

Abstract
As another clinical trial of a mitochondria-targeting cancer therapy faces failure, it calls for a thorough re-evaluation of the strategy; the time has come to go from the bedside back to the bench.
A century ago, Otto Warburg observed that tumors produce energy by converting vast amounts of glucose to lactate in the cytoplasm, a process known as aerobic glycolysis. This contrasts with the main pathways used by normal cells – namely oxidative phosphorylation or the tricarboxylic acid (TCA) cycle in mitochondria. Over time, this led to the misconception that mitochondrial metabolism is somewhat inconsequential for tumorigenesis1. But this may very much depend on the cancer type2,3. For example, the rate of dysfunctional mitochondrial DNA mutations is highest in thyroid cancer and lowest in leukemia3, suggesting that leukemias have functional mitochondria and may be sensitive to mitochondrial inhibitors. Hence, therapies that target the mitochondria may be attractive for certain types of cancer2. In this issue of Nature Medicine, Yap et al.4 report on a phase I trial evaluating a potent mitochondrial inhibitor5 (IACS-010759) in individuals with acute myeloid leukemia and solid tumors. Unfortunately, promising preclinical findings did not translate into the clinic as intolerable neurotoxicity precluded adequate dosing, prompings the question – what lessons can be learned from this disappointing outcome?
Understanding mitochondrial function in normal and tumor cells is crucial to addressing this question. The mitochondrion does not simply house the electron-transport chain for oxidative phosphorylation; rather, it is a complex organelle that supports many biosynthetic pathways, including de novo synthesis of nucleotides and porphyrin (a component of hemoglobin)2. It also provides intermediates that can be converted to amino acids for protein synthesis, and is central for one-carbon metabolism. Approaches to targeting the mitochondrion in cancers include inhibiting complex 1 of the electron-transport chain (as done by Yap et al. with IACS-010759), as well as inhibition of nucleotide synthesis or the TCA cycle1,2.
Previous efforts to target the mitochondria in cancer have faced challenges. For example, the drug CPI-613 (also known as devimistat) is purported to be a TCA-cycle inhibitor, and showed impressive phase I results6; however, two phase III trials failed to demonstrate enhanced efficacy in pancreatic cancer or acute myeloid leukemia. Nonetheless, epidemiologic studies suggest that metformin, which inhibits the mitochondrial complex 1, may have a preventive anti-tumor effect –although it is insufficiently potent to be used at acceptable clinical therapeutic doses2. On the basis of these results, a potent mitochondrial complex I inhibitor – IACS-010759 – has been developed, and showed impressive preclinical efficacy in models of brain cancer and acute myeloid leukemia5.
The tolerability of IACS-010759 in mouse models raised hope that targeting mitochondrial complex 1 is clinically feasible5. However, potential side effects of metabolic drugs in humans – particularly those targeting the mitochondrion – may be gleaned from the manifestations of heritable disorders of the mitochondrion, as documented in the Online Mendelian Inheritance of Man (OMIM)1. In particular, mutations affecting the integrity of mitochondrial complex 1 are associated with optic neuropathy. This has now been borne out in the clinical study of Yap et al.4, in which one individual with a solid tumor had visual impairment due to optic nerve neurotoxicity, reminiscent of the effect of heritable complex 1 mutations.
Overall, the work by Yap et al. – which enrolled 17 individuals with acute myeloid leukemia and 23 with solid tumors – revealed that IACS-010759 has a narrow therapeutic index with dose-limiting toxicities that include neurotoxicity and increased blood lactate. Lactic acidosis is an expected side effect, because inhibition of mitochondrial function will induce metabolic rewiring toward the conversion of pyruvate to lactate (rather than its oxidation in the mitochondrial TCA cycle). In fact, phenformin – a more potent inhibitor of complex 1 than metformin – was removed from the market for the treatment of diabetes because of clinically significant lactic acidosis. However, in contrast to phenformin, which is not linked to neurotoxicity, IACS-010759 was associated with a roughly 50% rate of neurotoxic side effects among all patients.
Surprisingly, despite the toxicities reported with IACS-010759, which suggest that the drug has pharmacodynamic effects, the clinical responses were disappointing for individuals with AML – although one patient with prostate cancer did show a partial clinical response4. In this study, the substantial toxicities clearly outweigh the limited benefits of IACS-010759 (Fig. 1), which, along with the failure of CPI-613, delivers the sobering message that targeting metabolism is challenging. Aside from the successful targeting of mutant isocitrate dehydrogenase with a wide therapeutic index in AML7, targeting cancer metabolism1 – particularly the pathways that are held in common with normal cells – is restricted by narrow therapeutic indices and toxicity.
Fig. 1 |. Targeting metabolism for cancer therapy comes with a cost when the scale is tipped toward intolerable on-target, off-tumor side effects.
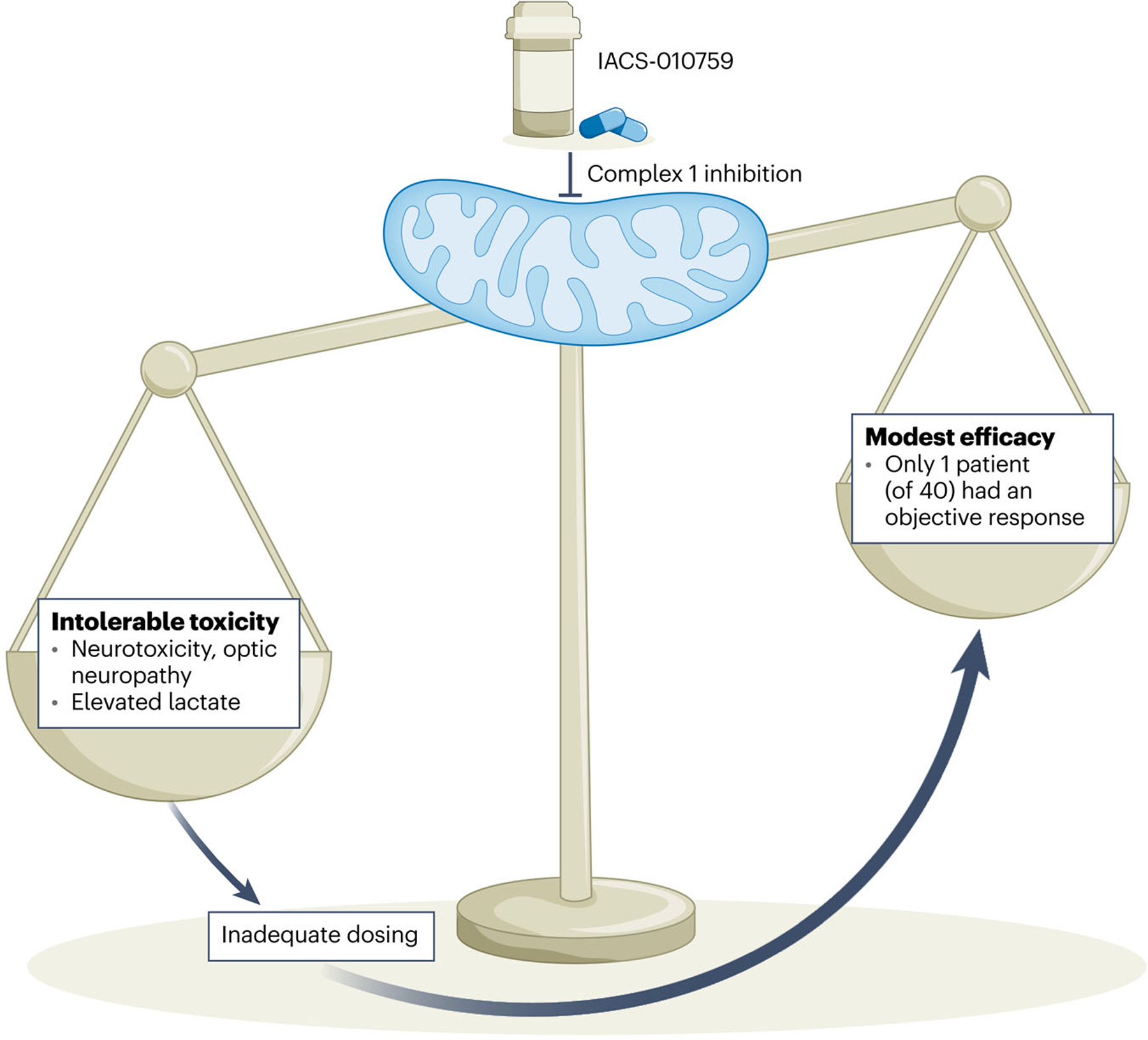
In a phase 1 study, Yap et al.4 have found that the mitochondrial complex 1 inhibitor IACS-010759 is associated with peripheral and optic neuropathy. These toxic side effects necessitate dose-limiting to the point of inadequacy, and hence only modest evidence of enhanced efficacy in patients with cancer.
Immune cells are among the normal cells that are affected by mitochondrial inhibitors. Although cytotoxic CD8+ T cells and Ml macrophages are glycolytic, pro-tumorigenic immune cells such as T regulatory cells, M2 macrophages and myeloid-derived suppressor cells all rely on oxidative phosphorylation; therefore, mitochondrial-targeting drugs might be expected to dampen their pro-tumor effects8. If this is indeed the case, we would expect IACS-010759 to be efficacious in solid tumors, but the study by Yap et al. proves otherwise – signaling a note of caution to the field of cancer metabolism. It is hoped that this report will give pause to strategies that target cancer-cell-autonomous metabolic vulnerabilities without thorough consideration of the potential adverse effects of metabolic inhibitors on normal cells, or on the anti-tumor arm of the immune system.
Despite the setbacks seen with IACS-010759 and CPI-613, there may still be viable ways of exploiting metabolism for cancer therapy. These strategies must, however, carefully and thoroughly consider how manipulating metabolism affects both the host and the tumor microenvironment. For example, the prodrug of a glutamine antagonist (D6-diazo-5-oxo-l-norleucine) appears to have features that favor anti-tumor immunity while inhibiting tumor-cell-intrinsic metabolism9. Whether these preclinical observations will sustain in clinical studies remains to be seen. There is also excitement in the area of immunometabolism10, which might be manipulated to enhance the anti-tumor effect of the immune system. Even here, however, there could be challenges and unforeseen consequences. For instance, acidity has been documented to blunt anti-tumor immunity, and it is thought that neutralizing the acidic tumor microenvironment with sodium bicarbonate could enhance immunotherapy by invigorating cytotoxic lymphocytes11. But in a recent study using a murine model of hepatocellular carcinoma, sodium bicarbonate paradoxically inhibited immune-checkpoint therapy by enhancing the activity of the signaling node mTORC1 in cancer cells12.
Given all of these observations, perhaps it is a good time for the field to pause and rigorously assess the side effects of metabolic drugs, as implored by the Hippocratic oath ‘Primum non nocere’, before embarking on further clinical studies.
Acknowledgements
C.V.D. is supported by the Ludwig Institute for Cancer Research and National Cancer Institute (NCI) grants R01CA252225, R01CA054197 and R01CA057341.
Footnotes
Competing interests
C.V.D. is consultant for the Barer Institute, Rafael Holdings, Inc. X.Z. declares no competing interests.
References
- 1.Stine ZE, Schug ZT, Salvino JM & Dang CV Nat. Rev. Drug Discov. 21, 141–162 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasan K, Werner M & Chandel NS Cell Metab. 32, 341–352 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorelick AN et al. Nat. Metab. 3, 558–570 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yap TA et al. Nat. Med. 10.1038/s41591-022-02103-8 (2023). [DOI] [Google Scholar]
- 5.Molina JR et al. Nat. Med. 24,1036–1046 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Alistar A et al. Lancet Oncol. 18, 770–778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yen K et al. Cancer Discov. 7, 478–493 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Wolpaw AJ & Dang CV Trends Cell Biol. 28, 201–212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leone RD et al. Science 366,1013–1021(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buck MD, Sowell RT, Kaech SM & Pearce EL Ce// 169,570–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piton-Thomas S et al. Cancer Res. 76,1381–1390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong Y et al. Cancer Res. Commun. 2, 842–856 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]