

Abstract
While large library docking has discovered potent ligands for multiple targets, as the libraries have grown the hit lists can become dominated by rare artifacts that cheat our scoring functions. Here, we investigate rescoring top-ranked docked molecules with orthogonal methods to identify these artifacts, exploring implicit solvent models and absolute binding free energy perturbation as cross-filters. In retrospective studies, this approach deprioritized high-ranking nonbinders for nine targets while leaving true ligands relatively unaffected. We tested the method prospectively against hits from docking against AmpC β-lactamase. We prioritized 128 high-ranking molecules for synthesis and testing, a mixture of 39 molecules flagged as likely cheaters and 89 that were plausible inhibitors. None of the predicted cheating compounds inhibited AmpC detectably, while 57% of the 89 plausible compounds did so. As our libraries continue to grow, deprioritizing docking artifacts by rescoring with orthogonal methods may find wide use.
Graphic Absttact
INTRODUCTION
In the last five years, the number of readily available molecules for ligand discovery has grown from several million to tens of billions. Structure-based docking of those new libraries has revealed new chemotypes with potent affinity for targets ranging from enzymes1–3 to GPCRS,4–11 to transporters,5,12 to kinases.6,13 As the libraries have grown, however, both simulations and experiments have shown that the very top of the docking-ranked list becomes populated with molecules that “cheat” the scoring function. These false positives can be classified into normal docking failures and cheating artifacts (Figure 1). Normal docking failures, while common, occur through well-known problems of balancing different energy terms in the scoring functions. They often inhabit the same chemical space as true ligands and spread relatively evenly through the docking results. What we call cheating artifacts, conversely, are rare molecules that, through failures of parameterization or adoption of unusual structures that are not properly assessed,1 rank among the very top molecules, often with scores offset from those of the overall distribution of docked molecules. As our virtual libraries continue to grow,8,14–19 such cheating molecules may have an ever greater impact on virtual screening hit lists.
Figure 1.
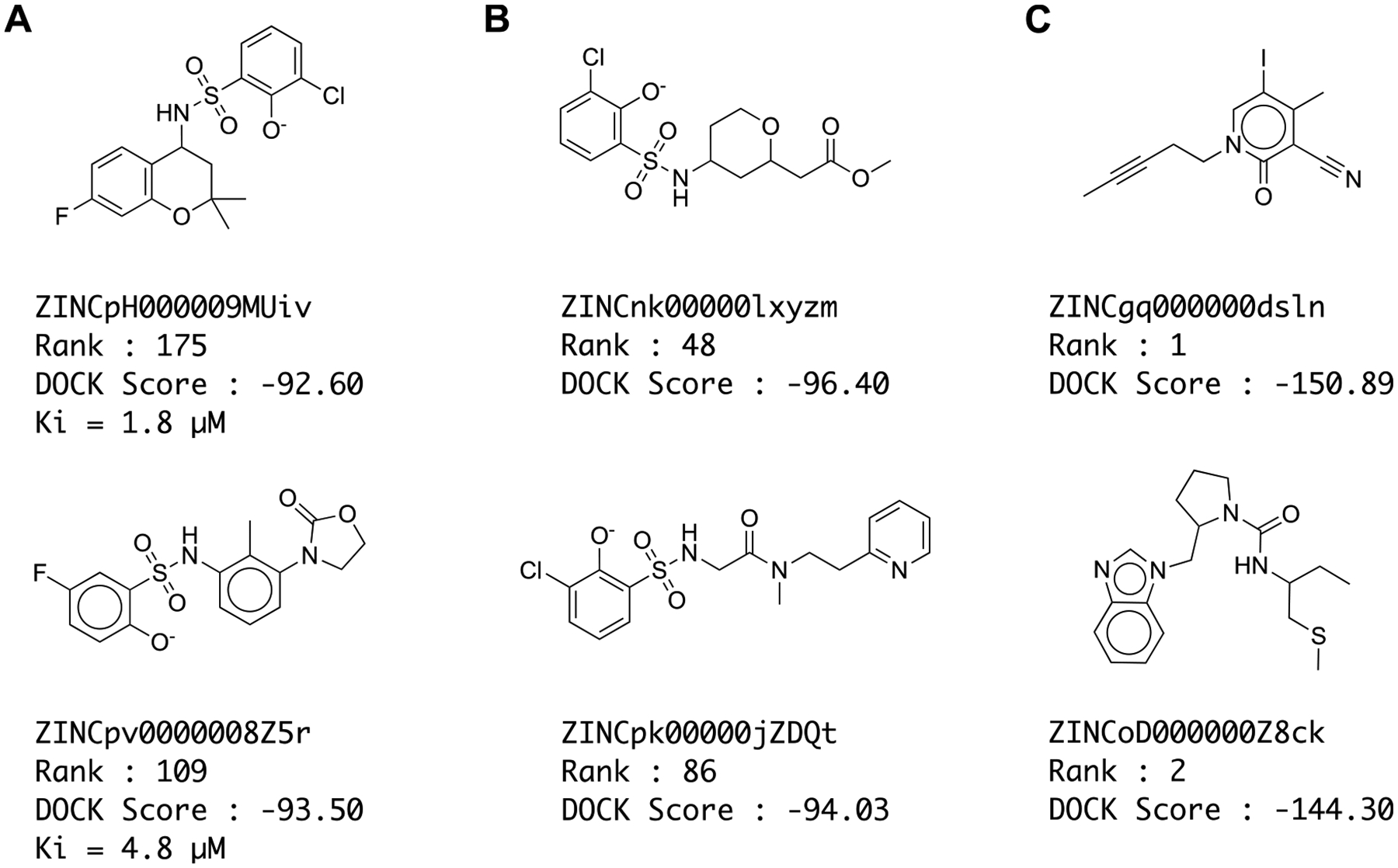
Examples of (A) true inhibitors, (B) normal docking failures, and (C) cheating compounds against AmpC. Ranks are out of 1.7 billion docked.
Since these cheaters arise from holes in a particular scoring function, rescoring high-ranking docked molecules with a second function may identify them as outliers. Rescoring high-ranking docked molecules has been used previously to select molecules more likely to fit well, either because of a consensus agreement of docking scores20 or reranking docked poses at a higher level of theory.21,22 Here, we seek not so much to pick winners but to eliminate problematic docking hits. We cross-filter high-ranking docked molecules with three different methods: we compare DOCK3.8 implicit solvation energies to those calculated by FACTS (fast analytical continuum treatment of solvation)23 and to those calculated by GBMV (generalized Born using molecular volume)24 and compare DOCK3.8 rankings to those from an AB-FEP (absolute binding free energy perturbation) calculation on the top-ranking molecules. These calculations are undertaken both retrospectively and prospectively, where 39 cheating artifacts identified by rescoring are synthesized and tested experimentally, as are 89 molecules judged to be likely ligands by the same rescoring strategy. The results of these experiments suggest that a rescoring approach may be useful to deprioritize cheating artifacts that can concentrate among the top-ranking molecules from large library docking.
RESULTS
Retrospective Study on a Target with Hundreds of Experimental Ligands and Decoys.
A good system to retrospectively optimize the cross-filtering approach is one where large libraries of molecules are docked and where hundreds of docking false and true positives are measured by experiment. There are only a handful of these, with among the richest sets being against the enzyme AmpC β-lactamase. In a recent study, 1.71 billion molecules were docked against AmpC, and over 1400 molecules were tested experimentally.25 As in earlier studies, the distribution of docking scores from this campaign suggested that cheating molecules might be present (Figure 2). As the library increased in size, there was a regular improvement in docking scores, until around 300 million molecules began to appear whose very favorable docking scores diverged from the rest of the distribution. As the library grew further, more of these molecules appeared, reaching ever better scores. Both simulation5 and experimentation with other systems1 suggested that these might be cheating molecules. Inspection of some of these suggested that they have unusual partial atomic charge distributions, particularly involving atoms lower in the periodic table (higher atomic numbers, like bromine or even sulfur) and nitriles (Supporting Information Figure 1). The high partial atomic charges lead to unusually favorable scores in AmpC’s highly polarized active site. We note that these particular cheaters did not appear as high-ranking in earlier campaigns where hundreds of molecules had been tested (e.g., the Sigma2 receptor1); thus far, our observation of cheating molecules has changed with the target.
Figure 2.
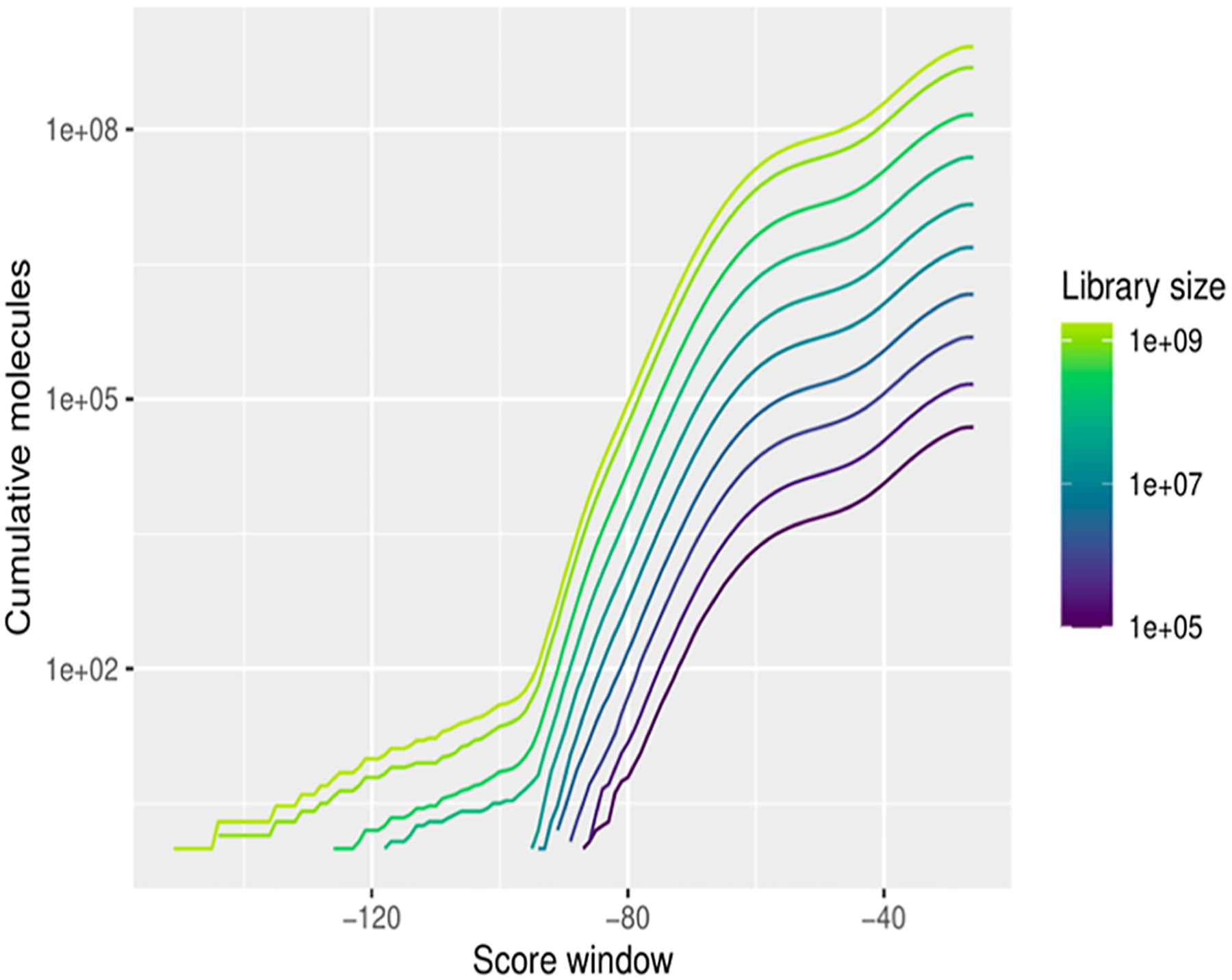
Distribution of docking scores with library growth against AmpC. As the library climbs toward a billion molecules, docking hits with unusually favorable (more negative) scores begin to appear.
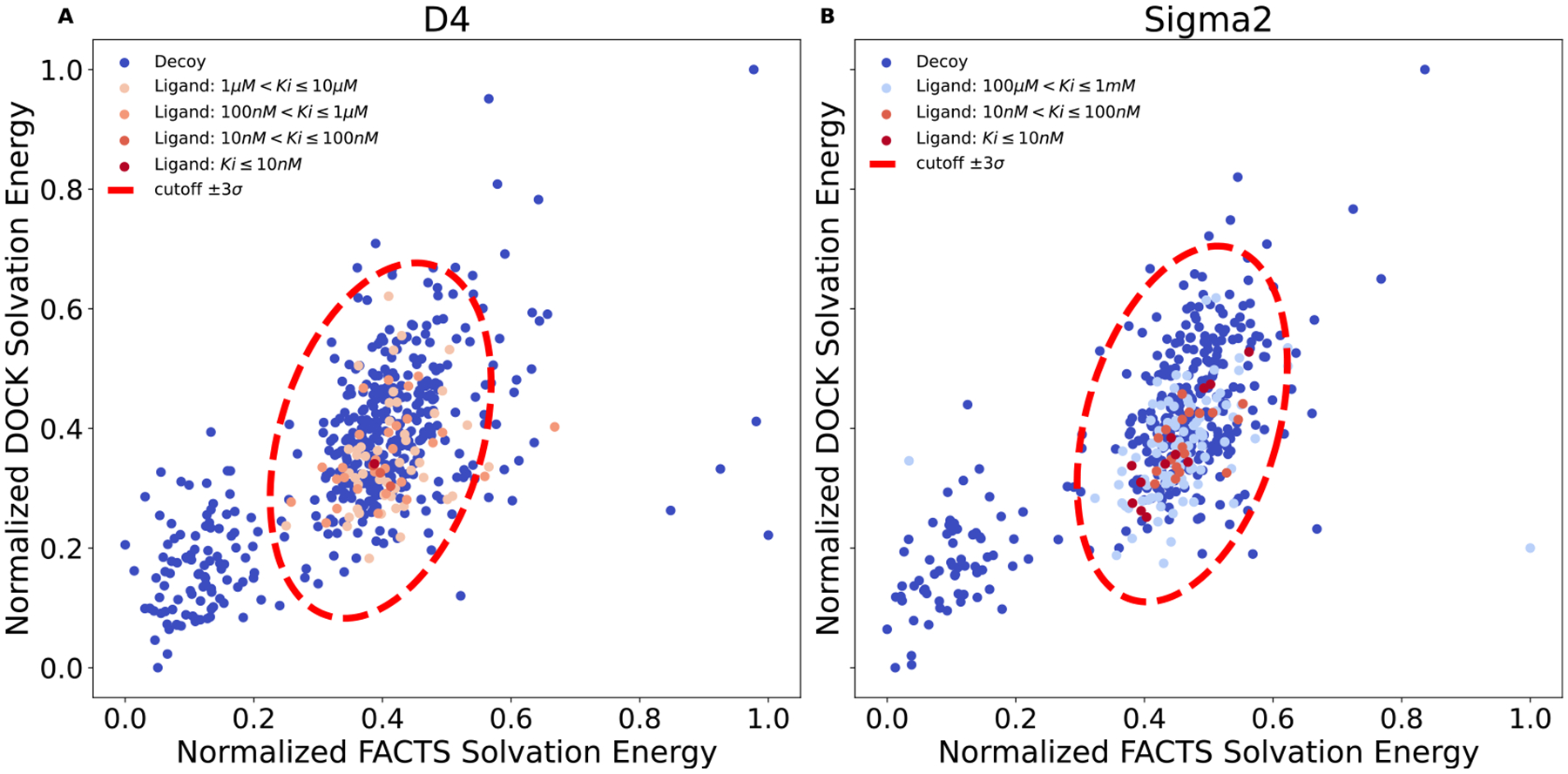
We set out to test the cross-filtering approach on the 1440 docked-and-experimentally tested AmpC hits, asking whether it could separate true ligands from nonbinders while minimizing false negatives. We began by calculating FACTS solvation energies for the 1439 compounds, which took an average of 90 s per molecule on a Linux CPU. Plotting the normalized DOCK3.8 and FACTS desolvation energies against each other revealed a bimodal normal distribution (Figure 3A). Clustering those molecules within 3σ of the mean of both distributions reveals an ellipse outside of which 268 of the molecules fall; these molecules are flagged as having unusual solvation energies (i.e., outliers of the solvation energy distribution) by one of the methods. Of these, 262 do not bind AmpC detectably up to 200 μM, while six were decent inhibitors, the best of which had an affinity of 9.7 μM. Thus, insisting that all molecules are within 3σ of both removed more than 22% of the nonbinders while only losing 3.8% of the true ligands, none of which was among the most potent (which ranged down to sub-μM Kd values). While we would prefer not to lose any of the true ligands, we do note that this was a stringent test of the method as it is applied to all the molecules tested in this campaign, not only the thin wedge of unusually well-scoring molecules where we expect the “cheaters” to concentrate.
Figure 3.

FACTS rescoring results for 1440 experimentally validated compounds against AmpC. (A) Joint distribution of normalized DOCK and FACTS solvation free energy contribution. Distribution of (B) DOCK and (C) FACTS solvation free energy contribution. (D) Percentage of ligands and decoys filtered.
Encouraged by these results, we applied the same protocol against the σ2 and D4 receptors, which had the advantage of having had about 500 docking hits synthesized and experimentally tested against them;1,6 in this sense, they were akin to the AmpC screen, though at a smaller scale. Here too, cross-filtering successfully flagged 22 and 28% nonbinders, respectively (Figure 4), while only losing four and three true ligands, respectively, none of which were among the more potent found in these docking campaigns. Many of the filtered compounds corresponded to molecules that had unusually favorable docking scores and unusual physical features for a hit and might thus be cheating molecules, a point to which we will return in the prospective testing part of this study.
Figure 4.
FACTS rescoring results for (A) D4 and (B) Sigma2. The total number of experimentally validated compounds is 537 and 495, respectively.
Cross-Filtering against Nine Receptors with Different Methods and Parameters.
To further investigate the robustness of this cross-filtering approach, we tested it against other docking targets with binding pocket environments different from those of AmpC, σ2, and D4. With the initial three studies establishing that this approach might be sensible, targeting a wider set of systems also afforded us the chance to try different rescoring methods and to vary the parameters with which we did so (Table 1).
Table 1.
Receptor Data Set
| receptor name (abbreviation) | protein class | docking hits rescored | experimentally tested compounds |
|---|---|---|---|
| AmpC β-lactamases (AmpC) | hydrolase | 300,000 | 1440 |
| serotonin transporter (SERT)12 | transporter | 300,000 | 40 |
| sigma-2 receptor (Sigma2)1 | membrane protein | 300,000 | 495 |
| SARS-CoV-2 macrodomain (Mac1)26 | MAR-hydrolase | 500,000 | 78 |
| SARS-CoV-2 main protease (Mpro)27 | cysteine hydrolase | 500,000 | 327 |
| melatonin receptor type 1A (MT1)11 | GPCR | 300,000 | 38 |
| cannabinoid receptor type 1 (CB1)28 | GPCR | 300,000 | 46 |
| dopamine receptor D4 (D4)6 | GPCR | 300,000 | 537 |
| α2a adrenergic receptor (alpha2a)9 | GPCR | 165,000 | 32 |
Here again, we sought cases where docked ligands had been tested, admittedly in the 40-molecule range rather than the 500 to 1500 as with σ2, dopamine D4, and AmpC, but nevertheless revealing both true ligands and false positives. There are by now close to 20 of these;1,4,6,8,9,11–13,26–30 we focused on 9 to which we had ready access (Table 1). Against each target, hundreds of millions to billions of compounds had been docked; we rescored the several hundred thousand top-ranking poses. For each of the nine targets, we explored rescoring not only with FACTS, as in the initial studies above, but also with a second implicit solvent method, GBMV, and several different calculation parameters (Table S1). As before, we note that this is not the true use case we envision for rescoring as it is applied to all docked-and-tested compounds, not only those very top-ranking ones where we might expect the cheaters to concentrate. Nevertheless, it provides a useful sanity check for the strategy.
We observed similar joint distributions for all nine targets using FACTS (Table 2 and Figure 5). Of the 300,000 to 500,000 high-ranking compounds rescored, between 7 and 19% were outliers by cross-filtering, sitting outside the bounds of the 3σ score distribution. For six of the nine targets, no potent ligands were among these outliers, while experimental nonbinders were captured as outliers, representing 3.85 to 13.51% of the total docking false positives across the screens (Table 2). For three targets, the Alpha2a adrenergic receptor, AmpC, and Sigma2, one potent ligand was categorized as an outlier, representing 12.5, 4.0, and 3.7% of the total ligands found for these targets, respectively; none are among the strongest binders found for their receptors. The best performance was for the CB1 and MT1 receptors and for serotonin transporter (SERT), where none of the true ligands are removed whatsoever. In these studies, we used FACTS with 1000 steps of minimization and ParamChem force-field parameters; the joint distributions for each of the other three parameter setups are plotted in Supporting Information Figures S2, S3, and S4, where similar distributions were observed. This suggests that the approach is robust to different parameters and different implicit solvent models.
Table 2.
Number/Percentage of Compounds Removed by Cross-Filtering
| receptor | Ki cutoff (μM)a | potent ligand | decoyb | compoundsc |
|---|---|---|---|---|
| alpha2a | 3.0 | 1/12.5% | 1/10.00% | 6239/7.29% |
| AmpC | 10.0 | 1/4.0% | 124/16.56% | 55592/18.82% |
| CB1 | 10.0 | 0/0.0% | 5/13.51% | 34197/11.65% |
| D4 | 0.3 | 0/0.0% | 18/14.29% | 24411/8.34% |
| MT1 | 3.0 | 0/0.0% | 3/11.54% | 30934/10.55% |
| Mac1 | 300.0 | 0/0.0% | 8/19.51% | 26954/9.35% |
| Mpro | 100.0 | 0/0.0% | 6/8.70% | 31276/10.83% |
| SERT | 10.0 | 0/0.0% | 1/3.85% | 29788/10.17% |
| sigma2 | 0.3 | 1/3.7% | 10/9.09% | 20734/7.08% |
The upper limit of the binding affinity for potent ligands.
Experimentally validated nonbinders.
All high-ranking compounds.
Figure 5.
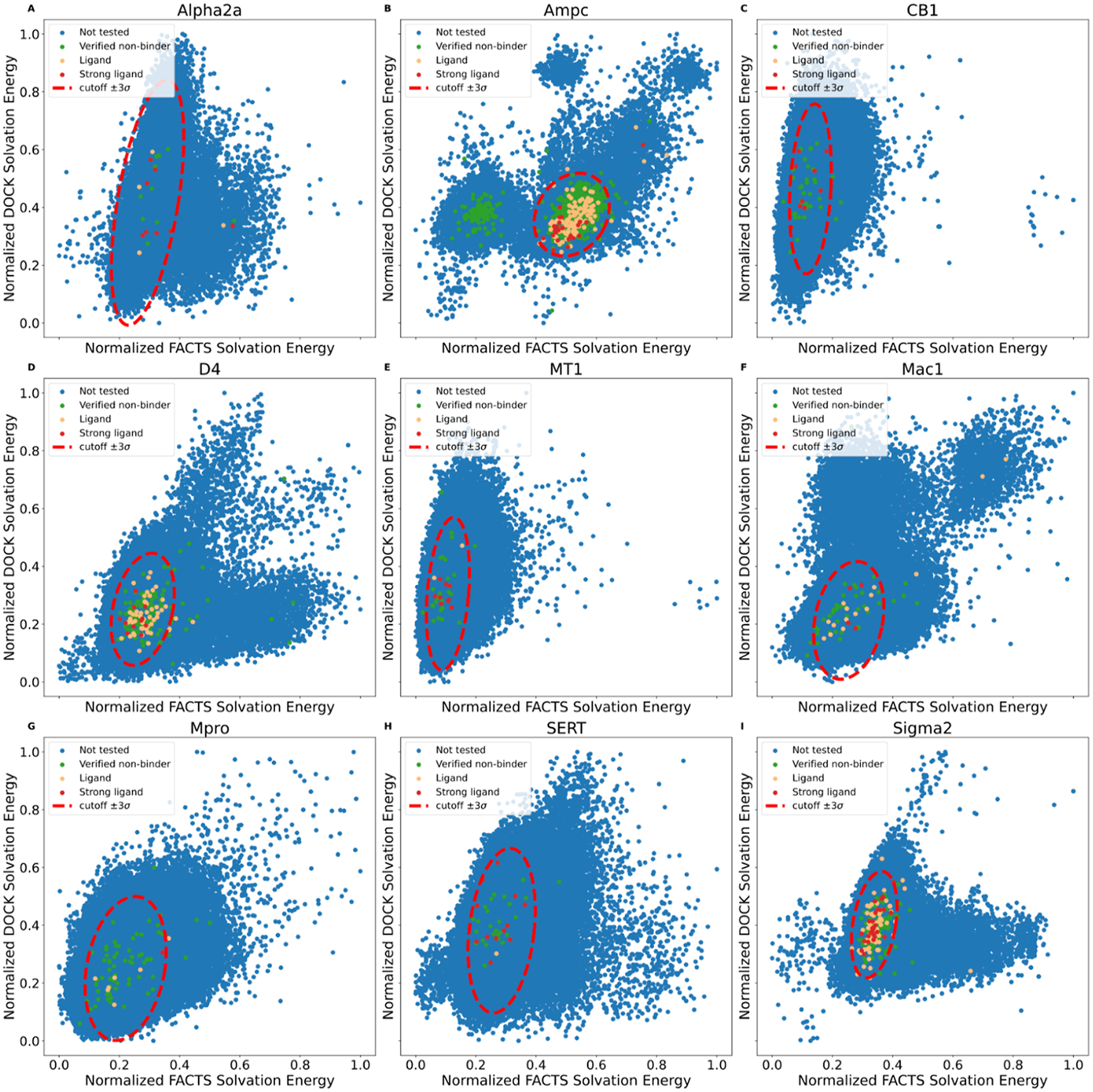
Joint distribution of the solvation free energy for receptor targets (A) Alpha2a, (B) AmpC, (C) CB1, (D) D4, (E) MT1, (F) Mac1, (G) Mpro, (H) SERT, and (I) Sigma2. Relatively potent ligands are colored red, and the rest of the ligands are colored orange. Experimentally tested nonbinders are colored green, and compounds that were not tested are colored blue.
More Compounds Are Flagged as Cheaters among Top-Ranking Results.
The true use case for cross-filtering is to remove molecules that cheat the scoring function, which experiment (Figure 2) and simulation5 suggest occur among the very highest-ranking compounds. It is thus interesting to understand how the number of docking hits captured as outliers changes with the docking rank. Overall, cross-filtering highlighted about 10% of the high-ranking docking hits from among the top 300,000 to 500,000 molecules (Table 2). If we plot the number of outliers captured by the cross-filtering across the rank distribution, however, we find higher fractions of outliers among the very top-ranking compounds for six of the nine targets (Supporting Information Figure 5).
Prospective Study—Are Cheaters Experimental Nonbinders?
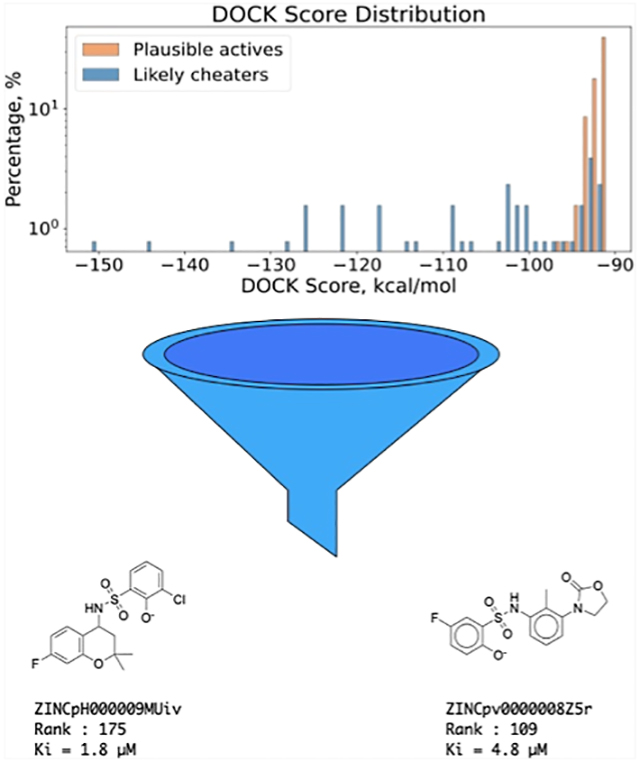
To test cross-filtering prospectively, we prioritized and synthesized the very top-ranking 128 de novo molecules from the AmpC screen25 (ranked 1 to 415 out of 1.7 billion, DOCK3.8 scores from −151 to −91 kcal/mol). None of these had been previously tested. Of these, solvation cross-filtering identified 39 as cheaters (ranked 1 to 407) falling outside of the 3σ radii of the mean scores of the joint distribution (Figure 6A) and 89 as noncheaters falling within that distribution. Given their good scores, these 89 were considered plausible AmpC inhibitors; their scores overlapped with those of the cheaters (Figure 6B).
Figure 6.
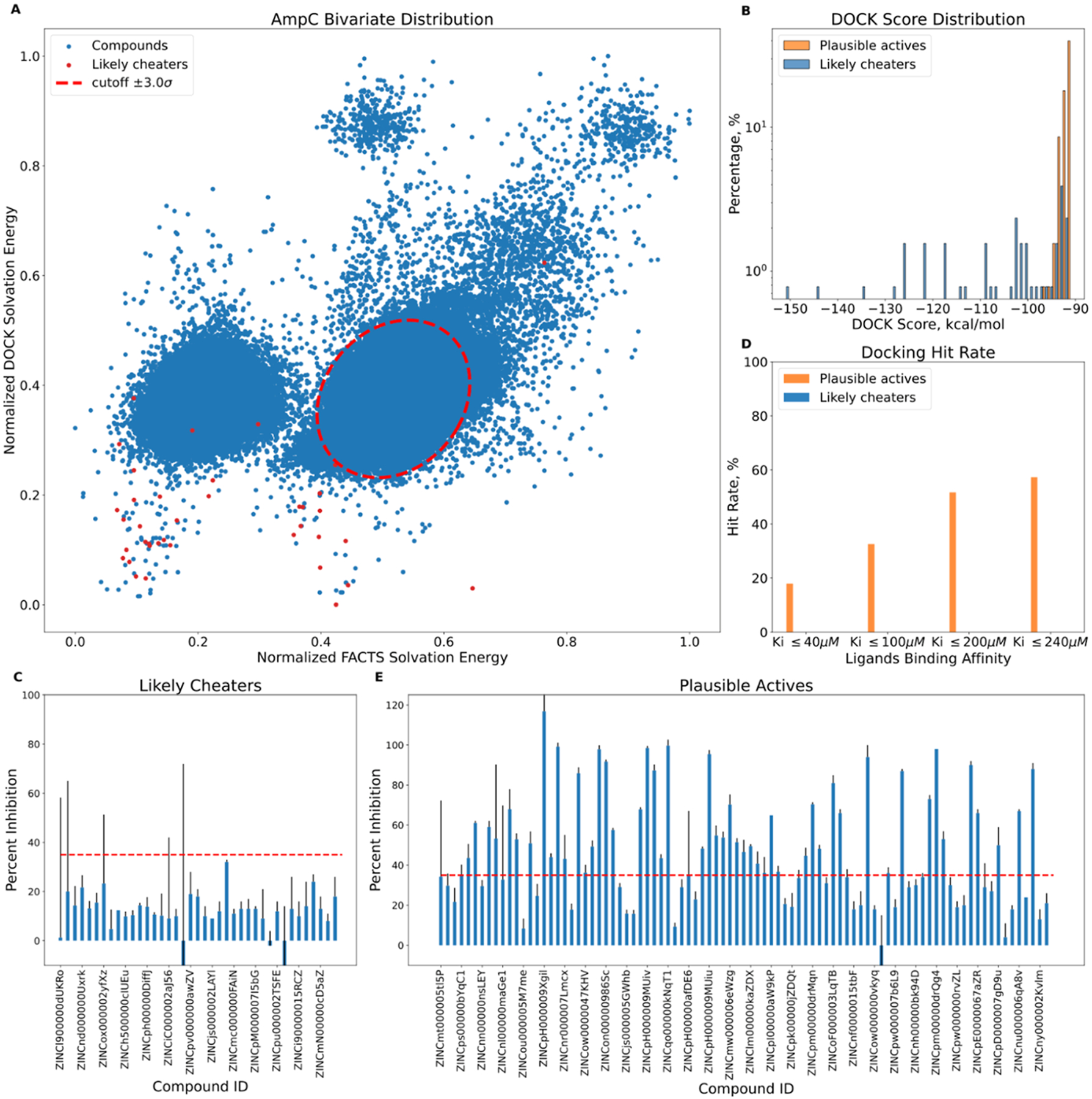
Experimental testing of predicted high-ranking docking cheaters and plausible true actives against AmpC. (A) Bivariate distribution of AmpC top 300 K ranking compounds. The predicted cheaters are highlighted in red. (B) DOCK score distribution for top-ranking putative cheaters and plausible true actives. (C) Percentage inhibition at 200 μM for likely cheaters. (D) Docking hit rate as a function of AmpC apparent Ki. (E) Percentage inhibition at 200 μM for plausible actives; for both panels (C,E), the ZINC numbers of only every fourth compound are shown for clarity.
All 128 molecules were tested for AmpC inhibition, initially at 200, 100, and 40 μM. None of the 39 outliers inhibited the enzyme substantially at even the top concentration, including compounds ranked 1 to 26 of the 1.7 billion docked; all were classified as nonbinders (Figure 6C). Conversely, of the 89 high-ranking plausible ligands, 51 (57%) inhibited meaningfully at the top concentration (Figure 6D), with inhibition at the lower concentrations consistent with apparent Ki values of 240 μM or better and with 19 inhibiting the enzyme with apparent Ki values between 1.8 and 50 μM (Figure 6E). Concentration–response curves for the eight most potent inhibitors were well-behaved (Figure 7). Their activities place them among the more potent inhibitors discovered for AmpC from docking or high-throughput screens6,25,31,32 (full compound results are listed in ampc-prospective-result.xlsx), while the gross hit rate of 57% (Figure 6D) is among the highest observed for this enzyme. The experimental results and corresponding SMILES string for each compound are recorded in the Supporting Information file (ampc-prospective-result.xlsx).
Figure 7.
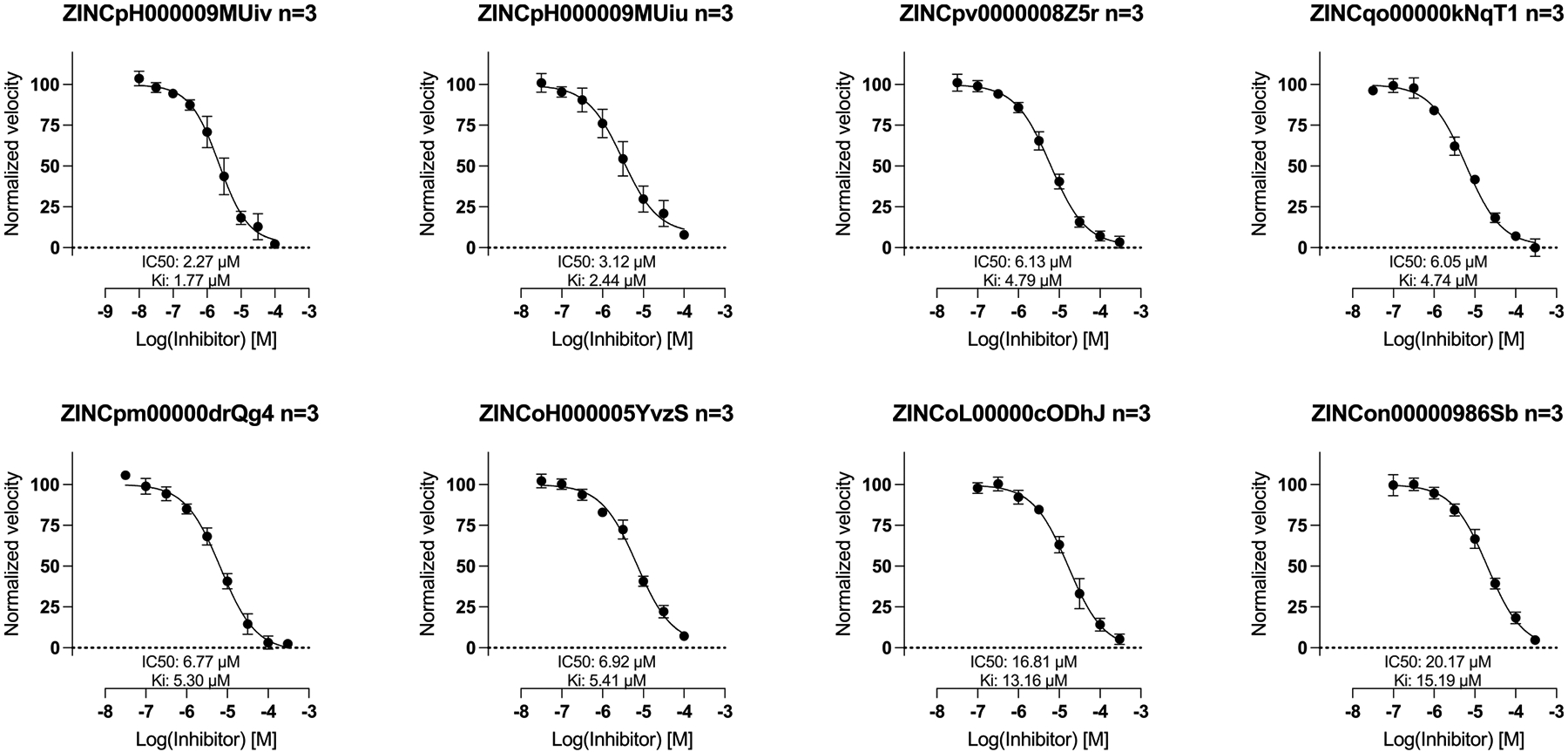
Concentration–response curves for the top eight docking hits. The curves were fit with Hill slopes fixed to 1; had we allowed them to vary, they would have been MUiv −1.0; MUiu −0.82; 8Z5r −0.96; NqT1−0.83; rQg4−0.96; YvzS −0.81; ODhJ-1.05; 86Sb −0.87.
The AmpC assays were run in 0.01% Triton X-100, reducing the likelihood of colloidal aggregation. Nevertheless, for the 10 most potent inhibitors, dynamic light scattering (DLS) and counter-screening against malate dehydrogenase (MDH) were used to investigate colloidal aggregation at concentrations 10-fold higher than their apparent AmpC Ki values (Supporting Information Figures S6 and S7). Only one compound, with an apparent Ki of 8.6 μM, formed colloid-like particles, while none were observed for other top hits, consistent with these latter molecules acting as classic, active-site-directed inhibitors.
Other Methods to Detect Cheaters—AB-FEP and MM/GBMV.
In principle, one should be able to use many orthogonal scoring functions to identify molecules that cheat docking scoring functions. We tried two more here: AB-FEP and molecular mechanics/GBMV (MM/GBMV). Both represent higher levels of theory than implicit solvation methods like FACTS, especially AB-FEP. We note that using these methods differs somewhat from comparing the two solvation energies (those from FACTS and DOCK3.8), which may be thought of as an outlier detection approach, whereas rescoring with MM/GBMV and AB-FEP are orthogonal scoring approaches where we apply a threshold to an energy to predict ligands and decoys. Both methods were applied to the same 128 top-ranking molecules described in the previous section (see Experimental Section).
While GBMV with only minimization had performed relatively well in retrospective calculations (Supporting Information Table 1), the addition of a molecular dynamics component seemed to diminish its ability to prospectively distinguish the 89 plausible inhibitors or the 51 true ones from the 39 cheating artifacts. A challenge here was knowing where to draw the cutoff between plausible inhibitors and likely cheating artifacts since the MM/GBMV energies were much higher in magnitude than the energies inferred from the experimental apparent Ki values (Figure 8a). Accordingly, we investigated different energy cutoffs to distinguish the cheaters from the plausible ligands. Irrespective of where we drew the boundary, however, either very few true cheaters (true negatives) were found or too many true inhibitors were (false negatives). For instance, when we set the cutoff to be worse (greater) than −75 kcal/mol, only 13 of the 39 cheaters were identified, as was one phenolate sulfonamide, a close analogue of true inhibitors and a plausible ligand (Figure 8b). As we raised the stringency of the cutoff, more cheaters were found, but so were more true inhibitors. Increasing the cutoff to −125 kcal/mol identified 24 of the true cheaters but at the cost of adding 13 of the 89 plausible ligands, of which 9 were ultimately shown to be true inhibitors by experiment. Thus, the higher sampling of MM/GBMV led to worse results than the simpler FACTS approach. This may reflect the increased noise on addition of the molecular dynamics and the large magnitude of the MM/GBMV energies. Retrospective calculations in other systems bore this out: against the nonbinders and what turned out to be true ligands from the large dopamine D4 and Sigma2 receptor screens, we observed the same inability to clearly distinguish between nonbinders and true ligands (Supporting Information Figure S8). A caveat here is that many of these nonbinders were what we consider normal-docking failures, though among the very best docking scores, about half of them were likely “cheaters”.
Figure 8.
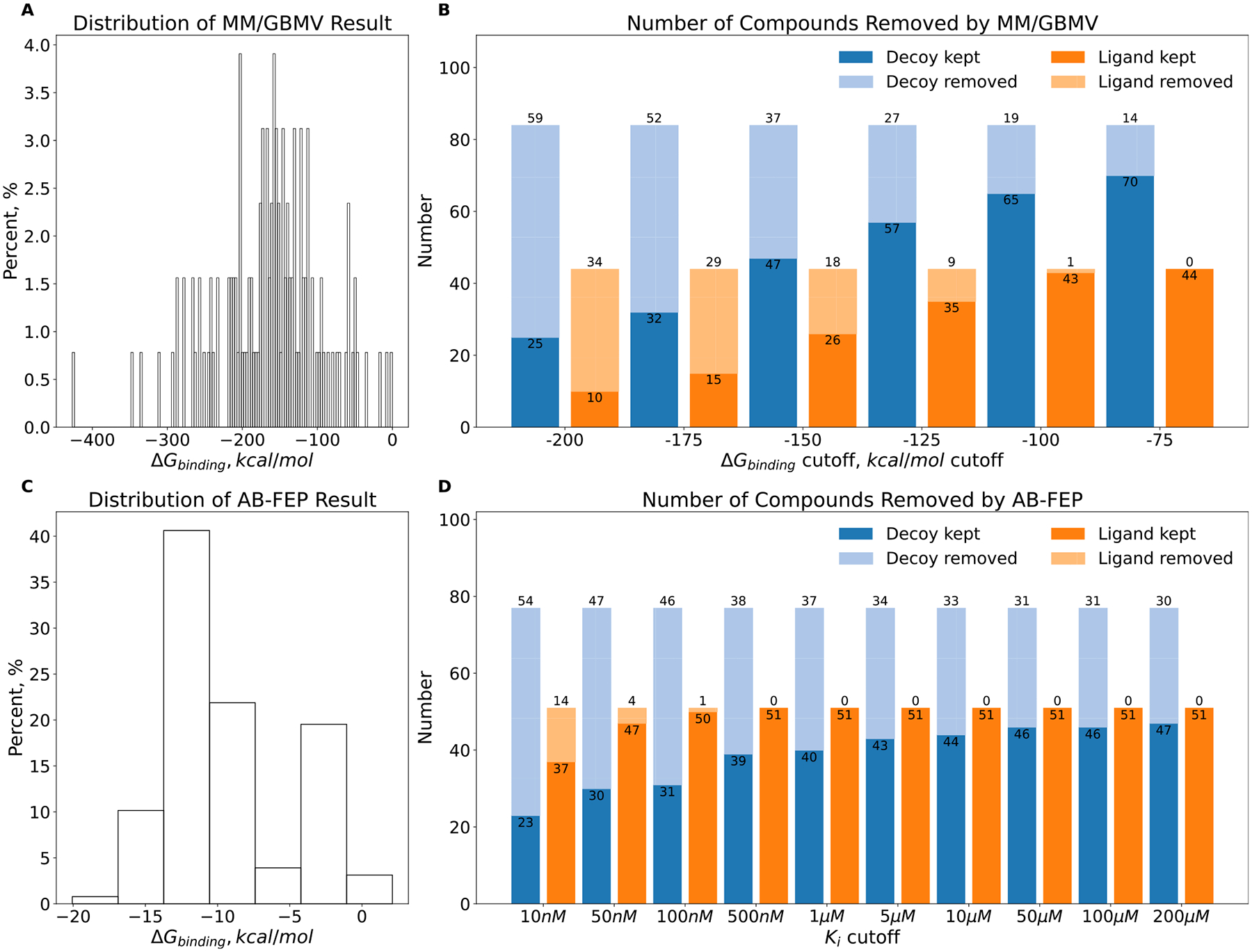
Rescoring the AmpC hits by MM/GBMV: (A) distribution of energy and (B) number of compounds removed as a function of energy. Rescoring results for AB-FEP: (C) distribution of energy and (D) number of compounds removed as a function of Kd.
AB-FEP performed better. Here too, the range of energies was larger than the experimental values (Figure 8c), reaching affinities in the midfemtomolar, and we considered two energy cutoffs to filter out putative cheating molecules: molecules calculated by AB-FEP to have Kd values worse than 200 μM (essentially the experimental cutoff) and molecules with calculated Kd values worse than 1 μM. At the 200 μM cutoff, AB-FEP removed 30 of the 39 FACTS-predicted cheating artifacts without flagging any false negatives (true inhibitors) (Figure 8d). At the 1 μM cutoff, it found 37 of the 39 cheating artifacts and classified another phenolate sulfonamide, a close analogue of true inhibitors so arguably a plausible ligand, as a cheater, with no experimentally confirmed false negatives found (Figure 8d). As the AB-FEP cutoff Kd became more potent than 1 μM, the number of false negatives began to rise. Detailed calculation results may be found in the Supporting Information (ampc-prospective- result.xlsx). We note that AB-FEP calculates binding affinities (expressed here as Kd values), while inhibition numbers are reported as enzymological Ki values; these two values represent the same thing, and both may be converted into free energies of binding.
DISCUSSION AND CONCLUSIONS
As our virtual libraries grow,2,8,19 both simulation5 and experiment25 suggest that docking results improve. Unfortunately, with this growth have emerged a small group of molecules that cheat our scoring functions and crowd the top scores of docking-ranked libraries.1,5 While rare, these molecules rise in sheer number as libraries grow; unchecked, they may come to dominate the top-ranking molecules. A key observation from this study is that many of these scoring function “cheaters” may be recognized and deprioritized by an orthogonal scoring function. Perhaps the most compelling evidence for this comes from a prospective study against the model enzyme AmpC β-lactamase. Cross-filtering identified 39 “cheaters” from the very top-ranks of a docking campaign, and on experimental testing none bound substantially. Meanwhile, the method found another 89 compounds, often interspersed among the 39 “cheaters”, that were plausible ligands. On experimental testing, 57% of these 89 docking hits inhibited the enzyme. These results suggest that it is possible to identify molecules that cheat our scoring functions by filtering them out of top-ranking lists to reveal the more plausible and interesting high-ranking molecules.
Several caveats merit mentioning. Although we suspect that molecules that exploit scoring-function holes may be ubiquitous in large-library docking, we have shown that they exist only for DOCK3.7/3.8. Correspondingly, we have focused on physics-based scoring to cross-filter docking results, and while we might expect many methods to perform well in this role, we have not shown that. Finally, we note that our strategy of rescoring the top 300,000 high-ranking molecules seems sensible for billion-molecule libraries, where we expect several hundred cheating artifacts concentrated at the highest ranks. As our libraries grow toward trillions of molecules, however, there may be hundreds of thousands of cheating artifacts, demanding a second criterion to find the region where the cheaters can be separated from the true ligands. One way to do this would be to rescore not so much a fixed number of high-ranking molecules but to consider the region where the docking scores diverge from the distribution for the overall library (Figure 2). Irrespective of library size, such a break in the scoring distribution may identify the occurrence of cheating artifacts.
These caveats should not obscure the key points of this study. We expect ever greater numbers of molecules to find holes in our scoring functions as docking libraries continue to grow and diversify.5 Rescoring top-ranking molecules with a second scoring function that, whatever its own holes, is unlikely to share those of the primary scoring function can help to eliminate these molecules, revealing the more interesting and plausible molecules that they rank among. This strategy thus may be generally useful in the field, and multiple scoring functions may be useful for rescoring. The FACTS method used here may be particularly well-suited to physics-based approaches; accordingly, we provide easy-to-use rescoring and analysis scripts in the supplementary files.
EXPERIMENTAL SECTION
Computational Details.
Unless otherwise specified, Open Babel33 was used to generate random ligand conformations, while ParamChem34,35 was used to prepare ligand topology and parameter files. The CHARMM C36 force fields36 were used, and molecular dynamics was performed in CHARMM.37 The general AMBER force field38 was used for comparison. Detailed molecular dynamics scripts are documented in the Supporting Information.
Rescoring with the Implicit Solvent Model.
We used the implicit solvent model FACTS23 and the GBMV24 for rescoring. Unless otherwise noted, for each of the docked poses, we performed a short minimization (1000 steps) using FACTS with the CHARMM force field. The protein atoms are fixed to reduce the computational cost. The total energy of the system is recorded (i.e., the sum of Eelec, Evdw, Esolvation, and the internal energy of the protein and ligand). The solvation energy is explicitly computed by the implicit solvent model. The free energy at the unbound state is computed by first generating 20 random ligand conformations using the Open Babel functionality (obrotamer), followed by minimization. Then, we computed the ensemble average of these 20 trials and considered it as Eligand free energy at unbound state because we are only interested in the same receptor structure. The protein free energy at the bound state is a constant and can be neglected. The detailed CHARMM script for rescoring can be found in Supporting Information.
| (1) |
Cross-Filtering with Bimodal Distribution.
For compounds with different topologies binding to the same receptor binding pocket, it is reasonable to assume that these compounds have similar physiochemical properties so that they could maintain similar key interactions with the binding pocket (i.e., normal distribution). However, the top-ranking artifacts cheat one or multiple energy terms in the scoring function mainly because of (1) incorrect parameterization of the force field and (2) the missing details in the scoring function. Therefore, using another docking method might help separate these artifacts.
As shown in Figure 3, one could draw an ellipse to determine the outliers. The width, height, and center of the ellipse are equivalent to the standard deviation and the mean of each of the normal distributions. While the orientation of the ellipse can be determined by the covariance of the two variants, the size of the ellipse is defined by the user (i.e., the σ cutoff). We determined the ellipse based on the mean and the standard deviation of each of the normal distributions and the covariance of the bivariate normal distribution (eq 2).
| (2) |
The detailed python script can be found in Supporting Information.
Calculating Absolute Binding Free Energy.
Absolute binding free energy (AB-FEP) calculations were performed with FEP+ on Schrodinger Web Services. The OPLS4 force field was used along with the Force Field Builder, where missing parameters were calculated. The total molecular dynamics simulation time for each ligand’s AB-FEP calculation was set to 1 ns. The docking poses used as the input for FEP were WScore docking results. All calculations were performed using Schrödinger Software Suite 2023–4.39
The MM/GBMV experiments were run up as follows:40 for each compound, we collected the top 5 docking poses. The bound state was minimized in vacuum with a maximum of 2000 steps before running molecular dynamics with the GBMV implicit solvent model. Hydrogen mass repartitioning was applied to reduce the computational cost. The total simulation length was 30 ns, and the last 1 ns of the simulation was used for the MM/GBMV calculation. Each simulation was repeated five times, and the average energy was computed. The lowest energy value among the five docking poses was considered the free energy of the bound state for that compound. The free energy for the unbound ligand state was computed with 25 random conformations generated by Open Babel, followed by the same MM/GBMV protocol.
AmpC β-Lactamase Enzymology.
AmpC was purified as described.41 All candidate inhibitors were dissolved in DMSO at 20 mM, and more dilute DMSO stocks were prepared as necessary so that the concentration of DMSO was held constant at 1% v/v in 50 mM sodium cacodylate buffer, pH 6.5. AmpC activity and inhibition were monitored spectrophotometrically using either CENTA or nitrocefin as substrates. All assays included 0.01% Triton X-100 to reduce compound aggregation artifacts. Active compounds were further investigated for aggregation by DLS and by detergent-dependent inhibition of the counter-screening enzyme MDH.
For initial screening, the docking hits were diluted such that the final concentrations in the reaction buffer were 200, 100, and 40 μM. In these assays, the AmpC substrate nitrocefin16 was used, with an [S]/Km ratio of 0.56 (Km nitrocefin 180 μM; [S] = 100 μM) and 0.16 ([S] = 28 μM). The colorimetric assay was carried out using a BMG Labtech CLARIOstar for kinetic measurements of 50 s in a 96-well format. IC50 values reflect the percentage inhibition fit to a dose–response equation in GraphPad Prism with a Hill coefficient set to one Ki was calculated using the Cheng–Prusoff equation For eight of the most potent compounds, based on the initial three concentration-point results, full dose–response curves were measured.
Docking Hits Tested.
All compounds were synthesized by Enamine and used without further purification. Of the 51 active compounds, all but 6 had purities of ≥95%. All of the 6 had purities ≥91%. The identities of all compounds are given in ampc-prospective-result.xlsx of the Supporting Information and are available from Enamine.
MDH Enzyme Inhibition Assay.
Compounds were diluted to 100 μM in 50 mM KPi buffer, pH 7, at a final concentration of 1% DMSO (v/v). Samples were incubated with MDH (Sigma, 442610) for 5 min. The reaction was initiated by adding 200 μM oxaloacetic acid (Sigma, 04126) and 200 μM nicotinamide adenine dinucleotide (NADH) (Sigma-Aldrich,10128023001). The reaction was monitored for 80 s at an absorbance of 340 nm. Sample rates were divided by the DMSO control rates. Compounds that showed less than 35% inhibition were not considered to be inhibitors. Compounds that surpassed the enzyme inhibition threshold were screened as a concentration–response curve from 100 to 0.1 μM. Identified inhibitors were rescreened at 100 μM in the presence of 0.01% (v/v) Triton-X 100 to determine detergent reversibility. All samples were screened in triplicate. Data were analyzed by using GraphPad Prism version 10.2.3 (Boston, MA).
Dynamic Light Scattering.
Compounds were diluted in a filtered 50 mM KPi buffer, pH 7, at a final concentration of 1% DMSO (v/v). All compounds were initially screened at a top concentration of 100 μM using a DynaPro Plate Reader III. Samples that had a scattering intensity >1 × 107 cnts/s scattering in this instrument were considered to form colloid-like particles and were rescreened as a concentration–response curve in eight-point half-log dilutions. Data were separated into two groups: aggregating concentrations (scattering >1 × 107 cnts/s) and nonaggregating concentrations (scattering <1 × 107 cnts/s). A line was generated for each group, and the point of intersection of the two lines serves as the critical aggregation concentration. All samples were screened in triplicate. Data were analyzed using GraphPad Prism.
Supplementary Material
Funding
This work is supported by US National Institute of Health grants R35GM122481 (to BKS) and GM130587 (to CLB).
ABBREVIATIONS USED
- AB-FEP
absolute binding free energy perturbation
- FACTS
fast analytical continuum treatment of solvation
- FEP
free energy perturbation
- GBMV
generalized born using molecular volume
- MM/GBMV
molecular mechanics/generalized born molecular volume
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01632.
Detailed description of bivariate distribution method development, filtering results with different implicit solvent models and force field parameters, DLS results, MDH enzyme inhibition assay results for the top 10 compounds, CHARMM and Python scripts for cross-filtering, and data set for reproducing AmpC retrospective results (PDF)
Measured experimental Ki, AB-FEP, MM/GBMV, and filtering results for all 128 prospectively tested compounds (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.4c01632
The authors declare the following competing financial interest(s): BKS is co-founder of BlueDolphin LLC, Epiodyne Inc, and Deep Apple Therapeutics, Inc., and serves on the SRB of Genentech, the SAB of Schrodinger LLC, and the SAB of Vilya Therapeutics. No other authors declare competing interests.
Contributor Information
Yujin Wu, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Fangyu Liu, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Isabella Glenn, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Karla Fonseca-Valencia, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States;.
Lu Paris, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Yuyue Xiong, Schrödinger, Inc., San Diego, California 92121, United States.
Steven V. Jerome, Schrödinger, Inc., New York, New York 10036, United States
Charles L. Brooks, III, Biophysics Program, University of Michigan, Ann Arbor, Michigan 48109, United States;.
Brian K. Shoichet, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States;
REFERENCES
- (1).Alon A; Lyu J; Braz JM; Tummino TA; Craik V; O’Meara MJ; Webb CM; Radchenko DS; Moroz YS; Huang X-P; et al. Structures of the σ2 receptor enable docking for bioactive ligand discovery. Nature 2021, 600, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gorgulla C; Boeszoermenyi A; Wang Z-F; Fischer PD; Coote PW; Padmanabha Das KM; Malets YS; Radchenko DS; Moroz YS; Scott DA; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ghahremanpour MM; Tirado-Rives J; Deshmukh M; Ippolito JA; Zhang C-H; Cabeza de Vaca I; Liosi M-E; Anderson KS; Jorgensen WL Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med. Chem. Lett 2020, 11, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kaplan AL; Confair DN; Kim K; Barros-Álvarez X; Rodriguiz RM; Yang Y; Kweon OS; Che T; McCorvy JD; Kamber DN; et al. Bespoke library docking for 5-HT2A receptor agonists with antidepressant activity. Nature 2022, 610, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lyu J; Irwin JJ; Shoichet BK Modeling the expansion of virtual screening libraries. Nat. Chem. Biol 2023, 19, 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; O’Meara MJ; Che T; Algaa E; Tolmachova K; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sadybekov AA; Brouillette RL; Marin E; Sadybekov AV; Luginina A; Gusach A; Mishin A; Besserer-Offroy E; Longpré JM; Borshchevskiy V; et al. Structure-based virtual screening of ultra-large library yields potent antagonists for a lipid GPCR. Biomolecules 2020, 10, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sadybekov AA; Sadybekov AV; Liu Y; Iliopoulos-Tsoutsouvas C; Huang X-P; Pickett J; Houser B; Patel N; Tran NK; Tong F; et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature 2022, 601, 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fink EA; Xu J; Hübner H; Braz JM; Seemann P; Avet C; Craik V; Weikert D; Schmidt MF; Webb CM; et al. Structure-based discovery of nonopioid analgesics acting through the α2A-adrenergic receptor. Science 2022, 377, No. eabn7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jones CA; Brown BP; Schultz DC; Engers J; Kramlinger VM; Meiler J; Lindsley CW Computer-Aided Design and Biological Evaluation of Diazaspirocyclic D4R Antagonists. ACS Chem. Neurosci 2024, 15 (12), 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Stein RM; Kang HJ; McCorvy JD; Glatfelter GC; Jones AJ; Che T; Slocum S; Huang X-P; Savych O; Moroz YS; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Singh I; Seth A; Billesbølle CB; Braz J; Rodriguiz RM; Roy K; Bekele B; Craik V; Huang X-P; Boytsov D; et al. Structure-based discovery of conformationally selective inhibitors of the serotonin transporter. Cell 2023, 186, 2160–2175. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Beroza P; Crawford JJ; Ganichkin O; Gendelev L; Harris SF; Klein R; Miu A; Steinbacher S; Klingler F-M; Lemmen C Chemical space docking enables large-scale structure-based virtual screening to discover ROCK1 kinase inhibitors. Nat. Commun 2022, 13, 6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Walters WP; Wang R New Trends in Virtual Screening. J. Chem. Inf. Model 2020, 60, 4109–4111. [DOI] [PubMed] [Google Scholar]
- (15).Ruddigkeit L; Van Deursen R; Blum LC; Reymond J-L Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model 2012, 52, 2864–2875. [DOI] [PubMed] [Google Scholar]
- (16).Coley CW; Green WH; Jensen KF Machine learning in computer-aided synthesis planning. Acc. Chem. Res 2018, 51, 1281–1289. [DOI] [PubMed] [Google Scholar]
- (17).Cavasotto CN; Di Filippo JI The impact of supervised learning methods in ultralarge high-throughput docking. J. Chem. Inf. Model 2023, 63, 2267–2280. [DOI] [PubMed] [Google Scholar]
- (18).Chen W; Cui D; Jerome SV; Michino M; Lenselink EB; Huggins DJ; Beautrait A; Vendome J; Abel R; Friesner RA; et al. Enhancing hit discovery in virtual screening through absolute protein–ligand binding free-energy calculations. J. Chem. Inf. Model 2023, 63, 3171–3185. [DOI] [PubMed] [Google Scholar]
- (19).Gorgulla C Recent developments in ultralarge and structure-based virtual screening approaches. Annu. Rev. Biomed. Data Sci 2023, 6, 229–258. [DOI] [PubMed] [Google Scholar]
- (20).Charifson PS; Corkery JJ; Murcko MA; Walters WP Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem 1999, 42, 5100–5109. [DOI] [PubMed] [Google Scholar]
- (21).Zhou T; Caflisch A High-throughput virtual screening using quantum mechanical probes: discovery of selective kinase inhibitors. ChemMedChem 2010, 5, 1007–1014. [DOI] [PubMed] [Google Scholar]
- (22).Graves AP; Shivakumar DM; Boyce SE; Jacobson MP; Case DA; Shoichet BK Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J. Mol. Biol 2008, 377, 914–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Haberthür U; Caflisch A FACTS: Fast analytical continuum treatment of solvation. J. Comput. Chem 2008, 29, 701–715. [DOI] [PubMed] [Google Scholar]
- (24).Lee MS; Salsbury FR Jr.; Brooks CL III Novel generalized Born methods. J. Chem. Phys 2002, 116, 10606–10614. [Google Scholar]
- (25).Liu F; Mailhot O; Glenn IS; Vigneron SF; Bassim V; Xu X; Fonseca-Valencia K; Smith MS; Radchenko DS; Fraser JS; Moroz YS; Irwin JJ; Shoichet BK The impact of Library Size and Scale of Testing on Virtual Screening. bioRxiv 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gahbauer S; Correy GJ; Schuller M; Ferla MP; Doruk YU; Rachman M; Wu T; Diolaiti M; Wang S; Neitz RJ; Fearon D; Radchenko DS; Moroz YS; Irwin JJ; Renslo AR; Taylor JC; Gestwicki JE; von Delft F; Ashworth A; Ahel I; Shoichet BK; Fraser JS Iterative computational design and crystallographic screening identifies potent inhibitors targeting the Nsp3 macrodomain of SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A 2023, 120 (2), e2212931120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Fink EA; Bardine C; Gahbauer S; Singh I; Detomasi TC; White K; Gu S; Wan X; Chen J; Ary B; et al. Large library docking for novel SARS-CoV-2 main protease non-covalent and covalent inhibitors. Protein Sci. 2023, 32, No. e4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tummino TA; Iliopoulos-Tsoutsouvas C; Braz JM; O’Brien ES; Stein RM; Craik V; Tran NK; Ganapathy S; Shiimura Y; Tong F; Ho TC; Radchenko DS; Moroz YS; Rodriguez Rosado S; Bhardwaj K; Benitez J; Liu Y; Kandasamy H; Normand C; Semache M; Sabbagh L; Glenn I; Irwin JJ; Kumar KK; Makriyannis A; Basbaum AI; Shoichet BK Large library docking for cannabinoid-1 receptor agonists with reduced side effects. Cold Spring Harbor Laboratory 2024, No. 2023.02.27.530254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tingle BI; Irwin JJ Large-scale docking in the cloud. J. Chem. Inf. Model 2023, 63, 2735–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gorgulla C; Padmanabha Das KM; Leigh KE; Cespugli M; Fischer PD; Wang Z-F; Tesseyre G; Pandita S; Shnapir A; Calderaio A; et al. A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening. iscience 2021, 24, 102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Powers RA; Shoichet BK Structure-Based Approach for Binding Site Identification on AmpC β-Lactamase. J. Med. Chem 2002, 45, 3222–3234. [DOI] [PubMed] [Google Scholar]
- (32).Babaoglu K; Simeonov A; Irwin JJ; Nelson ME; Feng B; Thomas CJ; Cancian L; Costi MP; Maltby DA; Jadhav A; et al. Comprehensive Mechanistic Analysis of Hits from High-Throughput and Docking Screens against β-Lactamase. J. Med. Chem 2008, 51, 2502–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).O’Boyle NM; Banck M; James CA; Morley C; Vandermeersch T; Hutchison GR Open Babel: An open chemical toolbox. J. Cheminf 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Vanommeslaeghe K; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Vanommeslaeghe K; Raman EP; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; et al. CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (39).Wang L; Chambers J; Abel R Protein–ligand binding free energy calculations with FEP+. Methods Mol. Biol 2019, 2022, 201–232. [DOI] [PubMed] [Google Scholar]
- (40).Lai TT; Eken Y i.; Wilson, A. K. Binding of per-and polyfluoroalkyl substances to the human pregnane X receptor. Environ. Sci. Technol 2020, 54, 15986–15995. [DOI] [PubMed] [Google Scholar]
- (41).Usher KC; Blaszczak LC; Weston GS; Shoichet BK; Remington SJ Three-Dimensional Structure of AmpC β-Lactamase from Escherichia coli Bound to a Transition-State Analogue: Possible Implications for the Oxyanion Hypothesis and for Inhibitor Design. Biochemistry 1998, 37, 16082–16092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.