

Abstract
Fatty acids are metabolized by β-oxidation within the “mitochondrial ketogenic pathway” (MKP) to generate β-hydroxybutyrate (BHB), a ketone body. BHB can be generated by most cells but largely by hepatocytes following exercise, fasting, or ketogenic diet consumption. BHB has been shown to modulate systemic and brain inflammation; however, its direct effects on microglia have been little studied. We investigated the impact of BHB on Aβ oligomer (AβO)-stimulated human iPS-derived microglia (hiMG), a model relevant to the pathogenesis of Alzheimer's disease (AD). HiMG responded to AβO with proinflammatory activation, which was mitigated by BHB at physiological concentrations of 0.1–2 mM. AβO stimulated glycolytic transcripts, suppressed genes in the β-oxidation pathway, and induced over-expression of AD-relevant p46Shc, an endogenous inhibitor of thiolase, actions that are expected to suppress MKP. AβO also triggered mitochondrial Ca2+ increase, mitochondrial reactive oxygen species production, and activation of the mitochondrial permeability transition pore. BHB potently ameliorated all the above mitochondrial changes and rectified the MKP, resulting in reduced inflammasome activation and recovery of the phagocytotic function impaired by AβO. These results indicate that microglia MKP can be induced to modulate microglia immunometabolism, and that BHB can remedy “keto-deficiency” resulting from MKP suppression and shift microglia away from proinflammatory mitochondrial metabolism. These effects of BHB may contribute to the beneficial effects of ketogenic diet intervention in aged mice and in human subjects with mild AD.
Keywords: Alzheimer's, amyloid, inflammation, ketone, microglia, mitochondria, phagocytosis
1 |. INTRODUCTION
Microglia survey and respond to their microenvironment to maintain brain homeostasis. Their cellular processes are highly mobile, and they respond to various environmental challenges with multiple actions, which include phagocytosis of debris and release of cytokines and chemokines.1 Such constant surveillance and responses require large energy expenditure and sometimes flexible means to operate in suboptimal energetic conditions.2 It is now widely accepted that microglia are equipped with metabolic flexibility, which underlies the repertoire of microglial states3,4 and allows them to utilize alternative nutrients other than glucose, such as the amino acid glutamine or fatty acids.2,5 However, despite recent advances, the impact of metabolic shift on microglial function is poorly understood.4 Metabolisms of energy substrates have been well studied in neurons and astrocytes, but much less so in microglia, especially human microglia, as most studies so far have used murine primary microglia or cell lines such as MG-6 or BV-2 as models.
It is not surprising that metabolic flexibility of microglia is foremost governed by mitochondria. The alternative energy sources used by microglia are metabolized by mitochondria, and glutamine metabolism has been shown to be intimately related to mitochondrial function in physiology and in diseases.6,7 Fatty acids are metabolized by β-oxidation within the “mitochondrial ketogenic pathway” (MKP) to generate β-hydroxybutyrate (BHB), a ketone body. In addition to being a passive energy carrier, BHB is increasingly recognized as a signaling molecule,8 but its signaling function in microglia is still poorly understood. Such knowledge is important as BHB may mediate the significant beneficial and inflammation suppressing effects to the brain by therapeutic ketosis such as ketogenic diet (KD) or fasting.9 We previously reported that mice on isocaloric KD not only lived ~12% longer, they showed significantly better cognitive, memory, and physical functions compared to mice at the same chronological age on the standard carbohydrate-rich control diet.10 During KD consumption, circulating BHB is mainly produced by the MKP in hepatocytes. Because BHB readily penetrates and could be an important energy source for the brain, microglia can be influenced by BHB from KD consumption.
In this study, we investigated the impact of BHB on Aβ oligomer (AβO)-stimulated human microglia, a model relevant to the pathogenesis of Alzheimer's disease (AD), the most common cause of dementia in elderly populations. In AD brains, microglia lose their homeostatic molecular signature and show profound functional impairments, such as increased production of proinflammatory cytokines, elevated reactive oxygen species (ROS), impaired phagocytosis, and increased inflammasome formation.11 Recently, the pivotal roles of microglia-orchestrated neuroinflammation in AD have been established.12-15 A generally accepted hypothesis states that activated microglia become functionally impaired and release cytotoxic substances and proinflammatory cytokines that cause neuronal damage and aggravate AD pathology.12 While multiple factors may cause microglial activation in AD, early studies have established that different species of Aβ aggregates are potent stimulants of microglia. Among them, we and others found that the small soluble AβO assembled from Aβ42 peptide provides far stronger stimulation to induce microglial activation.16,17 Aβ aggregates are recognized by a range of microglial pattern recognition receptors to induce mainly proinflammatory responses that could mediate Aβ-induced neurotoxicity, impair phagocytic function, and prime microglia to enhance their sensitivity and reactivity to inflammatory stimuli.18 Understanding mechanisms of such activations could reveal microglial therapeutic targets. From an interventional point of view, how to shift AD-associated microglial metabolic states toward beneficial immunometabolic outcomes is a critical question. We therefore intended to test the hypothesis that BHB provides immunometabolic countermeasures to AβO-activated inflammatory/neurotoxic microglia states. Moreover, to better model human microglial pathology, we employed human induced pluripotent stem cell (iP-SC)-derived microglia (hiMG) as a major model rather than the widely used primary cultures from neonatal rodents, based on the insight that rodents are generally a better model for neuronal pathology than they are for microglial pathology,19 and that for optimal translational validity, human microglia are recommended to be used to identify human-relevant molecular pathways and therapeutic targets.20
2 |. MATERIALS AND METHODS
2.1 |. Human iPSC culture, microglia differentiation, and BHB treatment
Human iPSCs were obtained from ALSTEM.INC. (Richmond, CA). The line used in this study was Human iPS Cell Line 26 (Episomal, CD34+, and ApoE3). Cells were plated onto Matrigel (Fisher) coated plates and cultured with mTeSR plus (Stemcell Technology). For microglia differentiation, we followed a previously described protocol.21,22 Briefly, 2 × 106 iPSCs were plated onto Aggrewell 800 plates (Stemcell Technology) to form embryoid bodies (EBs) in mTeSR1 supplemented with bone morphogenetic protein 4 (BMP4, 50 ng/mL)/vascular endothelial cell growth factor (VEGF, 50 ng/mL)/stem cell factor (SCF, 20 ng/mL), and culture for four days with daily medium change. On the fifth day, EBs were plated onto gelatin coated 6-well plates with 20 EBs per well in X-VIVO15 (Lonza) supplemented with M-CSF (100 ng/mL), IL-3 (25 ng/mL), glutamax (2 mM), penicillin/streptomycin (100 U/mL and 100 ug/mL), and β-mercaptoethanol (55uM), and the medium was changed weekly. After 3–4 weeks, floating primitive macrophage precursor (PMP) was collected and plated onto 12-well plates (5 × 104 cells/well), 6-well plates (3 × 105 cells/well), or 100 mm dishes (1.5 × 106 cells) and differentiated in microglia differentiation medium (Advanced DMEM/F12 supplemented with IL-34 [100 ng/mL], GM-CSF [10 ng/mL], N2 supplement [1×], glutamax [2 mM], penicillin/streptomycin [100 U/mL and 100 ug/mL], and β-mercaptoethanol [55 uM]) for two weeks. All experiments were conducted using microglia obtained following two-week differentiation. In this study, experiments using a batch of differentiated microglia are considered as independent replicates that generate independent data points. Most experiments were conducted with at least three replicates.
We tested the effects of BHB in AβO-stimulated hiMG in low glucose (1.5 mM), which would optimally show the metabolic and signaling function of BHB.23 For treatment, culture medium was replaced by glucose-free medium (SILAC Advanced DMEM/F-12 Flex Media, no glucose, no phenol red supplemented with L-lysine [0.499 mM], L-arginine [0.699 mM], IL-34 [100 ng/mL], GM-CSF [10 ng/mL], N2 supplement [1×], glutamax [2 mM], penicillin/streptomycin [100 U/mL and 100 ug/mL], and β-mercaptoethanol [55 uM]), with or without BHB. Cells were incubated for 30 min before treatment with AβO (3 μM) for 24 h. Following the addition of BHB, AβO, or control solutions that were prepared with regular glucose-containing DMEM/F12 medium (17.5 mM glucose), the final concentration of glucose in the incubation medium was 1.5 mM.
2.2 |. Aβ oligomer preparation
AβO composed of Aβ1-42 peptide was prepared following a standard procedure24 with a modification that the HFIP-treated Aβ1-42 peptide (Bachem) was dissolved in DMSO and then diluted with advanced DMEM/F12 culture medium instead of the F12 medium originally described, followed by incubation at 4°C for 24 h and 10 min centrifugation at 10 000 × rpm at 4°C. This preparation of AβO has been extensively characterized in our laboratory.16 Briefly, to ensure consistency of quality, a random sample from each batch was chosen and imaged using electron microscopy and atomic force microscopy to characterize the size and shape of the aggregates. The biological activities of each batch were confirmed by determining for AβO the neurotoxic activity, synaptic binding activity, and ability to rapidly induce exocytosis of MTT formazan, as described previously.16
2.3 |. qPCR
Total RNA from cultured cells was extracted using RNeasy Plus Mini Kit. cDNA was synthesized using 100 ng RNA and iScript Reverse Transcription Supermix (BioRad). Quantitative real-time polymerase chain reaction (qPCR) was performed using SsoFast™ EvaGreen Supermix and CFX96 qPCR system (BioRad). The forward/reverse primer sequences used are listed in Table 1. Gene expression was normalized to an endogenous gene, β-actin. Relative cDNA levels for the target genes were analyzed by the 2–ΔΔCt method.
TABLE 1.
Primers and primer sequences used for qPCR assays.
| Gene (Invitrogen) | Primer Sequence | |
|---|---|---|
| IL-1β (Human) | FW: GTGCAGTTCAGTGATCGTACAGG RV: CCACAGACCTTCCAGGAGAATG |
|
| IL-6 (Human) | FW: CCAGCTATGAACTCCTTCTC RV: GCTTGTTCCTCACATCTCTC |
|
| TNF-α (Human) | FW: CTCTTCTGCCTGCTGCACTTTG RV: ATGGGCTACAGGCTTGTCACTC |
|
| P2RY12 (Human) | FW: AAGAGCACTCAAGACTTTAC RV: GGGTTTGAATGTATCCAGTAAG |
|
| TMEM119 (Human) | FW: AGTCCTGTACGCCAAGGAAC RV: GCAGCAACAGAAGGATGAGG |
|
| CD206 (Human) | FW: CAGCGGTTGGCAGTGGA RV: CAGCTGATGGACTTTCCTGGTAAC |
|
| IGF-1 (Human) | FW: CTTCAGTTCGTGTGTGGAGACAG RV: CGCCCTCCGACTGCTG |
|
| NOS2 (Human) | FW: CAGCGGGATGACTTTCCAAG RV: AGGCAAGATTTGGACCTGCA |
|
| β-Actin (Human) | FW: TCAAGATCATTGCTCCTCCTGAG RV: ACATCTGCTGGAAGGTGGACA |
|
| Gene (Bio-Rad) | Primer | |
| AUH (Human) | qHsaCED0004657 | |
| HADH (Human) | qHsaCED0044872 | |
| IL-10 (Human) | qHsaCED0044704 | |
| IVD (Human) | qHsaCED0044239 | |
| MCU (Human) | qHsaCID0018492 |
2.4 |. Quantification of mitochondria number, mitochondrial reactive oxygen species (ROS), and mitochondrial Ca2+
After AβO treatment, cells were incubated with MitoTracker Deep Red FM (Invitrogen) for mitochondrial count or MitoSOX (Fisher) for mitochondrial ROS according to the manufacture's instruction. After staining, cells were harvested in in PBS with 1% BSA and reactivities quantified by using a BD Accuri™ C6 Plus flow cytometer (BD Biosciences). For mitochondrial Ca2+ quantification, cells were incubated with 200 nM MitoTracker and 3 μM Rhod-2 AM (Invitrogen) in Ca2+-free PBS with 1% BSA at 37°C. After 45 min incubation, the dye solution was removed and fresh 1%BSA was added, and the cells were incubated for another 30 min to allow complete de-esterification of intracellular AM esters according to the manufacture's instruction. After washing with PBS, cells were collected in 1%BSA and analyzed by flow cytometry using a BD Accuri C6 plus flow cytometer (BD Biosciences).
2.5 |. Mitochondrial permeability transition pore (mPTP) assay
mPTP opening was analyzed by MitoProbe™ Transition Pore Assay Kit (Molecular Probes) according to the manufacture's instruction with slight modifications. Briefly, 24 h after AβO and/or BHB treatment, cells (3 × 105 cell/well/6 wells) were incubated with Calcein-AM (1 μM)/CoCl2 (500 μM) in Hank's balance salt solution with calcium (HBSS/Ca2+) for 20 min at 37°. After incubation, cells were washed with HBSS/Ca2+ and PBS with Ca2+ (PBS/Ca2+) once each. 1% BSA in PBS/Ca2+ was added to the cultures, and cells were further stained with MitoTracker Deep Red FM (300 nM) for 45 min at 37°C. Cells were collected with 1% BSA in PBS/Ca2+ and analyzed by flow cytometry using a BD Accuri C6 plus flow cytometer (BD Biosciences).
2.6 |. Western blot
Cells were lysed in cell lysis buffer (140 mM NaCl, 10 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 1% Tx100, 0.1% SDS, 0.1% sodium deoxycholate) with protease inhibitor cocktail and phosphatase inhibitor (Sigma). Equivalent amounts of protein were analyzed by 4–20%Tris-Glycin gel electrophoresis (Invitrogen). Proteins were transferred to PVDF membranes and probed with antibodies at 4° overnight. Visualization was enabled using enhanced chemiluminescence (GE Healthcare Pharmacia). The following primary antibodies were used: anti-NLRP3 (1:700, Cell Signaling), anti-Pro-caspase 1 (1:700, cell Signaling), anticleaved caspase1 (1:700, Cell Signaling), anti-ASC/ TMS1 (1:700, Cell Signaling), anti-ProIL-1β (1:700. Cell Signaling), anti-MCU (1:700, Cell Signaling), anti-phos-pho-Shc (1:700, Cell Signaling, recognizing Tyr317 of phosphor-Shc), anti-Total-Shc (1:700, Abcam), and anti-β-actin (1:2000, Cell Signaling). Secondary antibodies for western blot were HRP-conjugated antirabbit IgG and antimouse IgG antibody (1:1000, Cell Signaling).
2.7 |. Phagocytosis assay
HiMG (7 × 104/well in 12-well plates) were treated with AβO (3 μM) with and without BHB (1 mM). Twenty-four hours later, cells were incubated with pHrodo Green BioParticles (5 μg/mL, Invitrogen) or Aβ1-42 monomer-FITC conjugated (500 nM, Bachem) for 1 h. Cells were then washed three times, 5 min each, with PBS. For cells being tested for phagocytosis of Aβ1-42 monomer-FITC conjugated, they were further incubated with 0.4% Trypan blue for 1 min to quench surface-bound Aβ. The cells were then analyzed by cell imaging. Fluorescence and bright images were taken by ImageXpress Pico scanner (Molecular devices). Fluorescence intensity was quantified by ImageJ. BioParticle-treated cells on a coverslip were fixed with 4% paraformaldehyde for 20 min, incubated with 5% BSA for 60 min, and then incubated with Iba1 antibody (1:700, Biocare Medical) overnight at 4°C followed by antirabbit IgG-Alexa594 conjugated (1:1000, Invitrogen) and Hoechst33342 (5 ug/mL, Invitrogen). Fluorescence images were taken by using a fluorescence microscope (Nikon).
2.8 |. RNAseq analysis
2.8.1 |. Library preparation for transcriptome sequencing
Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen). RNA quality evaluation (yield, purity, and integrity), cDNA library construction, and Illumina sequencing were performed by NovoGene (Sacramento, CA, USA). All RNA-seq reads were then aligned to the human reference genome (GRCh38.p13) using Hisat2 version 2.1.0. FeatureCounts version 1.4.6 was used to quantify reads counts.
2.8.2 |. Statistical analysis of RNAseq data
Data quality control, nonexpressed gene filtering, median ratio normalization (MRN) implemented in DESeq2 package, and identification of differentially expressed (DE) genes were done using the open source, web-based platform Galaxy (usegalaxy.org). To control false-positive rates, we applied the Benjamini–Hochberg false discovery rate (FDR) approach.
Genes that passed a threshold of FDR <0.05 in DE analysis were considered for further analysis. Gene Ontology (GO) and KEGG pathway enrichment analysis was performed in RStudio (R version 4.2.1) using ClusterProfiler package.
2.9 |. Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 software. All data are presented as means ± SEM. Sample sizes and statistical test used for each comparison were provided in corresponding figure legends. p < .05 was considered to be statistically significant.
3 |. RESULTS
3.1 |. BHB attenuates proinflammatory activation of human iPSC-derived microglia by AβO
We followed a standard procedure to differentiate a human iPS cell line into microglia-like cells.22 In a recent article, we confirmed their differentiation and maturation into microglia using specific markers and established the optimal concentration of AβO at 3 μM to activate proin-flammatory responses.21 We tested the effects of BHB in AβO-stimulated hiMG in low glucose (1.5 mM) medium which would optimally show the metabolic and signaling function of BHB.23 (A comparison of the hiMG response in culture with 1.5 and 17.5 mM glucose is shown in Figure S1.) We found that BHB dose-dependently suppressed AβO-induced expression of IL-1β and TNF-α genes, showing significant effects at as low as 0.1 mM (Figure 1A). In subsequent experiments, we used 1 mM BHB because this concentration gave optimal results and is close to blood BHB levels following KD consumption in our previous mouse studies10,25 and in others' human and mouse studies.8,26-28 1 mM BHB also suppressed other proinflammatory mediator genes such as those for IL-6 and iNOS (Figure 1B). Consistent with gene expression results, ELISA of the conditioned media from hiMG cultures confirmed the suppression of AβO-induced IL-1β and TNF-α release (Figure 1C). AβO also significantly downregulated anti-inflammatory marker genes CD206, IGF-1 and IL-10, but BHB did not rectify their expression (Figure 1D). In addition, AβO downregulated P2ry12 and Tmem119 genes that are specifically enriched in microglia (for example, relative to macrophages) and signatures for their homeostasis state.29 This result is consistent with the findings that in AD brains, there is a significant overall decrease of TMEM119-positive microglia, and, despite the increase of Iba1 positivity in microglia surrounding amyloid plaques, there is a loss of P2RY12 positivity.30 BHB co-treatment recovered P2ry12 and Tmem119 gene expression to the Mock levels (Figure 1E).
FIGURE 1.

BHB attenuates proinflammatory activation of hiMG by AβO. (A) Quantitative PCR shows that BHB dose-dependently suppresses AβO-induced expression of IL-1β and TNF-α. (B) Quantitative PCR shows that 1 mM BHB suppresses AβO-induced expression of IL-6 and Nos2. (C) ELISA of the conditioned media from hiMG cultures shows that 1 mM BHB reduces AβO-induced release of IL-1β and TNF-α. (D) Quantitative PCR shows that AβO suppresses the expression of the genes CD206, IGF-1, and IL10, the levels of which are not influenced by co-treatment with 1 mM BHB. (E) Quantitative PCR shows that AβO suppresses the expression of the genes for P2RY12 and TMEM119, the levels of which are recovered by co-treatment with 1 mM BHB. In all qPCR experiments, the values of mock (solvent only with no AβO and no BHB) group set as 1. *p < .05, **p < .01, ***p < .001; one-way ANOVA with Tukey's post hoc test.
3.2 |. BHB reduces AβO-induced IL-1β release through blockage of NLRP3 inflammasome activation
BHB is known to suppress NLRP3 inflammasome activation, which is consistent with the observed reduction of IL-1β release. However, in this regard, the published studies tend to use concentrations of BHB much higher than 1 mM concentration we used, for example, 10 mM,31,32 which are less physiologically feasible. In addition, the impact of BHB in amyloid-induced inflammasome activation in microglia, highly relevant to the pathogenesis of aging-related neurodegenerative disorders, has not been established. A report showed that BHB did not inhibit inflammasome activation induced by synuclein amyloid fibrils in primary mouse microglia, even at a high concentration of 10 mM.32 In our model of AβO-activated hiMG, as expected, AβO treatment enhanced NLRP3 inflammasome activity, evidenced by increased levels of NLRP3, pro-caspase-1, apoptosis-associated speck-like protein containing a CARD (ASC), and pro-IL-1β that are signals and components required for inflammasome formation, as well as increased levels of cleaved caspase-1 (p20) (Figure 2A) and IL-1β (Figure 1C) that are products of inflammasome activity. Co-treatment with BHB significantly reduced NLRP3 inflammasome activation, manifested by reduced levels of all the above proteins except ASC, which showed a trend of reduction by BHB that did not reach statistical significance (Figure 2A,B).
FIGURE 2.

BHB reduces AβO-induced IL-1β release through blockage of inflammasome activation. Representative Western blots probed with antibodies specific to the listed signals or component proteins of NLRP3 inflammasome (A) and bar graphs (B) summarizing band intensities, normalized to those of β-Actin, from 3 to 4 independent experiments. *p < .05, **p < .01, ***p < .001; one-way ANOVA with Tukey's post hoc test.
3.3 |. BHB may supplement the mitochondrial ketogenic pathway (MKP) deficits in AβO-stimulated hiMG
To comprehensively understand the molecular mechanisms of BHB effects, we conducted next-generation RNA-sequencing (RNA-seq)-based transcriptome analysis on three groups of samples: AβO, AβO + BHB, and Mock (vehicle control). Unsupervised analysis of the transcript count data using principal component analysis (PCA) showed clear distinctions in the variations of the transcriptomic expression between three groups (Figure 3A). Notably, the AβO + BHB samples were clustered intermediate between the AβO and Mock groups, suggesting amelioration of AβO-induced changes toward the control levels. In line with the PCA, differential expression analysis was performed by group-wise comparison. A heatmap plot of differentially expressed (DE) transcripts illustrate the distinct molecular signatures of the groups (Figure 3B). The results revealed 1371 DE transcripts between AβO vs Mock and 201 DE transcripts between AβO + BHB vs Mock (Figure 3C,D), suggesting that BHB treatment prevented a large portion of the DE genes induced by AβO stimulation. GO/KEGG-based functional enrichment analysis of the DE transcripts revealed multiple cellular and molecular pathways that were altered by AβO and were ameliorated by BHB treatment, including those known to be highly relevant to microglial functions (Figure S2).
FIGURE 3.

RNA-seq analysis shows amelioration of AβO-induced gene changes by co-treatment with 1 mM BHB. (A) Plot of principal component analysis (PCA) of gene expression, showing a clear separation between groups. (B) Heatmap plot showing gene expression Z-scores of differentially expressed (DE) genes between groups. (C) Volcano plots showing −log10 (p value) versus −log2 (fold change) of normalized counts between AβO and Mock and between AβO + BHB and Mock, respectively. Each dot represents a single transcript. Red dots denote significant DE genes (adjusted p < .05). (D) Venn diagrams showing up- and downregulated genes compared to Mock group. Blue circles represent the number of up- or downregulated genes in the AβO group compared to the Mock group, and red circles represent the number of up- or downregulated genes in the AβO + BHB group compared to the Mock group.
One viable therapeutic approach to reduce neuroinflammation is to induce a ketogenic state in microglia.33 However, how pathological conditions affect microglial MKP per se or ketogenic capacity is poorly understood. To address this question, we searched for altered genes under the GO term “Fatty Acid β-Oxidation” (Figure S3). Three genes, Hadh, Ivd, and Auh, coding for enzymes responsible for production of β-hydroxy β-methylglutaryl-CoA (HMG-CoA), a precursor of BHB, were found downregulated by AβO (p < .01) (Figure 4A), which were further confirmed by qPCR (Figure 4B). HADH (hydroxyacyl-CoA dehydrogenase, the translational product of Hadh) catalyzes the 3rd step of the β-oxidation of the fatty acids in the mitochondrial matrix. Although the transcript for 3-ketoacyl-CoA thiolase that catalyzes the 4th step of β-oxidation was not altered, AβO treatment upregulated the Shc proteins, including the mitochondrial isoform p46Shc,34 and their tyrosine phosphorylated forms (Figure 4A,C,D). p46Shc is known to bind thiolase with high affinity and function as a negative mitochondrial thiolase activity regulator. It was shown that increasing p46Shc expression inhibits enzymatic activity of thiolase.35 Thus, AβO stimulation may result in reduced β-oxidation of the fatty acids by suppression the 3rd and 4th enzymatic steps, and BHB treatment mitigated these alterations.
FIGURE 4.

AβO Induces microglial MKP deficits. (A) Key known enzymatic steps that lead to ketone body formation, with red letters highlighting molecules significantly regulated by AβO and BHB. The transcripts for three enzymes, IVD, AUH, and HADH, are identified to be downregulated by AβO. P46Shc, an adaptor protein that binds thiolase and negatively regulates its activity, is upregulated by AβO. (B) Quantitative PCR confirmed that IVD, AUH, and HADH genes are downregulated by AβO, which are at least partially recovered by BHB cotreatment. (C) Representative Western blot probed with antibodies specific to Tyr317 epitope of phosphor-Shc and total Shc. (D) Bar graph summarizing band intensities of p46 and p52 isoforms of Shc and their phosphorylated forms, normalized to those of β-Actin, from 3 independent experiments, as well as the ratios of the intensities of the phosphorylated form over the total protein. *p < .05, **p < .01, ***p < .001; one-way ANOVA with Student–Newman–Keuls test.
IVD (isovaleryl-CoA dehydrogenase, translational product of Ivd) and AUH (AU RNA binding methylglu-taconyl-CoA hydratase, translational product of Auh) catalyze the 3rd and 5th steps of the leucine metabolism pathway. Because leucine, an essential branched-chain amino acid, is metabolized to generate acetoacetate and BHB in glia36 and shift microglial immune responses toward anti-inflammatory states,37 this AβO-induced downregulation of leucine pathway enzymes may further aggravate the reduced MKP and the associated proinflammatory metabolic state. Again, BHB treatment mitigated these changes (Figure 4B, Figure S3). Taken together, our data suggest that AβO-stimulated hiMG assume a “ke-to-deficient” metabolic state due to MKP suppression, and BHB rectifies MKP deficits to promote a ketogenic state of microglia.
3.4 |. BHB mitigates AβO-induced mitochondrial changes in hiMG
The MKP deficits may be part of mitochondrial changes in AβO-stimulated hiMG. Indeed, our RNA-seq data showed that AβO repressed mitochondrial oxidation-related genes in all five complexes in the respiratory chain, the tricarboxylic acid cycle, and the antioxidation defense. BHB co-treatment by contrast tended to increase these mitochondrial transcripts, including those necessary for complete oxidation of lipids (Figures S3 and S4). We therefore investigated if the beneficial effects of BHB are related to enhanced mitochondrial integrity, using a combination of specific mitochondria-targeting fluorescent dyes and flow cytometry. First, we noticed that in AβO-stimulated hiMG, the gene coding for mitochondria calcium uniporter (MCU) was significantly upregulated (Figure 5A), which could disrupt mitochondrial Ca2+ homeostasis by allowing more Ca2+ entry into mitochondria matrix.38 The upregulation of MCU protein by AβO was confirmed by Western blotting (Figure 5B). BHB co-treatment significantly reduced levels of both MCU transcript (Figure 5A) and protein (Figure 5B,C). These alterations of MCU are consistent with prior findings that while glucose dose-dependently stimulates MCU transcription and activity, a general signal of cellular activation and inflammation,39-41 a reduction of MCU enhances β-oxidation.42
FIGURE 5.

BHB protects microglia from AβO-induced mitochondrial dysfunction. (A–C) Quantitative PCR (A) and Western blotting (B and C) show that BHB co-treatment rectifies AβO-induced upregulation of MCU gene expression and protein level. (D–H) Flow cytometry shows that BHB co-treatment ameliorates AβO-induced mitochondrial Ca2+ increase, mitochondrial oxidative stress, and activation of mPTP. (D) Representative flow cytometry plots of hiMG co-labeled with Rhod-2 AM for mitochondrial Ca2+ and the MitoTracker (tetramethylrhodamine methyl ester or TMRM). The bar graph summarizes the counts of cells in the right upper quadrants of the plots. (E) Representative traces and a bar graph summarizing fluorescence intensities of Rhod-2 AM from three independent experiments. (F) No change of mitochondria mass between the three groups. Shown are representative traces and a bar graph summarizing fluorescence intensities of MitoTracker from 5 to 6 independent experiments. (G) Shown are representative traces and a bar graph summarizing fluorescence intensities of MitoSOX from 3 independent experiments. (H) Shown are representative traces and a bar graph summarizing fluorescence intensities of Calcein AM from 5 to 6 independent experiments. *p < .05, **p < .01, ***p < .001; one-way ANOVA with Tukey's post hoc test.
Next, we used Rhod-2 AM to measure mitochondrial Ca2+ concentration as described43 and found a 40% increase in steady-state mitochondrial Ca2+ in AβO-stimulated hiMG compared to Mock-treated hiMG (Figure 5D,E). BHB co-treatment normalized the mitochondrial Ca2+ level (Figure 5D,E). The above changes were not due to alterations of mitochondrial mass as we found no changes of cellular mitoTracker (TMRM) fluorescence intensity in all conditions (Figure 5F).
Mitochondrial Ca2+ overload is associated with several mitochondrial alterations, notably increased oxidative stress.44 Using a mitochondrial superoxide indicator mitoSOX, we showed that AβO stimulation resulted in increased levels of mitochondrial reactive oxygen species (mtROS), and BHB co-treatment ameliorated this increase (Figure 5G). Mitochondrial Ca2+ overload and increased mtROS also could lead to enhanced activation or opening of the mitochondrial permeability transition pore (mPTP),45 resulting in a loss of mitochondrial membrane potential. Indeed, AβO stimulation resulted in activation of mPTP evidenced by enhanced cellular fluorescence intensities from mitochondrial calcein release.46 BHB co-treatment completely reversed this mPTP activation (Figure 5H). Taken together, in human microglia, AβO induced substantial mitochondrial abnormalities that could be rectified by BHB.
3.5 |. BHB corrects AβO-induced deficits in phagocytosis
Phagocytosis is the signature function of microglia to remove unwanted or offending material from the brain microenvironment. Mitochondrial metabolism appears to fuel phagocytosis in microglia, an energy-consuming process. Proinflammatory metabolic shift toward increased glycolysis is associated with compromised phagocytosis and impaired Aβ engulfment.47 In fact, a survival strategy for offending microorganisms in macrophages is to upregulate mitochondrial Ca2+ through the MCU, resulting in suppression of phagocytosis.48 Because AβO stimulation enhanced glycolysis (Figure S3), upregulated MCU, and caused mitochondrial Ca2+ overload (Figure 5A-E), we tested how it influences phagocytosis. We found that AβO impaired the ability of hiMG to phagocytose fluorescent BioParticles.49 Co-staining with the microglial marker Iba-1 showed numerous green fluorescent BioParticles within Mock-treated, Iba-1-positive cells, but not so in AβO-stimulated cells (Figure 6A). Co-treatment with BHB rectified this deficit, as quantified by an automated scanner (Figure 6B). We also tested the ability of hiMG to phagocytose FITC-labeled Aβ monomer and showed a similar result (Figure 6C,D), suggesting that BHB can recover the ability of hiMG to engulf and clear Aβ, a function highly relevant to AD pathogenesis.
FIGURE 6.
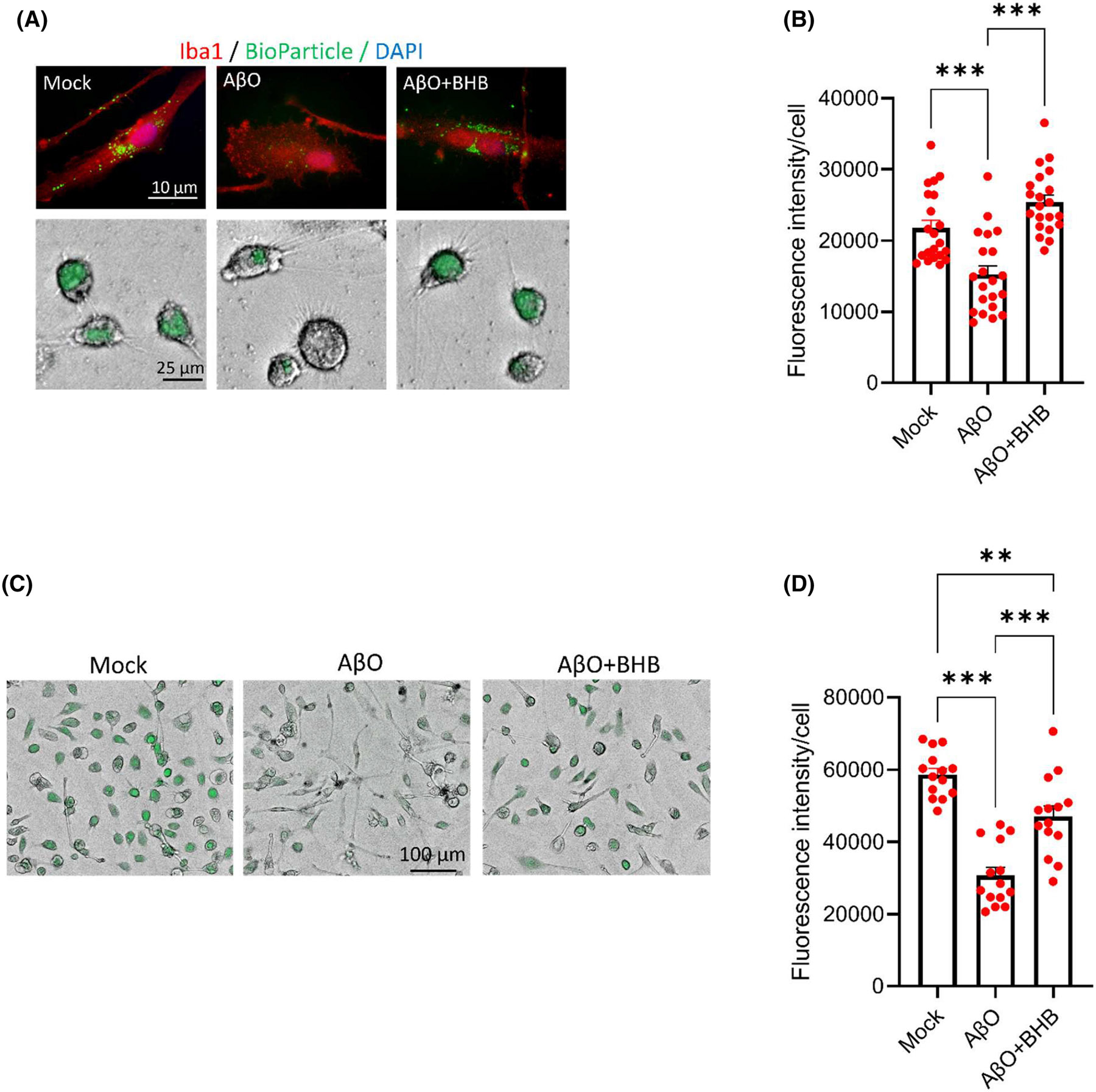
AβO impairs hiMG phagocytotic capacity, which is rescued by BHB co-treatment. (A and B) Phagocytosis of pHrodo Green BioParticles visualized by fluorescence microscopy. Green: BioParticles; red: Iba1. Shown are representative fluorescence images (upper panels) and combined fluorescence/brightfield images (lower panels) (A) and a bar graph (B) summarizing the fluorescence intensities of cells in 7 randomly selected fields (~100 cells per field) from three independent experiments. (C and D) Phagocytosis of FITC-labeled Aβ peptide. Shown are representative combined fluorescence/brightfield images (C) and a bar graph (D) summarizing the fluorescence intensities of cells in 5 randomly selected fields (~40 cells per field) from three independent experiments. **p < .01, ***p < .001; one-way ANOVA with Tukey's post hoc test.
4 |. DISCUSSION
BHB has been shown to modulate systemic and brain inflammation. However, direct effects of BHB on microglia have been little studied. To our knowledge, our study is the first to use human iPS-derived microglia to investigate the impact of BHB. This approach is important for translation as currently available few studies on BHB and microglia used microglial cell lines. Microglia cell lines would require cancer cell-like bioenergetics to sustain perpetual growth, appear to require much higher concentrations of BHB for efficacy, and may yield results divergent from differentiated human microglia used here. For example, BHB was shown to upregulate the expression of NOS2, a key proinflammatory gene in BV2 microglial cells,50 while our data showed BHB downregulates NOS2 (Figure 1B) and proinflammatory responses in general. Notably, we found significant effects of BHB at concentrations between 0.1 and 2 mM, a range equivalent to physiological blood levels when beneficial effects occur in human studies. In humans, serum levels of BHB rise from the basal levels of low μM to a few hundred μM after 12–16 h of fasting and 1–2 mM after two days of fasting. Serum levels of 1–2 mM BHB can also be reached after 90 min of intense exercise.8 As BHB readily enters the brain,51 we consider that the 1 mM BHB concentration we used in most of our experiments should approximate the actual brain BHB concentrations microglia are exposed to following ketogenic dietary regimens or exercise. This is in contrast to several prior studies, including those on mouse primary microglia,32 using higher concentrations up to 10 mM that are usually seen following prolonged starvation8,52 and likely not applicable to common physiological or pathological conditions.
Mitochondrial metabolic pathways in microglia dictate their functional or dysfunctional states.4,47,53 Their pivotal significance in early AD development was revealed by a recent large-scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid.14 RNA-seq showed AβO-induced overall increases in transcripts related to glycolysis, including those for glucose transporters (Figure S2), and decreases in those related to respiration (Figure S3), consistent with a proinflammatory state. Perhaps because AβO drives increased reliance of microglia on carbohydrates, multiple transcripts of mitochondrial lipid β-oxidation are significantly reduced by AβO, but by supplying BHB, which can only be oxidized in mitochondria, this functionality is rescued. Following AβO stimulation, transcriptomic data, confirmed by qPCR, showed that Hadh, Ivd, and Auh genes involved in the β-oxidation and leucin metabolism pathways were significantly downregulated, and Western blot showed that p46Shc, an inhibitor of thiolase in the MKP, was upregulated. The expected consequence is MKP suppression, hampering the ability of microglia to assume a ketogenic metabolic state.33 The observation that BHB stimulated the MKP and rescued microglial functions indicate that MKP may play a significant role in microglia immunometabolism and herein lies its pathological role from its suppression. This notion is consistent with the finding that BHB levels are lower in brain parenchyma of AD patients when compared with non-AD controls.54 Restricted metabolic flexibility from MKP suppression and low BHB production could result in diminished BHB signaling8 and contribute to microglia dysfunction in AD. This novel mechanism warrants further investigation.
A major contributing factor to the MKP deficiency state is Shc expression, which has been linked to AD.55,56 Shc is an adapter protein identified as a SH2 containing proto-oncogene existing in many isoforms and involved in transducing an extracellular signal into an intracellular signal. Our data showed that human microglia express p52Shc and p46Shc, two major Shc isoforms derived from the same gene. AβO stimulation not only increased the total protein levels of p52Shc and p46Shc but also their degrees of tyrosine phosphorylation (Figure 4C,D), which promotes their translocation to membrane where signaling transduction occurs.57 Among all Shc isoforms, p46Shc is localized to mitochondria, where it binds and inhibits thiolase.34,35,58 As such, our data suggests that in human microglia AβO-induced p46Shc (including its tyrosine-phosphorylated form) negatively regulates the MPK. This notion is also consistent with the in vivo finding that low Shc levels in mice resulted in increased liver and muscle β-oxidation enzyme activities (including those of HADH and thiolase in the MKP) in response to fasting.58 Interestingly, mice with Shc reduction showed improved mitochondrial function and resisted AD-like cognitive deficits in APP mice.55 Moreover, Shc was identified as one of the four hub genes for the conversion of mild cognitive impairment (a precursor syndrome to AD) to AD dementia.56 Taken together, p46Shc over-expression in microglia could make significant contributions to AD pathogenesis by impairing the MKP. BHB, in addition to supplement the MKP deficits, appears to initiate signals that lead to downregulation of both p46Shc and p52Shc (Figure 4C,D).
Deficits of the mitochondrial MKP are only one of the multifaceted mitochondrial abnormalities induced by AβO. These abnormalities appear centered on disrupted mitochondrial Ca2+ homeostasis, evidenced by the upregulation of MCU and a 40% increase in steady-state mitochondrial Ca2+ (Figure 5D,E), resulting in increased mtROS (Figure 5G) and activation of mPTP (Figure 5H). Notably, AβO was shown to upregulate MCU expression measured by immunofluorescence in “aged” cultured hippocampal neurons.59 Our data showed that in microglia AβO may also affect mitochondrial Ca2+ homeostasis via increasing MCU, and this involves at least transcriptional mechanisms. Mitochondrial dysfunction can be “sensed” by NLRP3 inflammasome60 to induce proinflammatory responses characterized by IL-1β production. Mitochondrial dysfunction can further disrupt the mechanism fueling phagocytosis,14,61 thereby impairing the clearance of toxic Aβ species as we showed here (Figure 6). Significantly, BHB was able to rectify these interwoven abnormalities. One may speculate that such a wide impact may not simply be explained by its passive role as an alternative fuel source. Rather, BHB has been increasingly recognized as an active signaling molecule with a wide range of direct actions and indirect actions via its metabolites.8 The known direct actions include its activation of cell surface receptors HCAR2 (hydroxycarboxylic acid receptor 2) and FFAR3 (free fatty acid receptor 3), inhibition of histone deacetylases (HDACs) to affect epigenetics, modulation of potassium channel function, and direct β-hydroxybutyrylation of proteins as a form of post-translational modification.8 Inhibition of potassium efflux through potassium channels may explain BHB's effects on the NLRP3 inflammasome.31 Notably, in the brain, HCAR2 is expressed selectively by microglia, and niacin, a high-affinity ligand for HCAR2, was shown to modulate microglial response and limits disease progression in a mouse model of AD.62 It is likely that BHB, also a ligand for HCAR2, induces a mechanism parallel to that of niacin. Our results warrant further investigations to parse out signaling pathways required for the pleotropic BHB effects in microglia.
In summary, using human microglia, our data show that BHB at physiological concentrations is highly potent in rectifying several AD-related microglial dysfunctions including impaired phagocytosis of Aβ. These effects of BHB may contribute to the beneficial effects of ketogenic diet intervention in aged mice10,25 as well as in human subjects with mild AD63 and supports it as amyloid-regulating treatment by enhancing microglial clearance of Aβ.64 Our data also suggests that the MKP could be a significant immunometabolic mechanism regulating microglia function. With the increasing realization of BHB being an endogenous signaling metabolite, BHB intervention may be considered an immunometabolic countermeasure not only balancing cellular energy but also actively enacting “drug-like” signaling activities.65
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institute of Health awards P01 AG025532 and in part by RF1 AG071665, P30 AG072972, and the MIND Institute IDDRC (P50 HD103526).
Funding information
HHS | NIH | Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), Grant/Award Number: P50 HD103526; HHS | NIH | National Institute of Neurological Disorders and Stroke (NINDS), Grant/Award Number: P01 AG025532; HHS | NIH | National Institute on Aging (NIA), Grant/Award Number: RF1 AG071665
Abbreviations:
- AD
Alzheimer's disease
- AβO
Aβ oligomer
- BHB
β-hydroxybutyrate
- hiMG
human iPSC-derived microglia
- iPSC
induced pluripotent stem cell
- KD
ketogenic diet
- LPS
lipopolysaccharide
- MCU
mitochondrial calcium uniporter
- MKP
mitochondrial ketogenic pathway
- mPTP
mitochondrial permeability transition pore
- mtROS
mitochondrial rective oxigen species
Footnotes
DISCLOSURES
All authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the methods and/or supplementary material of this article and are also available from the corresponding author upon reasonable request. The RNA sequencing data will be submitted to the NCBI Data or other depositories.
REFERENCES
- 1.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. [DOI] [PubMed] [Google Scholar]
- 2.Bernier LP, York EM, Kamyabi A, Choi HB, Weilinger NL, MacVicar BA. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun. 2020;11:1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110:3458–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernier LP, York EM, MacVicar BA. Immunometabolism in the brain: how metabolism shapes microglial function. Trends Neurosci. 2020;43:854–869. [DOI] [PubMed] [Google Scholar]
- 5.Sanjay Park M, Lee HJ. Roles of fatty acids in microglial polarization: evidence from in vitro and in vivo studies on neurodegenerative diseases. Int J Mol Sci. 2022;23:7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. 2020;52:1496–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin LW, Horiuchi M, Wulff H, et al. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J Neurosci. 2015;35:2516–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newman JC, Verdin E. β-Hydroxybutyrate: a signaling metabolite. Annu Rev Nutr. 2017;37:51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab. 2014;19:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roberts MN, Wallace MA, Tomilov AA, et al. A ketogenic diet extends longevity and Healthspan in adult mice. Cell Metab. 2017;26:539–546.e535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marschallinger J, Iram T, Zardeneta M, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23:194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson ECB, Dammer EB, Duong DM, et al. Large-scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellenguez C, Kucukali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. 2011;286:3693–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maezawa I, Nguyen HM, Di Lucente J, et al. Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer's disease: preclinical proof of concept. Brain. 2017;141:596–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–172. [DOI] [PubMed] [Google Scholar]
- 19.Penney J, Ralvenius WT, Tsai LH. Modeling Alzheimer's disease with iPSC-derived brain cells. Mol Psychiatry. 2019;25:148–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith AM, Dragunow M. The human side of microglia. Trends Neurosci. 2014;37:125–135. [DOI] [PubMed] [Google Scholar]
- 21.Jin LW, di Lucente J, Ruiz Mendiola U, et al. The role of FUT8- catalyzed core fucosylation in Alzheimer's amyloid-β oligomer-induced activation of human microglia. Glia. 2023;71:1346–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haenseler W, Sansom SN, Buchrieser J, et al. A highly efficient human pluripotent stem cell microglia model displays a neuronal-Co-culture-specific expression profile and inflammatory response. Stem Cell Reports. 2017;8:1727–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marosi K, Kim SW, Moehl K, et al. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J Neurochem. 2016;139:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pathak SJ, Zhou Z, Steffen D, et al. 2-month ketogenic diet preferentially alters skeletal muscle and augments cognitive function in middle aged female mice. Aging Cell. 2022;21:e13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crabtree CD, Kackley ML, Buga A, et al. Comparison of keto-genic diets with and without ketone salts versus a low-fat diet: liver fat responses in overweight adults. Nutrients. 2021;13:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kackley ML, Buga A, Crabtree CD, et al. Influence of nutritional ketosis achieved through various methods on plasma concentrations of brain derived neurotropic factor. Brain Sci. 2022;12:1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newman JC, Covarrubias AJ, Zhao M, et al. Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 2017;26:547–557.e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boche D, Gordon MN. Diversity of transcriptomic microglial phenotypes in aging and Alzheimer's disease. Alzheimers Dement. 2022;18:360–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kenkhuis B, Somarakis A, Kleindouwel LRT, van Roon-Mom WMC, Höllt T, van der Weerd L. Co-expression patterns of microglia markers Iba1, TMEM119 and P2RY12 in Alzheimer's disease. Neurobiol Dis. 2022;167:105684. [DOI] [PubMed] [Google Scholar]
- 31.Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deora V, Albornoz EA, Zhu K, Woodruff TM, Gordon R. The ketone body β-Hydroxybutyrate does not inhibit Synuclein mediated Inflammasome activation in microglia. J Neuroimmune Pharmacol. 2017;12:568–574. [DOI] [PubMed] [Google Scholar]
- 33.Lauro C, Limatola C. Metabolic reprograming of microglia in the regulation of the innate inflammatory response. Front Immunol. 2020;11:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ventura A, Maccarana M, Raker VA, Pelicci PG. A cryptic targeting signal induces isoform-specific localization of p46Shc to mitochondria. J Biol Chem. 2004;279:2299–2306. [DOI] [PubMed] [Google Scholar]
- 35.Tomilov A, Tomilova N, Shan Y, et al. p46Shc inhibits Thiolase and lipid oxidation in mitochondria. J Biol Chem. 2016;291:12575–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bixel MG, Hamprecht B. Generation of ketone bodies from leucine by cultured astroglial cells. J Neurochem. 1995;65:2450–2461. [DOI] [PubMed] [Google Scholar]
- 37.De Simone R, Vissicchio F, Mingarelli C, et al. Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim Biophys Acta. 2013;1832:650–659. [DOI] [PubMed] [Google Scholar]
- 38.Fan M, Zhang J, Tsai CW, et al. Structure and mechanism of the mitochondrial Ca(2+) uniporter holocomplex. Nature. 2020;582:129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casaril AM, Katsalifis A, Schmidt RM, Bas-Orth C. Activated glia cells cause bioenergetic impairment of neurons that can be rescued by knock-down of the mitochondrial calcium uniporter. Biochem Biophys Res Commun. 2022;608:45–51. [DOI] [PubMed] [Google Scholar]
- 40.Tarasov AI, Semplici F, Ravier MA, et al. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PloS One. 2012;7:e39722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W, Yang J, Chen S, et al. Importance of mitochondrial calcium uniporter in high glucose-induced endothelial cell dysfunction. Diab Vasc Dis Res. 2017;14:494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altamimi TR, Karwi QG, Uddin GM, et al. Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. J Mol Cell Cardiol. 2019;127:223–231. [DOI] [PubMed] [Google Scholar]
- 43.Cheng A, Yang Y, Zhou Y, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016;23:128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–188. [DOI] [PubMed] [Google Scholar]
- 45.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341(Pt 2):233–249. [PMC free article] [PubMed] [Google Scholar]
- 46.Petronilli V, Miotto G, Canton M, et al. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fairley LH, Wong JH, Barron AM. Mitochondrial regulation of microglial Immunometabolism in Alzheimer's disease. Front Immunol. 2021;12:624538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li T, Kong L, Li X, et al. Listeria monocytogenes upregulates mitochondrial calcium signalling to inhibit LC3-associated phagocytosis as a survival strategy. Nat Microbiol. 2021;6:366–379. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Kapellos TS, Taylor L, Lee H, et al. A novel real time imaging platform to quantify macrophage phagocytosis. Biochem Pharmacol. 2016;116:107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benito A, Hajji N, O'Neill K, Keun HC, Syed N. β-Hydroxybutyrate oxidation promotes the accumulation of Immunometabolites in activated microglia cells. Metabolites. 2020;10:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clarke K, Tchabanenko K, Pawlosky R, et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. 2012;63:401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cahill GF Jr. Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh S, Castillo E, Frias ES, Swanson RA. Bioenergetic regulation of microglia. Glia. 2018;66:1200–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shippy DC, Wilhelm C, Viharkumar PA, Raife TJ, Ulland TK. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer's disease pathology. J Neuroinflammation. 2020;17:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Derungs R, Camici GG, Spescha RD, et al. Genetic ablation of the p66(Shc) adaptor protein reverses cognitive deficits and improves mitochondrial function in an APP transgenic mouse model of Alzheimer's disease. Mol Psychiatry. 2017;22:605–614. [DOI] [PubMed] [Google Scholar]
- 56.Shigemizu D, Akiyama S, Higaki S, et al. Prognosis prediction model for conversion from mild cognitive impairment to Alzheimer's disease created by integrative analysis of multi-omics data. Alzheimers Res Ther. 2020;12:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sato K, Kimoto M, Kakumoto M, et al. Adaptor protein Shc undergoes translocation and mediates up-regulation of the tyrosine kinase c-Src in EGF-stimulated A431 cells. Genes Cells. 2000;5:749–764. [DOI] [PubMed] [Google Scholar]
- 58.Hagopian K, Tomilov AA, Tomilova N, et al. Shc proteins influence the activities of enzymes involved in fatty acid oxidation and ketogenesis. Metabolism. 2012;61:1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Calvo-Rodriguez M, Hernando-Perez E, Nuñez L, Villalobos C. Amyloid β oligomers increase ER-mitochondria Ca(2+) cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca(2+) remodeling. Front Cell Neurosci. 2019;13:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. [DOI] [PubMed] [Google Scholar]
- 61.Fairley LH, Lai KO, Wong JH, et al. Mitochondrial control of microglial phagocytosis by the translocator protein and hexokinase 2 in Alzheimer's disease. Proc Natl Acad Sci U S A. 2023;120:e2209177120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moutinho M, Puntambekar SS, Tsai AP, et al. The niacin receptor HCAR2 modulates microglial response and limits disease progression in a mouse model of Alzheimer's disease. Sci Transl Med. 2022;14:eabl7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor MK, Sullivan DK, Mahnken JD, Burns JM, Swerdlow RH. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer's disease. Alzheimers Dement (N Y). 2018;4:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor MK, Sullivan DK, Keller JE, Burns JM, Swerdlow RH. Potential for Ketotherapies as amyloid-regulating treatment in individuals at risk for Alzheimer's disease. Front Neurosci. 2022;16:899612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stubbs BJ, Koutnik AP, Goldberg EL, et al. Investigating ketone bodies as immunometabolic countermeasures against respiratory viral infections. Med (N Y). 2020;1:43–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available in the methods and/or supplementary material of this article and are also available from the corresponding author upon reasonable request. The RNA sequencing data will be submitted to the NCBI Data or other depositories.