

Abstract
Background:
Alzheimer’s disease (AD) is a complex neurodegenerative disease marked by increased amyloid-β (Aβ) deposition, tau hyperphosphorylation, impaired energy metabolism, and chronic ischemia-type injury. Cerebral microvascular dysfunction likely contributes to AD pathology, but its precise pathogenic role has been poorly defined.
Objective:
To examine microvascular reactivity to endothelium-dependent vasodilators and small conductance calcium-activated potassium (SK) channel activity in an intracerebral streptozotocin (STZ)-induced AD mouse model.
Methods:
Control and STZ-AD mice underwent Morris Water Maze and Barnes testing, after which cerebral microvascular and brain microvascular endothelial cells (MBMECs) were dissected to assess microvascular reactivity, responses to SK channel activator NS309, and ion-channel current recordings using whole-cell patch clamp methodology. Control mouse cerebral microvascular and human brain microvascular endothelial cells (HBMECs) were treated with soluble Aβ1–42 peptide to characterize microvascular reactivity and endothelial potassium currents.
Results:
STZ-AD mice exhibited impaired performance vs control mice in behavioral testing. STZ-AD mice also exhibited diminished cerebral microvascular responsiveness and MBMECs potassium current augmentation in response to NS309 compared with control mice. Incubation of control mouse cerebral micro-vessels and HBMECs with soluble Aβ (1 μM) for 2 h attenuated relaxation responses to NS309 and diminished NS309-sensitive endothelial potassium currents.
Conclusions:
STZ-AD mice exhibited impaired microvascular relaxation responses to endothelium-dependent vasodilators; SK/IK channel dysfunction may be involved in the mechanism of this impairment. Acute treatment with Aβ produced dysregulated cerebrovascular endothelial SK/IK channels. Further elucidation of the role of microvascular dysfunction in AD is needed to prevent the chronic ischemia-type injury that contributes to cognitive decline.
Keywords: Alzheimer’s disease, amyloid-β, cerebral endothelial dysfunction, cerebral microvascular dysfunction, Morris water maze, NS309, SK channels, streptozotocin
Introduction
In the United States it is estimated that 6.5 million people aged 65 and older live with Alzheimer’s disease (AD).1 In 2020, the estimated total healthcare costs for AD treatment (including skilled nursing care, home healthcare, medications, hospice) exceeded $305 billion. This number is also projected to increase to over $1 trillion in the near future.2
Clinical indications of AD onset range across a variety of cognitive domains including progressive impairment in higher-order executive functioning, learning and memory, visuospatial learning, and language.3 At the tissue level, hallmarks include extracellular amyloid-β (Aβ) peptide aggregates in plaques and in vessel walls, accumulations of hyperphosphorylated tau in neurofibrillary tangles, and dystrophic neurites and neuropil threads most abundantly manifesting in hippocampus, amygdala, and temporal cortex.4–6
Evidence suggests that cerebral microcirculatory impairment may accompany or precede neuronal loss in AD.7–11 Interventions targeting this dysfunction may help prevent AD pathology. The mechanism of Aβ-associated vasculopathy has been studied extensively and appears to be associated with decreased vascular endothelial integrity and impaired regulation of cerebral microvascular vasomotor tone.12–15 In one study, a human-amyloid precursor protein transgenic mouse model of AD showed increased endothelial sprouting in cerebral vessels.16 In another study, transgenic mouse models with high Aβ levels were shown to have elevated cortical microvascular density around 5 months that, by 27 months, significantly declined.17
To better understand the pathophysiology of AD with respect to Aβ and metabolic dysfunction, a disease model initiated by intracerebral injection of streptozotocin (STZ) was generated.18 STZ, originally identified as an antibiotic, is a glucosamine-nitrosourea compound toxic to insulin-producing beta cells within pancreatic islets and has historically been used for the study of type 1 diabetes mellitus (DM). Intracerebroventricular administration of STZ, however, has been shown to precipitate AD-like pathology, including cerebral Aβ plaques and vasculopathy, tau hyperphosphorylation, and spatial learning defects.19–21 Given its several biochemical and molecular parallels with DM and other disorders involving insulin resistance, AD has alternatively been classified as “type 3 diabetes”.18,22 The molecular basis of this connection involves the promotion by cerebral insulin resistance of brain oligodendrocyte and microvascular endothelial cell dysfunction, which subsequently facilitates the white matter degeneration seen in AD.8
In this study, we used a murine model of intracerebral STZ administration to investigate microvascular dysfunction in the setting of AD relative to vehicle-treated controls. We characterized the tissue effects of our model using immunohistochemical probing of amyloid and tau protein in cortical and hippocampal sections. Clinically, we employed the Morris water maze to evaluate spatial reference learning and memory. Finally, to delineate and quantify AD-associated vasculopathy, we examined cerebral microvascular vasodilatory responses along with brain endothelial cell responses to the selective SK/IK channel activator NS309, as well as the impact of Aβ peptide incubation on cerebral micro-vessels and cerebral microvascular endothelial cells.
Methods
Animals
C57BL/6J mice (6–8 months old, males and females) were used in this study (Jackson Laboratory, Bar Harbor, ME). The housing room was set to a 12 h light/dark cycle with lights off at 8 P.M., a temperature of 22°C, and a relative air humidity of 50%. Standard chow diet and water were given ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee of Rhode Island Hospital. A detailed description of the experimental design is depicted in Figure 1(A).
Figure 1.
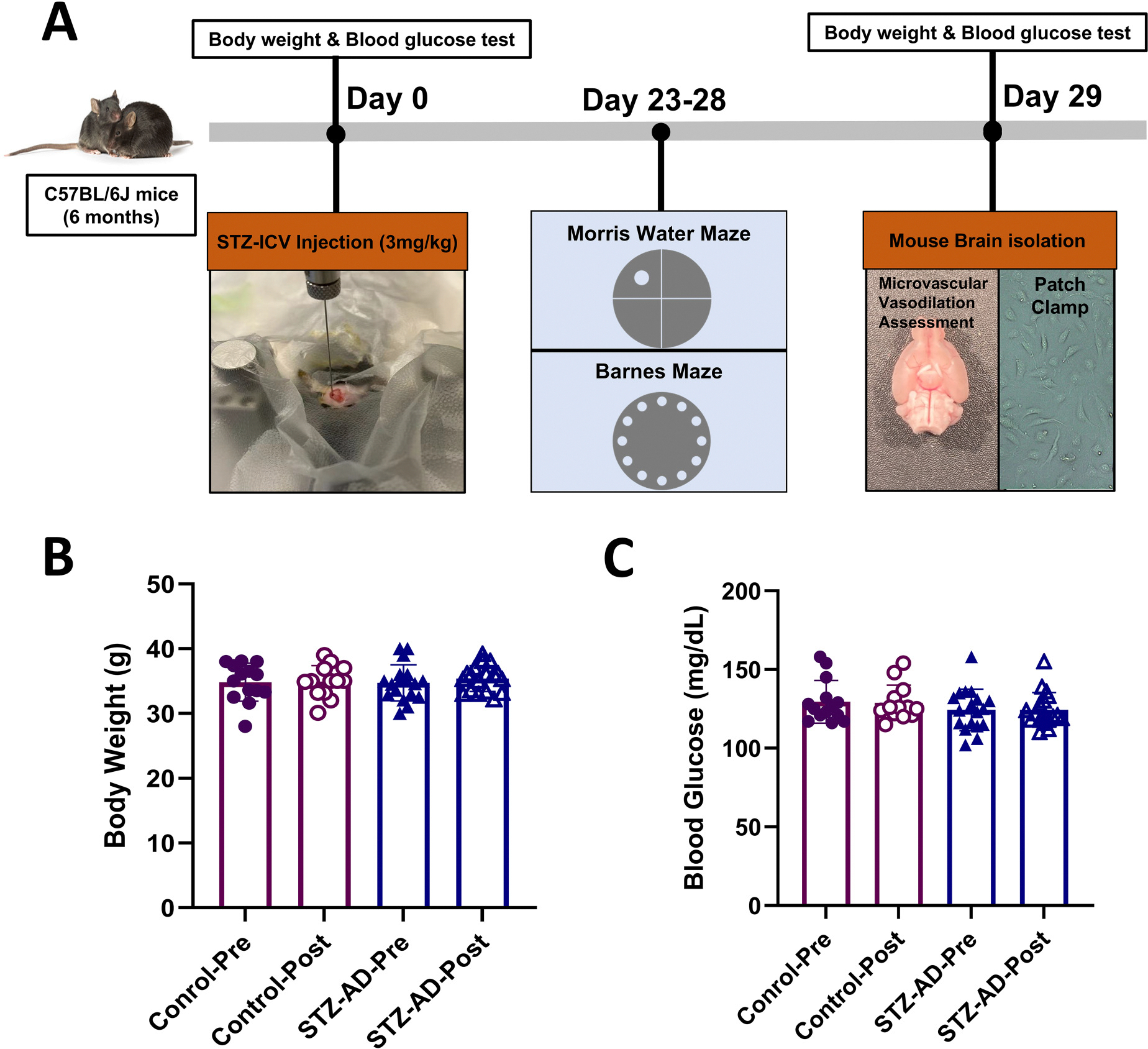
(A) Illustration of overall experimental design. STZ-AD mice received a single intracerebroventricular (ICV) injection of 3.0 mg/kg STZ in 3.0 μl cold citrate buffer (pH 4.5) into the right ventricle of the brain. Control mice also received ICV injection of 3.0 μl cold citrate buffer (pH 4.5). Twenty-three days after ICV injection, the mice were subjected to Morris water maze testing over the course of 5 days. Immediately after this, on day 29, all mice were anesthetized by inhaled isoflurane, after which a decapitation was performed, and brains were removed from the cranium. Brains were either placed in cold artificial cerebrospinal fluid (ACSF) buffer for in vitro microvascular experiments, or preserved in cell culture medium in preparation for endothelial cell isolation. (B) The mean body weights of control and STZ-AD mice were not significantly different before and after ICV-injection. (C) Blood glucose levels among control and STZ-AD mice were not significantly different before and after ICV-injection. Data are shown as mean ± SD.
STZ-AD mouse model
STZ-AD mice were produced by stereotactic injection of STZ (Sigma-Aldrich, St Louis, MO, USA) into the right lateral ventricle. Briefly, mice were anesthetized using 2.5% isoflurane (Sigma-Aldrich) and then secured onto a stereotactic apparatus. The bregma coordinates used for injection were 1.0 mm lateral, −0.3 mm posterior, and 2.25 mm inferior. Experimental mice received a single right intracerebroventricular (ICV) injection of 3.0 mg/kg STZ in 3.0 μl cold citrate buffer (pH 4.5), while control mice received an ICV injection of 3.0 μl cold citrate buffer (pH 4.5) alone. Twenty-three days after the ICV injection, all mice were subjected to Morris water maze testing over a 6-day period. On day 29 after the ICV injections, all mice were anesthetized by inhaled isoflurane and then decapitated. Following their immediate removal, brains were placed either in cold artificial cerebrospinal fluid (ACSF) buffer in preparation for in vitro microvascular experiments or preserved in cell culture medium in preparation for endothelial cell isolation. Spatial reference learning and memory were evaluated in Water maze and Barnes maze testing protocols.
Morris water maze
The test was performed in a white pool of 180 cm in diameter filled with water tinted with non-toxic white paint and maintained at room temperature (21 ± 2°C). During day 1 (visible platform), a platform (14 cm in diameter) was 1 cm above water surface, and during days 2–5 (hidden platform), the platform was submerged 1 cm below water surface. The starting position was randomized among four quadrants of the pool. For each trial, the animal was given 90 s to locate the hidden platform. If a mouse found the platform within 90 s, the latency time would be recorded. If a mouse failed to find the platform within 90 s, it was gently guided to it and left the mouse on the platform for 20 s. The latency to reach the platform (s), swim distance (cm), and swim speed (cm/s) were recorded. During day 6 (probe trail without platform), the platform was removed from the tank, the animal allowed to swim for 90 s, the time in target quadrant (%), swim speed (cm/s) and times of crossing the platform zone were recorded. At the end of each trial, the mice were dried and returned to the home cage until the next trial. The movement trajectory of the mouse was tracked by ANY-maze software (Stoelting Europe).
Barnes maze
The Barnes maze test was conducted using a grey circular platform (91 cm in diameter, 90 cm high) with 20 equally spaced holes (5 cm in diameter) around its circumference. An escape box was fitted under one of the holes, serving as the target location for the mice. The test was performed in a standard lit room with visual cues placed around the periphery to aid spatial navigation. The training trials were conducted over three consecutive days (Day 1 to Day 3). Each mouse was placed on the maze for a 2-min exploration session. If the mouse did not find the escape box within this period, it was gently guided to the box and allowed to stay there for 1 min. Each mouse received two training trials per day, with an intertrial interval of approximately 15 min. During these trials, the amount of time spent to locate the escape box (latency) were recorded. On Day 5, 48 h after the last training session, two probe trials were conducted to assess memory retention. During these trials, the escape box was removed from the platform, and the mice were allowed to explore the maze for 2 min each time. The time spent in the target quadrant, where the escape box had previously been located, and the distance was recorded. All sessions, including training and probe trials, were video recorded, and the data were analyzed using ANY Maze software (Stoelting, CO).
Micro-vessel dissection and assessment of vasodilation
Cerebral parenchymal arterioles (<70 μm diameter) were dissected from the isolated mouse brain as previously described.23 The ACSF solution consisted of 119 mM NaCl, 26.2 mM NaHCO3, 2.5 mM KCl, 1 mM NaH2PO4, mM 1.3 MgCl2, 10 mM glucose, and mM 1.8 CaCl2; micro-vessels were stabilized in this solution for 15–20 min at 37°C. Each micro-vessel was then pre-constricted with the thromboxane A2 analog U46619 (10−6 M). After stable constriction was achieved, microvascular relaxation was measured after exposure to vasodilators: first, the SK/IK channel activator NS309 (10−9–10−5 M), and then the endothelium-independent vasodilator sodium nitroprusside (SNP, 10−9–10−4 M). The order of drugs was random, with 1–2 interventions performed on each vessel.
Brain microvascular endothelial cells
Mouse brain microvascular endothelial cells (MBMECs) were isolated from harvested brains and then cultured and grown in a customized medium.24,25 Human brain microvascular endothelial cells (HBMECs) were harvested from healthy donors (Cell Systems Corporation, Kirkland, WA) and cultured and grown in EGM-2-MV medium (Lonza, Basel, Switzerland). Both MBMECs and HBMECs were incubated in a humidified incubator with 5% CO2 at 37° C. MBMECs were used as control cells, with STZ-AD mouse-derived cells comprising the experimental branch in potassium current recordings assessed through patch clamp studies. HBMECs were pre-treated with soluble Aβ peptides (1–42), and subsequently also probed using patch clamping to assess potassium currents.
Electrophysiologic study of endothelial cell K+ currents
Endothelial cells were washed in 0.05% trypsin and 0.02% EDTA for 1–2 min before patch clamp recordings. An Axopatch 200B amplifier and pClamp 10.6 (Molecular Devices, Foster City, CA) were used to record and analyze the potassium currents of MBMECs in whole-cell configuration in the voltage-clamp mode. The bath solution contained 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, and 30 mM glucose (pH 7.4), set to 36°C for MBMECs and to room temperature for HBMECs. Patch pipette resistance was 1–3 MΩ, and the pipette solution contained 110 mM K-aspartate, 20 mM KCl, 1 mM MgCl2, 8.5 mM CaCl2, 10 mM HEPES, 8 mM NaCl, 0.01 mM niflumic acid, and 10 mM BAPTA (pH 7.2, with calculated free Ca2+ 400 nM). The cells were examined every 5 s at the holding potential of −50 mV by 150 ms test pulses between −100 and +100 mV in 20-mV increments. The sampling rate was 10 kHz, with low pass filtering set at 2 kHz. The effect of administering NS309 (10−6 M) on the whole-cell K+ currents was examined using this configuration.
Congo red staining
Frozen brain tissue was obtained from mice and embedded in OCT. Ten-micrometer-thick coronal sections were for stained using Congo red staining: sections were incubated with hematoxylin for 5 min, followed by alkaline alcohol (Bluing Reagent) for 30 s, Congo red for 20 min, and finally mounted with synthetic resin. The total number of Congo red-stained plaques in hippocampal and cortical sections was recorded for statistical analysis.
Western blotting
Brain tissue homogenates from cortical and hippocampal tissue were harvested from STZ-AD and control mice in RIPA buffer. Total protein (10 μg) was fractionated by 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane using a wet transfer apparatus. Membranes were stained with Ponceau S and probed with antibodies to phospho-tau (Cell Signaling, Danvers, MA) that targeted Ser 396 (1:1000), Ser 202/Thr 205 (1:1000), and Thr 181 (1:1000), as well as antibodies to SK3 (1:200) and SK4 (1:200) (Alomone Labs, Jerusalem, Israel). Anti-GAPDH (Cell Signaling, Danvers, MA) was used as a sample loading control. Immunoreactivity was detected with species-appropriate secondary antibodies conjugated to horseradish peroxidase, followed by chemiluminescent enhancement reagents (Thermo Scientific, Rockford, IL). Chemiluminescent images corresponding to immunoreactivity were quantified with FIJI software.
ELISA
Brain tissue homogenates were utilized for Aβ levels in both cortex and hippocampus of control and STZ-AD mice. Soluble Aβ concentration was assessed per manufacturer instruction (Thermo Fisher Scientific, Waltham, MA).
Chemicals
Aβ peptides (1–42), NS309, and SNP were purchased from Sigma-Aldrich (St Louis, Mo).
Data analysis
Data graphing and analysis were performed using GraphPad Prism 9 (GraphPad Software, San Diego, CA). Graphs depict the mean ± standard deviation or standard error of results. Microvascular responses are expressed as the percent relaxation of the original diameter. The normality of data distribution was assessed by the Shapiro–Wilk test. Inter-group statistical comparison of protein expression was performed using Student’s t-test. Inter-group statistical comparisons of data derived from the water maze test, microvascular reactivity studies, and patch-clamp assays were done using two-way analysis of variance (ANOVA) testing accompanied post hoc by Tukey’s multiple comparisons test.
Results
Mouse characteristics
A total of 41 male and 10 female C57BL/6 mice, all initially 6–8 months of age, were used in this study. Previous studies have indicated that mice begin to exhibit initial signs of cognitive decline and pathological changes relevant to AD at this age. Mean body weight and blood glucose levels did not significantly differ between control mice and STZ-AD mice either before or after ICV injection (Figure 1(B) and (C)). The data for female mice are listed in the Supplemental Material, the mice mentioned below were male.
Spatial learning and memory
The Morris water maze test was conducted over six days. Track plots from the first trial during 5 consecutive days showed that mice showed significantly increased time spent searching for the platform (Figure 2(A)). On hidden platform days (2–5), there was a significant increase in escape latency of STZ-AD mice over time compared with control mice (p < 0.05, Figure 2(B)), indicating impaired spatial learning and memory in these animals. No significant differences were observed in mean speed, path efficiency, or the ratio of time spent in the target quadrant to total duration (p > 0.05, Figure 2(C)–(E)). Rotation times represent the number of times and the animal’s body completed an entire rotation of 360°; higher rotation times in STZ-AD mice suggested less certainty in platform localization compared with control mice (p < 0.05, Figure 2(F)). On day 6 of probe trial without platform, STZ-AD and control mice also exhibited significant differences with respect to time in target quadrant (%) and times of crossing platform zone (Figure 2(G)–(I)). However, the mean speed was not different between the control and STZ-AD groups.
Figure 2.

The Morris water maze (A–I) test: (A) Representative paths taken by mice on the first trial on days 1–5. (B) Escape latencies during days 1–5. (C) Mean speed during testing on days 1–5. (D) Path efficiency during days 1–5. (E) Ratio of time spent in the target quadrant to total duration in the maze during days 1–5. (F) Rotation times during days 1–5. (G–I) Time in target quadrant (%), mean speed (m/s), and times of crossing platform zone during day 6 of probe trial without platform. Visible platform, day 1; hidden platform, days 2–5. Barnes maze (J-M): (J) Escape latency across three consecutive days of training (Day 1, Day 2, and Day 3) measured in seconds, comparing Control and STZ-AD groups. (K) Representative probe trail images showing the path taken by Control and STZ-AD mice during the Barnes maze test. (L) Percentage of time spent in the target quadrant on Day 5 during the probe trials, with a significant reduction observed in STZ-AD mice compared to Control (*p < 0.05). (M) Distance traveled (in meters) during the probe trials on Day 5, showing no significant difference between Control and STZ-AD groups. Data are shown as the mean ± SEM, with a sample size of n = 14 for Male control and n = 18 for Male STZ-AD in the water maze test. For the Barnes maze test, there were n = 4 in the Male control and n = 5 in the Male STZ-AD group. p values were obtained using two-way ANOVA followed post hoc by Tukey’s test. **p < 0.01.
The Barnes maze test was conducted over five days. During the training phase (Days 1–3), there was a noticeable increase in escape latency in STZ-AD mice compared to controls, though this difference was not statistically significant (Figure 2(J)). During the probe trial, track plots revealed distinct differences in the search strategies between the two groups (Figure 2(K)). Control mice had a more concentrated search pattern towards the target quadrant, whereas STZ-AD mice exhibited a less focused search pattern. STZ-AD mice spent significantly less time in the target quadrant compared to control mice (p < 0.05, Figure 2(L)), indicating impaired spatial memory retention. Additionally, the distance traveled during the probe trial on Day 5 did not differ significantly between the two groups (Figure 2(M)), suggesting that the observed differences in spatial memory were not due to changes in general locomotor activity. These findings indicate that STZ-AD mice exhibit deficits in spatial memory as evidenced by their performance in the Barnes Maze, further supporting the presence of cognitive impairments in this model.
Quantitative assessment of phospho-tau and Aβ deposition in brain
Western blot analysis demonstrated a significant increase in phospho-tau levels (Ser 202/Thr 205) in STZ-AD relative to control mice, but no significant differences with respect to phospho-tau (Ser 396) and phospho-tau (Thr 181) between groups (Figure 3(A) and (B)). Histopathological analysis with Congo red staining of mouse hippocampal tissue from control and STZ-AD mice demonstrated significantly higher amyloid plaque burden in mice treated with STZ compared with control mice (p = 0.001, Figure 3(C) and (D)). There were increased levels of soluble Aβ in STZ-AD mice cortex and hippocampus lysate as compared to control by ELISA (p < 0.01, Figure 3(E) and (F)).
Figure 3.

Expression of p-tau and soluble amyloid in the cortex and hippocampus of mice across groups. (A) Western blot analysis of p-tau. (B) Quantification of p-tau Ser 396, p-tau Ser 202/Thr 205, and p-tau Thr 181 band density. (C, D) Demonstration of amyloid deposits by Congo red staining of mouse hippocampal sections. A representative sample is shown for normal mouse brains and for STZ-AD brains. Arrows indicate amyloid plaques. The bar graph represents the significantly higher amyloid plaque count in the STZ-AD group when compared with control animals. (E, F) Quantitative assessment of soluble Aβ levels in the lysates of the hippocampus and cortex using the Elisa. The levels of Aβ in STZ-AD were significantly higher than that in control (p < 0.01). Data are shown as the mean ± SEM, n = 4 in Control, n = 3 in STZ-AD; **p < 0.01. p values were obtained using two-tailed Student’s t tests.
STZ-AD-associated alterations in cerebral microvascular relaxation in response to the SK/IK channel activator NS309 and the endothelium-independent vasodilator SNP
STZ-treated mice exhibited markedly attenuated cerebral microvascular relaxation responses to increasing doses of NS309 when compared with control mice (10−6 M, p = 0.0226; 10−5 M, p = 0.0127; Figure 4(A)). By contrast, no significant differences in microvascular relaxation responses were observed with increasing concentrations of the endothelium-independent vasodilator SNP (p > 0.05, Figure 4(B)).
Figure 4.

(A) In vitro cerebral microvascular reactivity studies determined that the vascular relaxation response to increasing concentrations of the SK/IK channel activator NS309 was significantly impaired in STZ-AD male mice (n = 6) compared with control counterparts (n = 6). (B) In contrast, there was no substantial change in the vascular relaxation response to incubation with increasing concentrations of the endothelium-independent vasodilator SNP between STZ-AD (n = 6) and control mice (n = 6). (C–E) The selective small-/intermediate-conductance calcium-activated potassium (SK/IK) channel activator NS309 increased whole-cell K + currents in MBMECs from STZ-AD and control mice. (C) Representative current traces were obtained under control conditions in the presence of NS309 in STZ-AD (n = 8) and control (n = 6) animals. (D, E) Current-voltage plot of currents shown in C demonstrated reduced current in STZ-AD mice. Data are shown as mean ± SEM. *p < 0.05. p values were obtained using two-way ANOVA with post hoc Tukey’s test.
Endothelial SK/IK currents in MBMECs in response to NS309
MBMECs were isolated from eight mice (4 control and 4 STZ-AD) to compare NS309-sensitive currents across groups. Although treatment with NS309 increased total potassium currents in MBMECs isolated from both STZ-AD and control mice, the potassium current responses were significantly lower in the STZ-AD group (80 mV, p = 0.0375; 100 mV, p = 0.0397; Figure 4(C)–(E)).
Impact of soluble aβ pre-treatment on cerebral microvascular relaxation following NS309 administration in control mice
Microvascular vasodilation was evaluated after short-term (5-min) and long-term (2-h) incubation of control micro-vessels with soluble Aβ peptides. Short-term Aβ exposures had no significant effects on cerebral micro-vessel diameters (Figure 5(A)), but prolonged incubation with soluble Aβ significantly attenuated relaxation responses to NS309 in control brain micro-vessels (10−5 M, p = 0.0036, Figure 5(B)).
Figure 5.
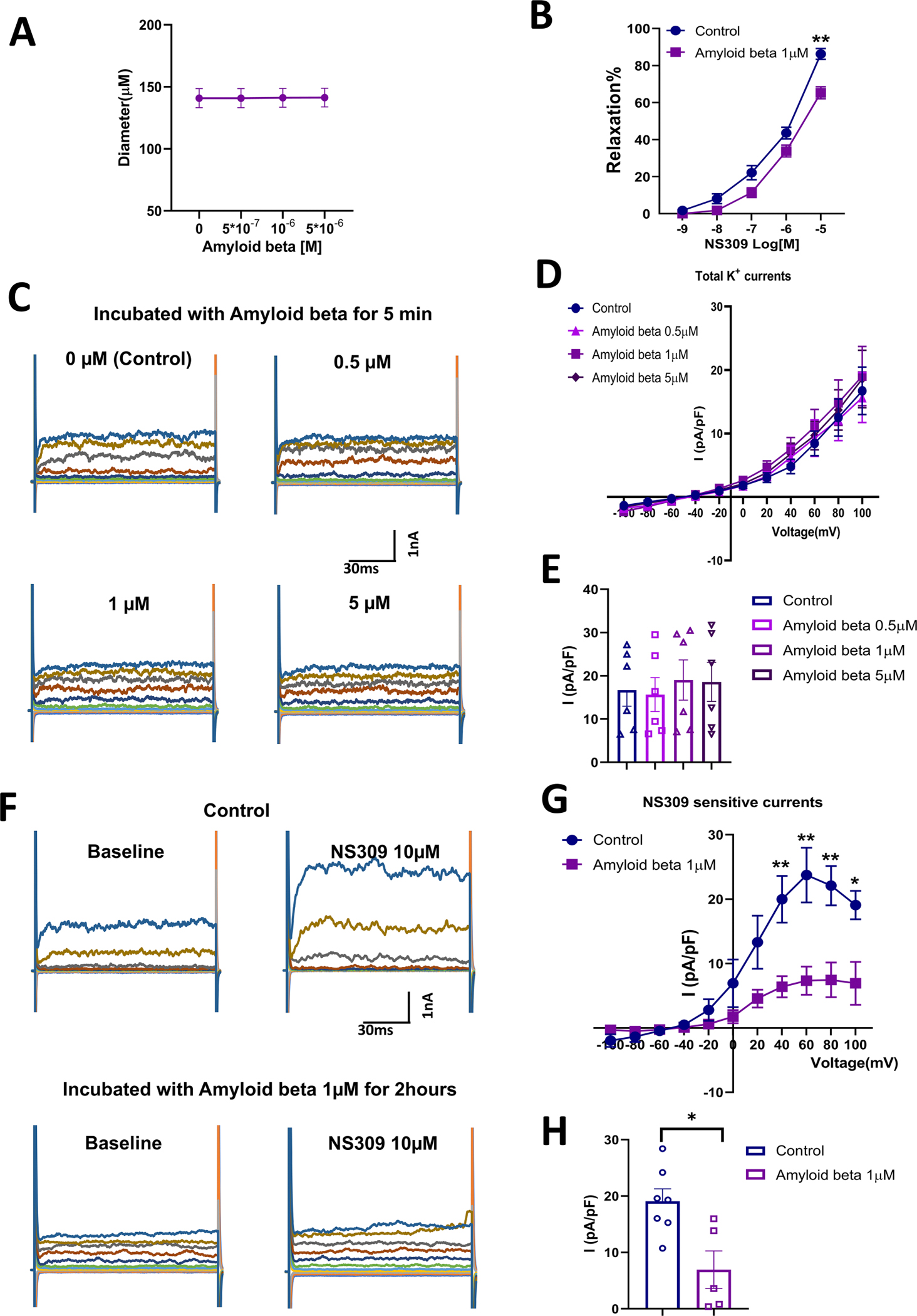
(A) Incubation of cerebral micro-vessels isolated from control male mice (n = 6) with soluble Aβ for 5 min did not result in a significant change in vessel diameter. (B) Soluble Aβ (1 μM) pre-treatment of cerebral micro-vessels isolated from mice resulted in an attenuated relaxation in response to the SK/IK channel activator NS309 (n = 6 in control group and n = 6 in Aβ treatment group). (C–E) Total K+ currents were not significantly altered following a five-minute incubation with soluble Aβ at various concentrations (0 μM, 0.5 μM, 1 μM, 5 μM) in HBMECs (n = 6). (F–H) Incubation of HBMECs in soluble Aβ (1 μM) for 2 h resulted in impaired SK currents (n = 7) when compared to controls that were not pre-treated with soluble Aβ (n = 5). Data are shown as mean ± SEM. *p < 0.05, **p < p-values were obtained using two-way ANOVA with post hoc Tukey’s test.
Potassium currents of HBMECs after pre-treatment with soluble Aβ
Whole-cell potassium currents of HBMECs were evaluated after a 5-min incubation with soluble Aβ (Figure 5(C)–(E)), and NS309-sensitive currents were assessed after a 2-h incubation with soluble Aβ in HBMECs (Figure 5(F)–(H)). After 5-min incubations of HBMECs with various concentrations of soluble Aβ (0 μM, 0.5 μM, 1 μM, 5 μM), total potassium currents were not significantly different relative to pre-treatment (Figure 5(C)–(E)). In contrast, longer incubations with soluble Aβ significantly impaired NS309-sensitive potassium currents relative to untreated HBMEC controls (40 mV, p = 0.0073; 60 mV, p = 0.0069; 80 mV, p = 0.0049; 100 mV, p = 0.0123; Figure 5(F)–(H))
SK channel protein expression in STZ-AD mice and HBMEC’s pre-treated with soluble Aβ
There were no significant differences in SK3 and SK4 protein expression between the control and STZ-AD groups in cortex and hippocampal tissue (Figure 6(A)–(C); p > 0.05). Additionally, pre-treatment with soluble Aβ did not alter HBMEC SK3 and SK4 expression compared to control (Figure 6(D)–(F); p > 0.05).
Figure 6.

Expression of SK3 and SK4 across different groups. (A) Western blot analysis of SK3 and SK4 in mouse cortical and hippocampal tissue. (B, C) Quantification of SK3 and SK4 band density derived from this tissue. (D) Western blot analysis of SK3 and SK4 in control and Aβ-treated HBMECs. (E, F) Quantification of SK3 and SK4 band density in control and Aβ treated HBMECs. Data are shown as mean ± SEM. N = 6 in each group.
Discussion
Growing evidence supports the concept that AD is fundamentally a metabolic disease with molecular and biochemical features that correspond with DM and other peripheral insulin resistance disorders.8,18,19,20,22 AD is thus sometimes referred to as “Type 3 Diabetes”.18,19,20,22 Thus, STZ induced AD is an ideal model for AD related metabolic changes and this model has been commonly used for AD research.
This study produced several important findings related to microvascular angiopathy in AD. First, we confirmed that mice administered intracerebral STZ exhibit significantly higher levels of phospho-tau, and thus exhibited histopathological hallmarks of AD. These findings were associated with significantly impaired spatial reference learning and memory in mice in a manner consistent with the induction of AD-like cognitive dysfunction, as evidenced by impaired performance of STZ-AD mice in Morris water maze and Barnes maze tests. With model efficacy established, we then demonstrated impairments in endothelium-dependent cerebral microvascular relaxation in STZ-AD mice, as evidenced by decreased vascular relaxation responses to the endothelial SK/IK channel activator NS309, together with decreased potassium currents in MBMECs derived from STZ-AD mice. Finally, we showed that pretreatment of mouse cerebral micro-vessels with Aβ attenuated NS309 responses, and that 2-h pre-treatment of HBMECs with Aβ decreased SK-mediated potassium currents compared to controls. Taken together, these findings suggest that SK channel dysfunction may play an important role in the vasculopathy of AD and is characterized by impaired microvascular endothelium-dependent vasomotor responses.
Several different types of SK channels have been identified, with SK1 channels predominantly localized in neurons; SK2 channels in cardiomyocytes, neurons, and endothelial cells; and SK3/SK4(IK) channels in endothelial cells.26 Endothelial SK channels are important contributors to endothelium-dependent vascular smooth muscle relaxation through a mechanism regulated by calcium and calmodulin.27 A growing body of literature implicates SK channel pathophysiology in a variety of disease/physical stress states, including diabetes, atrial fibrillation, hypertension, and ischemia-reperfusion injury.26,28,29
The decreased cerebral microvascular relaxation responses to NS309 and impaired potassium currents from MBMECs of STZ-AD mice seen in this study bear striking resemblance to studies of human and mouse coronary arterioles in the setting of diabetes mellitus and cardioplegic ischemia-reperfusion.28,30,31 Interestingly, pre-treatment of mouse coronary micro-vessels with NS309 prior to cardioplegic hypoxia-reperfusion has been shown to confer protection against post-procedure microvascular dysfunction, raising the question of whether a similar strategy may prove effective in our STZ-AD mouse model.31
There are several candidate explanations for why STZ-AD micro-vessels exhibited SK channel dysfunction in our study, particularly as STZ is a pro-diabetic toxin, and many aspects of AD pathophysiology are reminiscent of diabetic changes localized to the brain.18,19 Given this resemblance, recent research in diabetes showing that chronic inhibition of mitochondrial reactive oxygen species improves endothelial SK currents and responses to NS309 in type 2 diabetic mice may suggest a promising future direction for our work in the brain.30 The promise of this strategy is further reinforced mechanistically by the finding that STZ promotes extensive oxidative stress along-side neuroinflammation.32
Another candidate mechanism is that Aβ itself may affect SK channel activity, supported by our finding that incubation of control mouse cerebral micro-vessels with Aβ resulted in attenuated NS309 responses and reduced SK channel currents. Curiously, HBMECs incubated with soluble Aβ did not exhibit significant differences in SK channel expression relative to controls. This suggests that impaired potassium currents in treated HBMECs may depend more on altered signaling downstream of the SK channel than on altered cell membrane expression of the SK channels themselves.
As with all studies, ours is not without its limitations. For example, SK channel protein expression was not measured directly in STZ-treated mouse cerebral micro-vessels, and previous studies have demonstrated that SK channels may assist with the regulation of brain parenchymal arteriolar diameter and cortical blood flow.33 The impact of this limitation was mitigated, however, by our quantification of SK channel protein expression in mouse cortical and hippocampal tissue as well as HBMECs, in which we found no significant differences in expression between control and STZ-AD groups. Another important limitation to this study was its use of a mouse model; it will be important to verify our results regarding microvascular dysfunction in AD using isolated human cerebral vessels, and to test whether incubation of human cerebral micro-vessels with Aβ produces similar results. Furthermore, although increases in mouse brain Aβ and p-tau were observed in the early stages of our STZ-AD model, we cannot conclude that the increased Aβ and p-tau directly and mainly are responsible for cerebral microvascular dysfunction in-vivo. Therefore, factors other than Aβ and p-tau related to early stages of STZ-AD-induced cerebral endothelial dysfunction should be further investigated. Finally, due to resource constraints, we did not investigate the role of hyperphosphorylated tau to the same extent as Aβ on cerebral endothelial SK channel function in this study.
Additional studies should expand upon these advances, both in mechanistic breadth and through augmentation of translational potential. The former goal will be achieved through the application of a greater breadth of candidate molecular components of the STZ-AD-initiated pathophysiological cascade to this and equivalent models. Such efforts should incorporate, for instance, assays relevant to revealing the role of hyperphosphorylated tau, differentiation between the multiplicity of SK channel isoforms and additional inhibitors, and the use of other agents known to intervene within STZ-AD pathogenesis, such as protein kinase C inhibitors. Building on the substantial foundation provided by this experiment and supplemented by these elucidations, future experiments should verify the operation of these mechanisms within the human cerebral vasculature, with the ultimate goal of uncovering an agent capable of interrupting Alzheimer’s-associated angiopathy in patients prior to the onset of irreversible cortical pathology
Supplementary Material
Acknowledgments
The authors thank Julie Braza of the CPVB COBRE Service Cores (Ocean State Research Institute, Providence VA Medical Center) for assistance with cerebral endothelial cell isolation and culture. The authors also thank Dr. Ming Tong (Department of Medicine, Rhode Island Hospital) for assistance with the Morris water maze test. The authors thank Dr Richard Clements’s help on providing the apparatus for Barnes Maze Test.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research project was mainly supported by the National Institute of Health (NIH) R01HL136347-04S1, and 1R01 HL136347-01 to J.F. This work was supported in part by 1R01HL127072-01A1, National Institute of General Medical Science (NIGMS) of 5P20-GM103652 (Pilot Project) to J.F., 5P20-GM103652 to E.O.H., 1R01AA011431 to S.M.dl.M., and 1R01 HL136347-01, and R01HL46716 to F.W.S.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
Data availability
All data are available upon reasonable request.
References
- 1.Alzheimer’s disease facts and figures. Alzheimer’s Disease and Dementia. https://www.alz.org/alzheimers-dementia/facts-figures#prevalence (accessed 18 March 2023)
- 2.Wong W Economic burden of Alzheimer disease and managed care considerations. Am J Manag Care 2020; 26: S177–S183. [DOI] [PubMed] [Google Scholar]
- 3.Digma LA, Madsen JR, Reas ET, et al. Tau and atrophy: domain-specific relationships with cognition. Alzheimer Res Ther 2019; 11: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiwari S, Atluri V, Kaushik A, et al. Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomed 2019; 14: 5541–5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan L, Mao C, Hu X, et al. New insights into the pathogenesis of Alzheimer’s disease. Front Neurol 2020; 10: 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song C, Shi J, Zhang P, et al. Immunotherapy for Alzheimer’s disease: targeting β-amyloid and beyond. Transl Neurodegener 2022; 11: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Monte SM and Grammas P. Insulin resistance and oligodendrocyte/microvascular endothelial cell dysfunction as mediators of white matter degeneration in Alzheimer’s disease. In: Wisniewski T (ed.) Alzheimer’s disease. Brisbane (AU): Codon Publications, 2019, pp.123–145. [PubMed] [Google Scholar]
- 8.Erdener SE and Dalkara T. Small vessels are a big problem in neurodegeneration and neuroprotection. Front Neurol 2019; 10: 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai Z, Wang C, He W, et al. Cerebral small vessel disease and Alzheimer’s disease. Clin Interv Aging 2015; 10: 1695–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorelick PB, Scuteri A, Black SE, et al. American Heart Association stroke council, council on epidemiology and prevention, council on cardiovascular nursing, council on cardiovascular radiology and intervention, and council on cardiovascular surgery and anesthesia (2011) vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American stroke association. Stroke 2011; 42: 2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheffer S, Hermkens DMA, van der Weerd L, et al. Vascular hypothesis of Alzheimer disease: topical review of mouse models. Arterioscler Thromb Vasc Biol 2021; 41: 1265–1283. [DOI] [PubMed] [Google Scholar]
- 12.Koizumi K, Wang G and Park L. Endothelial dysfunction and amyloid-beta-induced neurovascular alterations. Cell Mol Neurobiol 2016; 36: 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iturria-Medina Y, Sotero RC, Toussaint PJ, et al. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 2016; 7: 11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gentile MT, Vecchione C, Maffei A, et al. Mechanisms of soluble beta-amyloid impairment of endothelial function. J Biol Chem 2004; 279: 48135–48142. [DOI] [PubMed] [Google Scholar]
- 15.Thomas T, Thomas G, McLendon C, et al. Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature 1996; 380: 168–171. [DOI] [PubMed] [Google Scholar]
- 16.Durrant CS, Ruscher K, Sheppard O, et al. Beta secretase 1-dependent amyloid precursor protein processing promotes excessive vascular sprouting through NOTCH3 signaling. Cell Death Dis 2020; 11: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giuliani A, Sivilia S, Baldassarro V, et al. Age-related changes of the neurovascular unit in the cerebral cortex of Alzheimer disease mouse models: a neuroanatomical and molecular study. J Neuropathol Exp Neurol 2020; 78: 101–112. [DOI] [PubMed] [Google Scholar]
- 18.Steen E, Terry BM, Rivera EJ, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis 2020; 7: 63–80. [DOI] [PubMed] [Google Scholar]
- 19.Lester-Coll N, Rivera EJ, Soscia SJ, et al. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis 2020; 9: 13–33. [DOI] [PubMed] [Google Scholar]
- 20.Kamat PK. Streptozotocin induced Alzheimer’s disease like changes and the underlying neural degeneration and regeneration mechanism. Neural Regener Res 2015; 10: 1050–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao C, Liu Y, Jiang Y, et al. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3β pathway in streptozotocin-induced Alzheimer rat model. Brain Pathol 2014; 24: 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen TT, Ta QTH, Nguyen TKO, et al. Type 3 diabetes and its role implications in Alzheimer’s disease. Int J Mol Sci 2020; 21: 3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pires PW, Dabertrand F and Earley S. Isolation and cannulation of cerebral parenchymal arterioles. J Vis Exp 2016; (111): 53835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Assmann JC, Müller K, Wenzel J, et al. Isolation and cultivation of primary brain endothelial cells from adult mice. Bio Protoc 2017; 7: e2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan MA, Schultz S, Othman A, et al. Hyperglycemia in stroke impairs polarization of monocytes/macrophages to a protective noninflammatory cell type. J Neurosci 2016; 36: 9313–9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kant S, Sellke F and Feng J. Metabolic regulation and dysregulation of endothelial small conductance calcium activated potassium channels. Eur J Cell Biol 2022; 101: 151208. [DOI] [PubMed] [Google Scholar]
- 27.Ledoux J, Werner ME, Brayden JE, et al. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006; 21: 69–78. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Kabakov AY, Xie A, et al. Metabolic regulation of endothelial SK channels and human coronary microvascular function. Int J Cardiol 2020; 312: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burashnikov A, Barajas-Martinez H, Hu D, et al. The small conductance calcium-activated potassium channel inhibitors NS8593 and UCL1684 prevent the development of atrial fibrillation through atrial-selective inhibition of sodium channel activity. J Cardiovasc Pharmacol 2020; 76: 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xing H, Zhang Z, Shi G, et al. Chronic inhibition of mROS protects against coronary endothelial dysfunction in mice with diabetes. Front Cell Dev Biol 2021; 9: 643810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Shi G, Liu Y, et al. Coronary endothelial dysfunction prevented by small-conductance calcium-activated potassium channel activator in mice and patients with diabetes. J Thorac Cardiovasc Surg 2020; 160: e263–e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park J, Won J, Seo J, et al. Streptozotocin induces Alzheimer’s disease-like pathology in hippocampal neuronal cells via CDK5/Drp1-mediated mitochondrial fragmentation. Front Cell Neurosci 2020; 14: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hannah RM, Dunn KM, Bonev AD, et al. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab 2011; 31: 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available upon reasonable request.