNA Biol. 2017; 14:914–25. 10.1080/15476286.2017.1306175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gupta S, Pal D Clusters of hairpins induce intrinsic transcription termination in bacteria. Sci Rep. 2021; 11:16194. 10.1038/s41598-021-95435-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fan Y, Wang W, Zhu Q iterb-PPse: identification of transcriptional terminators in bacterial by incorporating nucleotide properties into PseKNC. PloS One. 2020; 15:e0228479. 10.1371/journal.pone.0228479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nadiras C, Eveno E, Schwartz A et al. A multivariate prediction model for Rho-dependent termination of transcription. Nucleic Acids Res. 2018; 46:8245–60. 10.1093/nar/gky563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Millman A, Dar D, Shamir M et al. Computational prediction of regulatory, premature transcription termination in bacteria. Nucleic Acids Res. 2017; 45:886–93. 10.1093/nar/gkw749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gardner PP, Barquist L, Bateman A et al. RNIE: genome-wide prediction of bacterial intrinsic terminators. Nucleic Acids Res. 2011; 39:5845–52. 10.1093/nar/gkr168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dar D, Prasse D, Schmitz RA et al. Widespread formation of alternative 3′ UTR isoforms via transcription termination in archaea. Nat Microbiol. 2016; 1:16143. 10.1038/nmicrobiol.2016.143. [DOI] [PubMed] [Google Scholar]
- 55. Takada H, Mandell ZF, Yakhnin H et al. Expression of Bacillus subtilis ABCF antibiotic resistance factor VmlR is regulated by RNA polymerase pausing, transcription attenuation, translation attenuation and (p)ppGpp. Nucleic Acids Res. 2022; 50:6174–89. 10.1093/NAR/GKAC497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mandell ZF, Oshiro RT, Yakhnin AV et al. NusG is an intrinsic transcription termination factor that stimulates motility and coordinates gene expression with NusA. eLife. 2021; 10:e61880. 10.7554/ELIFE.61880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lalanne JB, Taggart JC, Guo MS et al. Evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell. 2018; 173:749–61. 10.1016/J.CELL.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Johnson GE, Lalanne JB, Peters ML et al. Functionally uncoupled transcription-translation in Bacillus subtilis. Nature. 2020; 585:124–8. 10.1038/S41586-020-2638-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hoon MJLD, Makita Y, Nakai K et al. Prediction of transcriptional terminators in Bacillus subtilis and related species. PLoS Comput Biol. 2005; 1:e25. 10.1371/JOURNAL.PCBI.0010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fuchs M, Lamm-Schmidt V, Sulzer J et al. An RNA-centric global view of Clostridioides difficile reveals broad activity of Hfq in a clinically important gram-positive bacterium. Proc Natl Acad Sci USA. 2021; 118:e2103579118. 10.1073/PNAS.2103579118/-/DCSUPPLEMENTAL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Forquet R, Jiang X, Nasser W et al. Mapping the complex transcriptional landscape of the phytopathogenic bacterium Dickeya dadantii. mBio. 2022; 13:e0052422. 10.1128/MBIO.00524-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dar D, Sorek R High-resolution RNA 3’-ends mapping of bacterial Rho-dependent transcripts. Nucleic Acids Res. 2018; 46:6797–805. 10.1093/NAR/GKY274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Santos-Zavaleta A, Salgado H, Gama-Castro S et al. RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res. 2019; 47:D212–20. 10.1093/NAR/GKY1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Choe D, Kim K, Kang M et al. Synthetic 3′-UTR valves for optimal metabolic flux control in Escherichia coli. Nucleic Acids Res. 2022; 50:4171–86. 10.1093/NAR/GKAC206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thomason MK, Voichek M, Dar D et al. A rhlI 5′ UTR-derived sRNA regulates RhlR-dependent quorum sensing in Pseudomonas aeruginosa. mBio. 2019; 10:e02253-19. 10.1128/MBIO.02253-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mediati DG, Wong JL, Gao W et al. RNase III-CLASH of multi-drug resistant Staphylococcus aureus reveals a regulatory mRNA 3′UTR required for intermediate vancomycin resistance. Nat Commun. 2022; 13:3558. 10.1038/S41467-022-31177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bastet L, Bustos-Sanmamed P, Catalan-Moreno A et al. Regulation of heterogenous lexA expression in Staphylococcus aureus by an antisense RNA originating from transcriptional read-through upon natural mispairings in the sbrB intrinsic terminator. Int J Mol Sci. 2022; 23:576. 10.3390/IJMS23010576/S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Slager J, Aprianto R, Veening JW Deep genome annotation of the opportunistic human pathogen Streptococcus pneumoniae D39. Nucleic Acids Res. 2018; 46:9971–89. 10.1093/NAR/GKY725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Warrier I, Ram-Mohan N, Zhu Z et al. The transcriptional landscape of Streptococcus pneumoniae TIGR4 reveals a complex operon architecture and abundant riboregulation critical for growth and virulence. PLoS Pathog. 2018; 14:e1007461. 10.1371/JOURNAL.PPAT.1007461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lee Y, Lee N, Hwang S et al. Genome-scale analysis of genetic regulatory elements in Streptomyces avermitilis MA-4680 using transcript boundary information. BMC Genomics. 2022; 23:68. 10.1186/S12864-022-08314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lee Y, Lee N, Hwang S et al. Genome-scale determination of 5′ and 3′ boundaries of RNA transcripts in Streptomyces genomes. Sci Data. 2020; 7:436. 10.1038/S41597-020-00775-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hwang S, Lee N, Choe D et al. System-level analysis of transcriptional and translational regulatory elements in Streptomyces griseus. Front Bioeng Biotechnol. 2022; 10:844200. 10.3389/FBIOE.2022.844200/FULL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lee Y, Lee N, Jeong Y et al. The transcription unit architecture of Streptomyces lividans TK24. Front Microbiol. 2019; 10:2074. 10.3389/FMICB.2019.02074/FULL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. kadi MA, Ishii E, Truong DT et al. Direct RNA sequencing unfolds the complex transcriptome of Vibrio parahaemolyticus. mSystems. 2021; 6:e0099621. 10.1128/MSYSTEMS.00996-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vera JM, Ghosh IN, Zhang Y et al. Genome-scale transcription-translation mapping reveals features of Zymomonas mobilis transcription units and promoters. mSystems. 2020; 5:e00250-20. 10.1128/MSYSTEMS.00250-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rosinski-Chupin I, Sauvage E, Sismeiro O et al. Single nucleotide resolution RNA-seq uncovers new regulatory mechanisms in the opportunistic pathogen Streptococcus agalactiae. BMC Genomics. 2015; 16:419. 10.1186/S12864-015-1583-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cho SH, Jeong Y, Hong SJ et al. Different regulatory modes of Synechocystis sp. PCC 6803 in response to photosynthesis inhibitory conditions. mSystems. 2021; 6:e0094321. 10.1128/MSYSTEMS.00943-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jeong Y, Hong SJ, Cho SH et al. Multi-omic analyses reveal habitat adaptation of marine cyanobacterium Synechocystis sp. PCC 7338. Front Microbiol. 2021; 12:667450. 10.3389/FMICB.2021.667450/FULL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Berkemer SJ, Maier LK, Amman F et al. Identification of RNA 3′ ends and termination sites in Haloferax volcanii. RNA Biol. 2020; 17:663–76. 10.1080/15476286.2020.1723328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li J, Yue L, Li Z et al. aCPSF1 cooperates with terminator U-tract to dictate archaeal transcription termination efficacy. eLife. 2021; 10:e70464. 10.7554/ELIFE.70464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Majdandzic A, Rajesh C, Koo PK Correcting gradient-based interpretations of deep neural networks for genomics. Genome Biol. 2023; 24:109. 10.1186/s13059-023-02956-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Robinson JT, Thorvaldsdóttir H, Winckler W et al. Integrative genomics viewer. Nat Biotechnol. 2011; 29:24–6. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software and data are available at https://github.com/BioinformaticsLabAtMUN/BacTermFinder and https://doi.org/10.5281/zenodo.14498242.