

Abstract
Understanding genetic regulation of metabolism is critical for gaining insights into the causes of metabolic diseases. Traditional metabolome-based genome-wide association studies (mGWAS) focus on static associations between single nucleotide polymorphisms (SNPs) and metabolite levels, overlooking the changing relationships caused by genotypes within the metabolic network. Notably, some metabolites exhibit changes in correlation patterns with other metabolites under certain physiological conditions while maintaining their overall abundance level. In this manuscript, we develop Metabolic Differential-coordination GWAS (mdGWAS), an innovative framework that detects SNPs associated with the changing correlation patterns between metabolites and metabolic pathways. This approach transcends and complements conventional mean-based analyses by identifying latent regulatory factors that govern the system-level metabolic coordination. Through comprehensive simulation studies, mdGWAS demonstrated robust performance in detecting SNP-metabolite-metabolite associations. Applying mdGWAS to genotyping and mass spectrometry (MS)-based metabolomics data of the METabolic Syndrome In Men (METSIM) Study revealed novel SNPs and genes potentially involved in the regulation of the coordination between metabolic pathways.
Keywords: metabolism, changing correlation, genotyping
Introduction
Genome-wide association study (GWAS) is a powerful tool for examining the genetic etiology of metabolic diseases, but does not elucidate the metabolic changes that lead to disease. To understand the latter, it is necessary to investigate the association between genotype and the regulation of the network of metabolites.
Metabolite-based GWAS (mGWAS) jointly considers genotyping data and measurements of metabolites to identify genetic variants associated with metabolic traits [1]. The most common approach is to quantify the association between individual metabolite abundance levels and single nucleotide polymorphisms (SNPs). mGWAS have successfully uncovered and replicated associations between the abundance of individual metabolites and SNPs at thousands of genetic loci [2–5]. However, these approaches primarily focus on static, univariate relationships, which fail to capture the higher-order coordination between metabolite levels, and thus do not reveal the full picture of genetic regulation of metabolic pathways.
Genetic variations can change the co-expression levels of metabolism-related genes, which in turn may lead to changes in the correlation between sets of metabolites. Further, changing activity levels of metabolic pathways may modulate the correlation patterns between metabolites without changing their average abundance levels, and thus may go undetected by mean-based mGWAS analyses. For example, [6] documented pairs of thorax metabolites whose mean values were constant but whose correlations were significantly different in participants with different dietary conditions. Under controlled conditions in experimental animals, correlation between some metabolite pairs can be manipulated by changing the level of exposure to the environmental chemical PCB [7]. These studies point to the existence of important second-order relations in metabolic regulations. It is conceivable that genetic factors could affect the level of correlations between some metabolites, but such associations have not been explored on the genome–metabolome scale.
There are major obstacles in the computation and interpretation of changing correlations between metabolites in association with genotypes. In general, identifying metabolite correlation differences between two or more groups defined according to some genetic measure (e.g. the absence/presence of a minor allele) involves computations on the order of 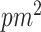 , where
, where  is the number of SNPs and
is the number of SNPs and 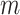 is the number of metabolites. This is an ultra high-dimensional problem with low statistical power and high computational cost. More importantly, given the complexity of metabolic regulation and its sensitivity to nutritional and environmental conditions, genotypes may only affect a small portion of the variation within metabolite sets. Finding SNP-metabolite-metabolite triplets alone may not identify genotypes associated with global regulation of metabolic coordination. Rather, our first focus should be on studying the global regulation of metabolic coordination at the system level and identifying SNPs that contribute to this regulation.
is the number of metabolites. This is an ultra high-dimensional problem with low statistical power and high computational cost. More importantly, given the complexity of metabolic regulation and its sensitivity to nutritional and environmental conditions, genotypes may only affect a small portion of the variation within metabolite sets. Finding SNP-metabolite-metabolite triplets alone may not identify genotypes associated with global regulation of metabolic coordination. Rather, our first focus should be on studying the global regulation of metabolic coordination at the system level and identifying SNPs that contribute to this regulation.
We propose a new approach, named metabolic differential-coordination GWAS (mdGWAS). The main idea is to pivot around latent factors that control the changing correlation of large groups of metabolite pairs and find SNPs and metabolic pathways that are associated with these factors, thereby linking SNPs to the regulated pathways (Fig. 1). The key steps of the pipeline consist of detecting latent factors that control the correlation changes for the metabolomics data, summarizing the changing correlation patterns at the metabolic pathway level, and performing a GWAS of the selected latent factors. We illustrate our approach on metabolite and genotype data from the METabolic Syndrome In Men (METSIM) Study and compare results to mean-based mGWAS of metabolic features. We show that mdGWAS can detect signals that are not captured by mGWAS, leading to important new findings.
Figure 1.

Overview of mdGWAS. (a) An illustration of the type of relation the analysis is designed to identify. (b) The logic behind using the latent factor approach: GWAS identifies associations between SNPs and a latent factor that governs the changing correlation between metabolic features. The network represents change of metabolite-metabolite associations when the genotype changes on a single representative SNP. Each node corresponds to a metabolite, and edges represent pairwise associations between metabolites. The edge colors indicate genotype-specific effects on the metabolite pair associations. (c) Flowchart of the data analysis: latent factors are identified from metabolomics data by Dynamic Correlation Analysis (DCA), and then analyzed by GWAS. Results include networks of differentially coordinated metabolites and metabolic pathways, functional annotation of associated SNPs and association plots.
Methods
Detection of Latent Factors that Govern Changing Correlation in Metabolomics Data
To identify latent factors governing changing correlation patterns among metabolite pairs, we used the Dynamic Correlation Analysis (DCA) framework [8]. This method captures second-order regulatory features in metabolic networks by identifying Dynamic Components (DCs). These DCs serve as phenotypes for subsequent GWAS.
Selecting metabolite pairs with potential changing correlations
Let  represent the metabolomics dataset, where
represent the metabolomics dataset, where 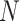 denotes the number of samples, and
denotes the number of samples, and 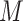 is the number of metabolites. To capture nonlinear dependencies and changing correlation patterns between metabolite pairs, we used the Liquid Association Coefficient (LAC), defined as:
is the number of metabolites. To capture nonlinear dependencies and changing correlation patterns between metabolite pairs, we used the Liquid Association Coefficient (LAC), defined as:
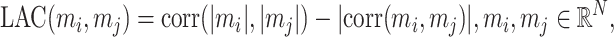 |
(1) |
where  and
and  are the normalized abundance vectors of metabolites
are the normalized abundance vectors of metabolites  and
and 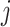 , respectively. The resulting LAC matrix
, respectively. The resulting LAC matrix 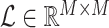 contains LAC values for all metabolite pairs. To focus on biologically significant relationships, we retained the top
contains LAC values for all metabolite pairs. To focus on biologically significant relationships, we retained the top 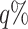 of LAC values, thereby enriching for metabolite pairs exhibiting strong patterns of changing correlation.
of LAC values, thereby enriching for metabolite pairs exhibiting strong patterns of changing correlation.
Feature matrix construction
For each selected metabolite pair  , we calculate their element-wise product across samples to construct pairwise association profiles:
, we calculate their element-wise product across samples to construct pairwise association profiles:
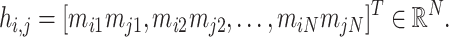 |
(2) |
These association profiles are subsequently stacked into a feature matrix  , where each row corresponds to a metabolite pair’s association profile across all samples. Here,
, where each row corresponds to a metabolite pair’s association profile across all samples. Here, 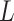 denotes the number of selected metabolite pairs.
denotes the number of selected metabolite pairs.
Dynamic component (DC) extraction
Principal patterns of changing correlations are identified by performing eigenvalue decomposition on the covariance matrix of 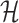 :
:
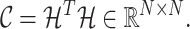 |
(3) |
The eigenvectors 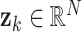 corresponding to the largest eigenvalues of
corresponding to the largest eigenvalues of  , are extracted as Dynamic Components (DCs). The DCs are initially ordered based on their proportion of variance explained (PVE), enabling prioritization of components that capture the most prominent patterns of changing correlation.
, are extracted as Dynamic Components (DCs). The DCs are initially ordered based on their proportion of variance explained (PVE), enabling prioritization of components that capture the most prominent patterns of changing correlation.
Biological Validation and Refinement
While eigenvalue decomposition orders the components by PVE, components with high variance may be associated with non-specific global signals such as batch effects or variations in sample handling, rather than biologically meaningful patterns. To address this, we applied a local false discovery rate (lfdr) procedure to refine and validate the biological relevance of DCs. For each DC 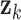 , we calculate the Liquid Association (LA) score between all metabolite pairs
, we calculate the Liquid Association (LA) score between all metabolite pairs 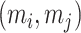 conditioned on
conditioned on 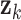 :
:
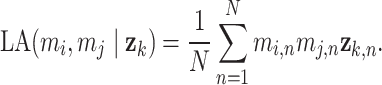 |
(4) |
Under the null hypothesis, these scores follow a normal distribution [9], allowing for statistical significance testing of metabolite pair associations using a mixture model-based lfdr estimation approach [10]. The DCs are then re-ordered based on the number of metabolite pairs significantly associated with each component, prioritizing biologically meaningful patterns.
GWAS on the latent factors
Using the investigator’s preferred model and implementation [e.g. 11] and specification of parameters (e.g. minimum minor allele count), we iteratively use each latent factor as the outcome in a GWAS. Standard checks, such as Manhattan plots and Quantile-Quantile plots, can be used to verify proper genomic control. We also computed the genomic inflation factor 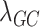 for all SNPs and for common and rare variants separately [12, 13]. Finally,
for all SNPs and for common and rare variants separately [12, 13]. Finally, 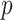 -values are thresholded to select a set of SNPs significantly associated with the latent factors. Given we study several latent factors, we used a significance threshold of
-values are thresholded to select a set of SNPs significantly associated with the latent factors. Given we study several latent factors, we used a significance threshold of 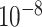 , which is slightly more stringent than the conventional threshold. We also further investigated more weakly associated SNPs using a threshold of
, which is slightly more stringent than the conventional threshold. We also further investigated more weakly associated SNPs using a threshold of 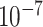 . This set likely contains more false positives, but may also contain true associations that do not meet a more stringent
. This set likely contains more false positives, but may also contain true associations that do not meet a more stringent 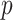 -value threshold due to the potential weak signals of second-order associations.
-value threshold due to the potential weak signals of second-order associations.
Functional interpretation of results
The analyses described above make connections between SNPs to latent factors of changing correlations, hence to the metabolite pairs associated with the factors. To further consolidate and interpret the results, we propose a framework to analyze it at the level of functional pathways. We first consider the metabolic pathways. When a pair of metabolites are found to be significantly associated with the latent factor, they add a changing correlation link between the pathways they belong to. We use the hypergeometric test to determine whether a pair of metabolic pathways is over-represented by comparing the total number of possible links given their respective metabolites, the number of links we find to be associated with the latent factor, and the total number of links established by the latent factor.
We next consider the functional pathway from the latent factors to the SNPs identified by our GWAS of latent factors. We combine haplotype block analysis and GTEx database data (https://www.gtexportal.org/home/) to identify genes associated with the selected SNPs. First, we identify relevant haplotype blocks, i.e. clusters of co-inherited alleles, and select all genes in the haplotype blocks of the selected SNPs. Second, recognizing that many SNPs influence gene transcription, as confirmed by the GTEx project [14], we find genes linked to the latent factors through the select SNPs by using all the SNP-gene association results from GTEx (version 8). We then conduct gene set enrichment analysis to find biological processes over-represented by the genes associated with the latent factors.
Data from the METSIM study
We analyzed a subset of the non-diabetic men in the METSIM study. A total of 6490 participants had matching covariates, including age, BMI, lipid medication usage status, and experimental batch information. We then removed participants that had a high proportion of missing values or belonged to more than one experimental batch, and 4269 participants remained. Finally, after removing participants without matching genotypes, the dataset contained 4220 participants.
Genetic data pre-processing
METSIM participant samples were genotyped on the Human OmniExpresss-12v1_C BeadChip (OmniExpress) and Infinium HumanExome-12 v1.0 BeadChip (Exome Chip) platforms. Genotyping and imputation methods are described in detail in [15]. A resulting 16 607 533 variants with high imputation quality (i.e. minimac RSQ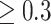 ) were carried forward.
) were carried forward.
Based on the sample quality controls described in [15], we removed 3 samples with sex chromosome anomalies, 6 with evidence of participant duplication, 7 population outliers and 10 samples with non-Mendelian inheritance inconsistencies. In addition, we removed one individual from each of 7 monozygotic twin pairs.
Metabolic feature pre-processing
The metabolomics data were preprocessed by Metabolon. In metabolomics data, the missing values are typically not Missing Completely At Random or Missing At Random [16]. Imputation strategies for spectroscopy data were compared in [17, 18] and [19], to name a few, and the K-nearest neighbors (KNN) algorithm was shown to perform well. We imputed metabolic features using the KNN algorithm with  chosen after investigation of missing patterns in the METSIM metabolic feature data. After imputation, each metabolic feature is regressed on the confounders age, age2, and BMI. The residual metabolic feature is then normalized through a rank-based inverse normal transformation using Bliss’ transformation [20]. We conducted additional sensitivity analyses to verify robustness to the transformation.
chosen after investigation of missing patterns in the METSIM metabolic feature data. After imputation, each metabolic feature is regressed on the confounders age, age2, and BMI. The residual metabolic feature is then normalized through a rank-based inverse normal transformation using Bliss’ transformation [20]. We conducted additional sensitivity analyses to verify robustness to the transformation.
Simulation Study
To evaluate the performance of mdGWAS framework in detecting SNP-metabolite-metabolite associations, we conducted a simulation study to assess detection accuracy, focusing on false positives rates (FPR) and false negative rates (FNR) error rates.
Genomic Data Simulation
We simulated high-dimensional genomic data by generating a SNP genotype matrix 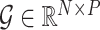 , where
, where  represents the number of SNPs and
represents the number of SNPs and 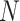 is the sample size. Each SNP genotype was independently sampled from a standard normal distribution:
is the sample size. Each SNP genotype was independently sampled from a standard normal distribution:
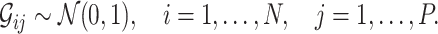 |
(5) |
A small proportion of SNPs (0.01% to 0.1% of the total SNPs) were designated as causal variants influencing latent regulatory factors, while the remaining SNPs were treated as unassociated noise.
The latent regulatory factors 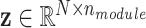 were modeled as dynamic components (DCs) in metabolic networks, where
were modeled as dynamic components (DCs) in metabolic networks, where 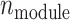 is the number of regulatory modules. These factors were influenced by the causal SNPs, defined as:
is the number of regulatory modules. These factors were influenced by the causal SNPs, defined as:
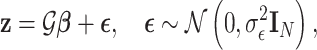 |
(6) |
where 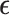 represents normally distributed noise with variance
represents normally distributed noise with variance 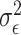 . The effect sizes of causal SNPs were encoded in the sparse vector
. The effect sizes of causal SNPs were encoded in the sparse vector 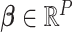 , with non-zero elements corresponding to causal variants. The sparsity of
, with non-zero elements corresponding to causal variants. The sparsity of 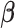 ensures that only a small proportion of SNPs contribute to the latent factors. The non-zero elements of
ensures that only a small proportion of SNPs contribute to the latent factors. The non-zero elements of 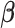 were drawn from:
were drawn from:
 |
(7) |
where 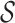 denotes the set of causal SNPs,
denotes the set of causal SNPs, 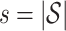 is the number of causal variants, and
is the number of causal variants, and 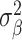 controls the total genetic variance explained.
controls the total genetic variance explained.
Metabolomic Data Simulation
To emulate the modular structure of biological metabolic networks, we simulated metabolomic data governed by latent regulatory factors. Each regulatory module consisted of metabolites whose pairwise correlations were modulated by a latent factor 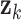 . The correlation between metabolites
. The correlation between metabolites 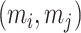 in the
in the  -th module was defined by the changing correlation matrix:
-th module was defined by the changing correlation matrix:
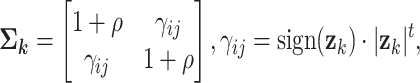 |
(8) |
where  is the changing correlation coefficient modulated by
is the changing correlation coefficient modulated by 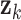 ,
, 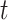 is a scaling parameter controlling regulatory strength, and
is a scaling parameter controlling regulatory strength, and 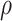 introduces a noise component accounting for random fluctuations in metabolite correlations.
introduces a noise component accounting for random fluctuations in metabolite correlations.
Log-metabolite abundances in the  -th module were then sampled from a multivariate normal distribution:
-th module were then sampled from a multivariate normal distribution:
 |
(9) |
where 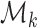 represents the log-metabolite abundance vector for the
represents the log-metabolite abundance vector for the  -th module.
-th module.
In addition to regulatory modules, we introduced noise metabolites that were uncorrelated with any latent factor. These noise metabolites were sampled independently from a normal distribution:
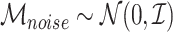 |
(10) |
where  is the identity matrix. The final metabolomic dataset consisted of
is the identity matrix. The final metabolomic dataset consisted of 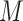 metabolites across
metabolites across  samples, capturing both structured regulation and unstructured background variation. Here,
samples, capturing both structured regulation and unstructured background variation. Here, 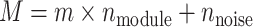 ,
, 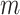 represents the number of metabolites in each regulatory module,
represents the number of metabolites in each regulatory module, 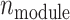 is the number of regulatory modules, and
is the number of regulatory modules, and  is the number of noise metabolites.
is the number of noise metabolites.
Dynamic Component Detection and Evaluation
DCA was applied to the simulated metabolomic dataset to extract latent dynamic components (DCs). The top three DCs were selected based on the number of significant metabolite pairs identified, which were compared with the true metabolite pairs to calculate the FPR and FNR score for recovering true associations.
To assess genetic associations, we performed marginal regression analysis between the detected dynamic components 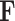 and individual SNPs:
and individual SNPs:
 |
(11) |
where  represents the effect size of
represents the effect size of  , and
, and 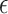 denotes the residual error. The statistical significance of each association was assessed using p-values, and a False Discovery Rate (FDR) procedure was applied to identify SNPs significantly associated with the dynamic components.
denotes the residual error. The statistical significance of each association was assessed using p-values, and a False Discovery Rate (FDR) procedure was applied to identify SNPs significantly associated with the dynamic components.
The mdGWAS framework demonstrated robust performance in identifying metabolite pairs with changing correlation under varying causal SNPs and total SNP sizes (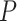 ), metabolite counts (
), metabolite counts (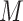 ), genetic variance (
), genetic variance ( ), and noise (
), and noise ( ). In the experiments, we set the sample size to
). In the experiments, we set the sample size to 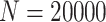 , the metabolite count to
, the metabolite count to 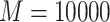 , and the noise parameters
, and the noise parameters 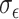 and
and 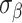 to 0.5, ensuring a realistic and challenging simulation scenario to evaluate the framework’s efficacy.
to 0.5, ensuring a realistic and challenging simulation scenario to evaluate the framework’s efficacy.
The simulation results in Fig. 2 show that as the total SNP count and causal SNP count increase, the error rates (FPR and FNR) exhibit a slight upward trend, reflecting the increasing complexity of the genetic architecture. Despite this, mdGWAS performs consistently well, highlighting its accuracy in detecting regulatory signals. Furthermore, mdGWAS demonstrates strong robustness to noise, with its performance remaining stable under varying noise levels, indicating the method’s reliability in challenging data conditions.
Figure 2.

Simulation results. (a) Error rates (FPR and FNR) for SNP detection under different total SNP numbers. The x-axis indicates the number of causal SNPs, and the y-axis shows the error rates. Red lines represent the true latent factors (solid: FPR, dashed: FNR), while blue lines represent mdGWAS-detected DCs (solid: FPR, dashed: FNR). (b-c) Error rates for SNP detection under varying noise levels (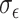 and
and  , ranging from 0.1 to 0.9, respectively.) (d) Error rates for metabolite-pair detection across varying sample sizes. (e) Error rates for metabolite-pair detection under different
, ranging from 0.1 to 0.9, respectively.) (d) Error rates for metabolite-pair detection across varying sample sizes. (e) Error rates for metabolite-pair detection under different 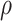 settings.
settings.
In metabolite pair detection, mdGWAS achieves near-zero FNR, regardless of increasing sample sizes or noise levels. This result underscores the framework’s ability to reliably identify true metabolite associations. Overall, these findings validate mdGWAS as a robust and accurate method, capable of handling noisy and complex datasets effectively.
Results
We conducted DCA on the metabolomics data, and selected the first 10 factors, or dynamic components (DCs), that accounted for 72% of PVE cumulatively. For each of these 10 factors, the number of significant metabolic feature pairs associated with the factor was computed. We then selected the top 5 factors with the largest number of significant pairs of metabolic features. The number of metabolic feature pairs are listed in Table 1, where the ordering of the DCs is based on the number of metabolite pairs associated.
Table 1.
Number of significantly associated metabolite pairs and significantly associated SNPs for the top dynamic components
| DC 1 | DC 2 | DC 3 | DC 4 | DC 5 | |
|---|---|---|---|---|---|
metabolite pairs with 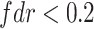
|
4039 | 2334 | 2055 | 822 | 738 |
SNPs with 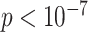 ( (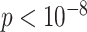 ) ) |
163(99) | 1(0) | 508(227) | 5(1) | 127(18) |
genes associated with the SNPs at level 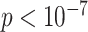
|
35 | 2 | 122 | 3 | 47 |
We conducted GWAS using EPACTS v3.3.0 (https://github.com/statgen/EPACTS). We examined Quantile-Quantile plots of the 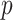 -values and computed
-values and computed 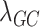 for rare and common variants, which confirmed that we have adequate genomic control.
for rare and common variants, which confirmed that we have adequate genomic control.
For each DC, we collected all the significant SNPs and the genes associated with the SNPs based on haplotype blocks or GTEx database. We then examined whether the genes point to certain biological functions collectively, using the gene set analysis method GOstat. We summarize the counts of SNPs and genes in Table 1. Interestingly, we find that two of the DCs are associated with very few SNPs, indicating that the collective changing correlation behavior captured by those two DCs are not governed at the genetic level. This is understandable given the complexity of metabolic regulation. In the following discussions, we focus on the three DCs associated with a substantial number of SNPs and genes.
Dynamic Component 1
Figure 3a shows the interplay of metabolic pathways in association with DC1. The connecting lines represent significant relations between pathways. The size of a pathway’s label is proportional to its number of connections. Within this network of metabolic pathways with changing coordination, ‘Food components/plants’ and ‘Urea cycle; Arginine and proline metabolism’ emerge as pivotal pathways. Dietary choices and external interventions significantly impact metabolic states [22]. Amino acids, integral to cellular signaling and nutrient metabolism, assume a central role. Lysine, for instance, an essential amino acid, requires dietary intake or supplementation. Similarly, arginine and citrulline intricately regulate hemodynamics, systemic homeostasis, and various physiological functions [23]. Tryptophan metabolism, governing the breakdown of tryptophan, is pivotal for serotonin and niacin synthesis, making it susceptible to dietary choices and hormonal fluctuations [24]. Additionally, ‘androgenic steroids’ in Fig. 1c orchestrate interactions with several pathways. Androgenic steroids, known to trigger hepatic injury with consequential effects on bile acid and benzoate metabolism, up-regulate bile acid synthesis genes and transporters, leading to cholestasis [25]. Steroid use also perturbs benzoate metabolism, which detoxifies dietary compounds.
Figure 3.

Biological results of dynamic component 1. (a) Metabolic pathway pairs associated with DC1. (b) Top biological processes of genes associated with significant SNPs. (c) Brief summary of the functions of key genes in the top biological processes based on Genecards [21].
In tandem with the metabolic network exploration, we delve into the associations between SNPs and metabolic pathways. All the significant SNPs are listed in Supplementary File 1. Here we focus our discussion on the biological interpretations of the genes linked to the SNPs significantly associated with DC1 by investigating the biological processes implicated by genes in the haploblocks of, or linked by GTEx database to the significant SNPs.
For DC1, there are 27 significant biological processes with 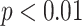 . Table 3b presents the top 5 biological pathways after manual removal of highly redundant ones with over 80% genes overlapping. The top biological processes include ‘negative regulation of viral processes’, ‘negative regulation of interleukin-8 production’, ‘glutamate biosynthetic process’, ‘MHC class I biosynthetic process positive adjustment’, and ‘neurotrophin production’. These terms intricately relate to pathways associated with immune system regulation, inflammation, type 2 diabetes, and development/maintenance of the nervous system. The regulation of viral processes is pivotal in thwarting viral replication within the body. The negative regulation of interleukin-8, a pro-inflammatory cytokine, tempers the extent of inflammatory responses. Proper control of interleukin-8 levels can mitigate chronic inflammation. The positive regulation of MHC class I biosynthetic processes enhances the machinery responsible for the production of MHC class I molecules, central to immune surveillance [26]. Glutamate, integral to energy metabolism, and its perturbation, associated with insulin resistance, have a profound link with type 2 diabetes. Insulin, which plays a pivotal role in immune cell function, is critical for antiviral T cell proliferation and cytokine production [27].
. Table 3b presents the top 5 biological pathways after manual removal of highly redundant ones with over 80% genes overlapping. The top biological processes include ‘negative regulation of viral processes’, ‘negative regulation of interleukin-8 production’, ‘glutamate biosynthetic process’, ‘MHC class I biosynthetic process positive adjustment’, and ‘neurotrophin production’. These terms intricately relate to pathways associated with immune system regulation, inflammation, type 2 diabetes, and development/maintenance of the nervous system. The regulation of viral processes is pivotal in thwarting viral replication within the body. The negative regulation of interleukin-8, a pro-inflammatory cytokine, tempers the extent of inflammatory responses. Proper control of interleukin-8 levels can mitigate chronic inflammation. The positive regulation of MHC class I biosynthetic processes enhances the machinery responsible for the production of MHC class I molecules, central to immune surveillance [26]. Glutamate, integral to energy metabolism, and its perturbation, associated with insulin resistance, have a profound link with type 2 diabetes. Insulin, which plays a pivotal role in immune cell function, is critical for antiviral T cell proliferation and cytokine production [27].
Recent studies underscore the active interplay between the immune system and systemic metabolism, influenced by endocrine factors. This interplay can disrupt blood sugar regulation and weaken the immune system’s response to infections [28]. Infection and inflammation contribute to the onset of type 2 diabetes, particularly in individuals with pre-existing metabolic dysfunction [27]. Notably, patients with type 2 diabetes exhibit heightened susceptibility to infectious diseases [29, 30].
Table 3c presents the functional descriptions of the genes involved in the top biological processes based on GeneCards [21]. The LY6E gene, linked to ‘negative regulation of viral processes’, exerts a critical influence on T cell physiology, tumorigenesis, and immune regulation. The research on LY6E has primarily focused on its role in virus-host interactions rather than involvement in metabolic regulation. Likewise, CIITA is the master regulator of class II major histocompatibility complex gene transcription. Their functions suggests a potential link between these genes and the ‘androgenic steroid’ metabolic pathway, which is prominent in 3(a), as androgens play a role in immune modulation [25]. CIITA interacts with HIC1 to repress SIRT1 transcription, leading to a skewed metabolic phenotype mimicking insulin resistance in response to interferon gamma (IFN-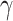 ) in skeletal muscle cells [31].
) in skeletal muscle cells [31].
The LARP7 gene is a major regulator of Pol II activity, which synthesizes mRNA [32]. In addition, studies have found that LARP7 can protect against heart failure by enhancing mitochondrial function [33], indicating its broader role in metabolic regulation.
The GLUD1 gene, partaking in ‘glutamic acid biosynthesis’, aligns with the ‘urea cycle’ metabolic pathway, impacting insulin homeostasis. Glutamate, a pivotal neurotransmitter, affects insulin regulation and glucose metabolism. Genes tied to insulin and glucose metabolism also connect with the ‘long-chain saturated fatty acid’ metabolic pathways, influencing energy utilization.
Proprotein convertase subtilisin/kexin 6 (PCSK6) is a key modulator in cardiovascular biology, including heart formation and lipoprotein metabolism [34]. It controls the function of smooth muscle cells in vascular remodeling [35]. Its function in enhancing inflammation can also affect metabolic balance [36].
Dynamic Component 3
Among the metabolic pathway pairs associated with DC3 shown in Fig. 4a, important pathways include ‘lysophospholipid’ and ‘phospholipid metabolism’. Additionally, we observe the relevance of ‘sphingomyelin’ and ‘phosphatidylcholine’. Lysophospholipids assume a vital role in phospholipid metabolism, serving as key intermediates in the biosynthesis of complex phospholipids. They actively participate in cell signaling, contribute to membrane structure, and facilitate lipid transport [37]. A robust connection exists between fatty acid metabolism and phospholipid metabolism, with fatty acids being fundamental components of phospholipids that constitute a crucial part of cell membranes [38]. Notably, phosphatidylcholine, the most abundant phospholipid in cell membranes, plays a pivotal role in regulating lipid and energy metabolism. Its synthesis is crucial for overall cell development and health. Alterations in phospholipid levels can significantly impact functional metabolism, including aspects like blood lipids and insulin resistance.
Figure 4.

Biological results of dynamic component 3. (a) Metabolic pathway pairs associated with DC3. (b) Top biological processes of genes associated with significant SNPs. (c) Brief summary of the functions of key genes in the top biological processes based on Genecards [21].
Furthermore, Fig. 4a highlights the changing relations between ‘hemoglobin and porphyrin metabolism’, ‘urea cycle’, and ‘lysophospholipids’. The hemoglobin and porphyrin metabolic pathways are intrinsically linked to heme synthesis, a critical component of hemoglobin responsible for oxygen transport in red blood cells. This complex metabolic process involves various steps, including porphyrin biosynthesis and iron binding, ensuring the efficient transport of oxygen in the bloodstream [39]. Additionally, it is essential to recognize the connection between blood oxygen delivery and the urea cycle. Notably, ornithine, an intermediate in this pathway, plays a vital role in the urea cycle, a crucial detoxification process converting ammonia into urea for excretion [40].
Table 4b presents top biological processes over-represented by the genes linked to significant SNPs associated with DC3. These terms encompass ‘cardiac muscle tissue morphogenesis’, ‘antigen processing and presentation of peptide antigens’, ‘muscle cell differentiation’, ‘positive regulation of supramolecular fiber organization’, and ‘T helper cell lineage commitment’. These biological processes are clearly focused on heart structure and function, as well as immune system regulation.
Proper cardiac muscle tissue morphogenesis is vital for the efficient functioning of the heart [41, 42]. Similarly, muscle cell differentiation is a critical function for the continuous renewal and repair of cardiac muscle tissue [43]. Supramolecular fiber organization pertains to the arrangement of protein fibers in the heart muscle. Proper regulation of this process ensures the correct alignment of these fibers, which is essential for effective heart contractions [44].
Immune cells, including macrophages and T cells, play essential roles in cardiac tissue repair and regeneration. They are instrumental in combating infections that may lead to conditions like endocarditis and myocarditis, thereby safeguarding the heart from long-term damage. On the other hand, an aberrant immune response can result in autoimmune diseases. Conditions such as systemic lupus erythematosus can affect heart structures and impede cardiac pumping capacity, potentially leading to myocarditis and related diseases [45].
Proper antigen processing and presentation also aid in maintaining immune system balance [46]. T-helper (Th) cells are immune cells that orchestrate immune responses. The balance of T-helper cell subtypes can influence cardiac inflammation and immune responses. Research indicates that T-helper cell polarization can serve as an indicator of the risk of cardiovascular diseases. Individuals with coronary artery syndrome and advanced atherosclerosis tend to exhibit a Th1 bias [47]. Patients with autoimmune diseases like asthma and systemic lupus erythematosus often show increased Th1/Th2 cell ratios [48].
Table 4c shows the summary of the selected genes involved in the top biological processes. FOXC1, HEY2, BMP10, and XIRP2, the first three of which are transcription factors, are involved in regulation of cardiac tissue morphogenesis. Beside their roles in cardiac development, which has a profound impact on the overall metabolic status, BMP signaling can influence the differentiation and function of adipocytes [49], affecting adipose tissue development and insulin sensitivity.
Among other examples, MTOR is a well known key regulator of metabolism [50]. Dysregulation in the mTOR signaling pathway can cause metabolic disorders such as obesity and type 2 diabetes. PYCARD is a major component of the NLRP3 inflammasome, which is directly linked to metabolic regulation in response to inflammation [51].
Dynamic Component 5
In the metabolic pathway pairs related with DC5 shown in Fig. 5a, the majority of the pathways are related to lipid metabolism. One of the highly connected pathway is ‘short-chain fatty acid’. Short-chain fatty acids are produced by anaerobic bacteria in the intestine through the fermentation of dietary fiber and peptides and are related to cellular energy metabolism [52]. Short-chain fatty acids are used by intestinal mucosal cells for energy, producing heat for the organism and acting as signaling molecules—an acetate of short fatty acids, which serves as a prerequisite for cholesterol synthesis [53].
Figure 5.

Biological results of dynamic component 5. (a) Metabolic pathway pairs associated with DC5. (b) Top biological processes of genes associated with significant SNPs. (c) Brief summary of the functions of key genes in the top biological processes based on Genecards [21].
Two other highly connected pathways are ‘phosphotidylcholine (PC)’ and ‘sphingomyelins’, both of which are critical to the structural integrity of cell membranes, and are connected with ‘short-chain fatty acid’. Sphingomyelin is the main sphingolipid in cell membranes, and its distribution is related to cholesterol distribution. The interaction between cholesterol and sphingomyelin results in sphingomyelin being one of the risk factors for cardiovascular development[54].
Another prominant pathway is ‘benzoate metabolism’, which is connected to ‘tryptophan metabolism’. Benzoate metabolism is a key factor in the detoxification of certain dietary compounds. Sodium benzoate, a type of benzoate, is one of the most commonly used food additives. It is conjugated with glycine to form hippuric acid, which is in turn excreted through the urinary system [55].
Table 5b presents top biological processes over-represented by the genes linked to significant SNPs associated with DC5. The top terms include ‘optic nerve development’, ‘positive regulation of steroid metabolic process’, ‘androgen receptor signaling pathway’, ‘coronary vasculature development’, and ‘muscle organ development’.
Steroids assume regulatory roles in various metabolic processes within the body, including glucose metabolism [56]. Disturbances in lipid metabolism and elevated glucose and insulin levels contribute to atherosclerosis development in type 2 diabetes patients [57]. In some cases, elevated free fatty acid levels suggest the onset of insulin resistance before hyperglycemia manifests [58]. Androgens, a subclass of steroids, also bear metabolic implications. Imbalanced androgen-to-estrogen ratios and low testosterone plasma concentrations correlate with an increased risk of type 2 diabetes and vascular disease [59]. Supplementing testosterone has been shown to improve the balance of glucose and lipid metabolism in the body [60].
The development of coronary vasculature and muscle system globally influence the metabolic status of the organism. Coronary vasculature development encompasses the formation and expansion of the coronary arteries and veins within the heart. This process is important in ensuring the heart muscle receives an adequate blood supply. Well-developed coronary arteries guarantee that the heart has ample nutrients for sustaining normal energy metabolism. Additionally, they are responsible for pumping blood within the heart muscle, making damaged coronary arteries a potential precursor to heart disease [61].
The term ‘optical nerve development’ does not appear straight-forward in its relation with metabolic regulation. However, research demonstrates that diabetes-induced vascular complications encompass cardiovascular disease and diabetic retinopathy [62]. Furthermore, patients with diabetes often exhibit abnormal eye blood vessels, rendering them susceptible to cardiovascular diseases [63]. Retinal dilation is a common occurrence in diabetes [64]. Concurrently, the influence of CVD and various metabolic disorders adversely affects retinal small blood vessels and capillaries, resulting in pathological changes [65].
Table 5c shows the summary of the selected genes involved in the top biological processes. EPHB1 plays an important role in nervous system development. It was also found to regulate cancer stem cell enrichment [66]. NAV2 is required for cranial nerve development, and blood pressure regulation in animal model [67]. It is also found to be involved in rheumatoid arthritis [68].
DAB2, FGF1, and TMF1 are associated with regulation of steroid metabolism and androgen signaling pathway. Interestingly, ‘androgen steroids’ is a metabolic pathway found in Fig. 5a, indicating a direct functional association at the gene level and the metabolite level. DAB2, in addition to its role in clathrin-mediated endocytosis, is implicated in circulatory cholesterol homeostasis [69]. FGF1 is known as an important regulator of lipid, bile acid, and carbohydrate metabolism [70]. The production of FGF1 is upregulated in adipose tissue in response to dietary stress, which is critical for adipose tissue plasticity [71]. TMF1 also plays a role in insulin-regulated glucose homeostasis [72].
Discussion
Uncovering the intricate genetic regulation of metabolism is pivotal to a deeper understanding of metabolic diseases. In the field of gene expression, ample evidence suggests that gene network dysregulation can result in shifts in correlations between gene pairs without affecting average expression levels [73]. Various methods have been developed to investigate differential correlations within observed [74, 75] or unobserved biological conditions [8, 76] that influence co-expression patterns. In the field of metabolomics, traditional metabolome-based genome-wide association studies (mGWAS) have effectively identified associations between individual metabolite abundance and specific single nucleotide polymorphisms (SNPs). These associations primarily focus on alterations in average metabolite levels. Nevertheless, studies in both domains are constrained as they struggle to fully capture the genome’s role in orchestrating the complex regulation of metabolic networks.
Our mdGWAS approach addresses these limitations by identifying genetic loci that regulate metabolic relationships. Unlike traditional mGWAS, which focuses on average metabolite levels, our method captures the genome’s influence on the relation between metabolites and metabolic pathways, providing a deeper understanding of metabolic regulation. Among the significant findings of regulators of metabolic coordination, several genes are known for their direct implications for type 2 diabetes (T2DM) and its complications. MTOR, a central regulator of insulin signaling and glucose metabolism, showed strong associations with metabolic dynamics. Its dysregulation is linked to key T2DM features such as insulin resistance, 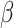 -cell dysfunction and impaired autophagy [77, 78]. Similarly, CIITA, involved in immune-metabolic interactions, was downregulated in diabetic patients [79], highlighting the complex interplay between immune function and metabolic regulation. Additionally, the identification of BMP10 and DAB2 further elucidates the connection between metabolic coordination and cardiovascular complications in diabetes [80–82].
-cell dysfunction and impaired autophagy [77, 78]. Similarly, CIITA, involved in immune-metabolic interactions, was downregulated in diabetic patients [79], highlighting the complex interplay between immune function and metabolic regulation. Additionally, the identification of BMP10 and DAB2 further elucidates the connection between metabolic coordination and cardiovascular complications in diabetes [80–82].
Our findings complement and extend previous metabolic genetic studies. For example, traditional mGWAS approaches have identified associations between SNPs and the levels of nine specific amino acids in METSIM [15], revealing five novel loci, including genes such as LOC157273/PPP1R3B, WWOX, LIPC, TRIB1, and ZFHX3. These findings highlighted the intricate relationship between lipid metabolism genes and amino acid levels, particularly in the context of insulin resistance and obesity. Our approach identifies a broader spectrum of metabolic regulation by identifying changing relationships. The overlap between our findings and known metabolic regulators, coupled with the discovery of novel associations, validates the biological relevance of our approach while expanding our understanding of metabolic network regulation. This suggests that integrating both mGWAS and mdGWAS approaches provides a more comprehensive view of the genetic architecture underlying metabolic diseases.
From a translational perspective, these findings offer several opportunities for experimental validation. Future functional studies in T2DM-relevant systems, such as  -cells, adipocytes, or animal models, could investigate how these genetic variants influence metabolite correlations and their effects on insulin sensitivity and metabolic homeostasis. Such studies would not only validate the biological significance of our findings but also potentially identify new therapeutic targets for diabetes and its complications.
-cells, adipocytes, or animal models, could investigate how these genetic variants influence metabolite correlations and their effects on insulin sensitivity and metabolic homeostasis. Such studies would not only validate the biological significance of our findings but also potentially identify new therapeutic targets for diabetes and its complications.
Key Points
Our framework is the first to analyze genetic influences on the changing correlation of metabolites and metabolic pathways.
The new framework detects genotypes that influence the correlation between groups of metabolites.
Our method enhances the understanding of genetic regulation of metabolism by finding SNPs associated with the second order relations between metabolic pathways.
Supplementary Material
Acknowledgments
The authors thank Professor Michael Boehnke for his invaluable guidance and support.
Contributor Information
Emily C Hector, Department of Statistics, North Carolina State University, Raleigh, NC 27695, United States.
Daiwei Zhang, Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, United States; Department of Biostatistics and Department of Genetics, University of North Carolina, Chapel Hill, NC 27599, United States.
Leqi Tian, School of Data Science, the Chinese University of Hong Kong, Shenzhen, Guangdong 518172, P.R.China.
Junning Feng, School of Data Science, the Chinese University of Hong Kong, Shenzhen, Guangdong 518172, P.R.China.
Xianyong Yin, Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, United States.
Tianyi Xu, School of Data Science, the Chinese University of Hong Kong, Shenzhen, Guangdong 518172, P.R.China.
Markku Laakso, School of Medicine, University of Eastern Finland, FI-70211 Kuopio, Finland.
Yun Bai, School of Medicine, the Chinese University of Hong Kong, Shenzhen, Guangdong 518172, P.R.China.
Jiashun Xiao, Shenzhen Research Institute of Big Data, Shenzhen, Guangdong 518172, P.R.China.
Jian Kang, Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, United States.
Tianwei Yu, School of Data Science, the Chinese University of Hong Kong, Shenzhen, Guangdong 518172, P.R.China.
Conflict of interest: The authors declare no competing interests.
Funding
This work was partially supported by NIH grants (R01DA048993, R01MH105561, R01GM124061, R01GM087964), NSF grant IIS2123777, Guangdong Talent Program (2021CX02Y145), Guangdong Provincial Key Laboratory of Big Data Computing, and Shenzhen Key Laboratory of Cross-Modal Cognitive Computing (ZDSYS20230626091302006).
Code availability
The R implementations of mdGWAS is available on GitHub https://github.com/Seven595/mdGWAS.
References
- 1. Luo J. Metabolite-based genome-wide association studies in plants. Curr Opin Plant Biol 2015;24:31–8. 10.1016/j.pbi.2015.01.006 [DOI] [PubMed] [Google Scholar]
- 2. Gieger C, Geistlinger L, Altmaier E. et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet 2008;4:e1000282. 10.1371/journal.pgen.1000282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Raffler J, Friedrich N, Arnold M. et al. Genome-wide association study with targeted and non-targeted NMR metabolomics identifies 15 novel loci of urinary human metabolic individuality. PLoS Genet 2015;11:1–28. 10.1371/journal.pgen.1005487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shin S-Y, Fauman EB, Petersen A-K. et al. An atlas of genetic influences on human blood metabolites. Nat Genet 2014;46:543–50. 10.1038/ng.2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou S, Kremling KA, Bandillo N. et al. Metabolome-scale genome-wide association studies reveal chemical diversity and genetic control of maize specialized metabolites. Plant Cell 2019;31:937–55. 10.1105/tpc.18.00772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Laye MJ, Tran V, Jones DP. et al. The effects of age and dietary restriction on the tissue-specific metabolome of drosophila. Aging Cell 2015;14:797–808. 10.1111/acel.12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whitfield Åslund ML, Simpson AJ, Simpson MJ. 1H NMR metabolomics of earthworm responses to polychlorinated biphenyl (PCB) exposure in soil. Ecotoxicology 2011;20:836–46. 10.1007/s10646-011-0638-9 [DOI] [PubMed] [Google Scholar]
- 8. Tianwei Y. A new dynamic correlation algorithm reveals novel functional aspects in single cell and bulk rna-seq data. PLoS Comput Biol 2018;14:e1006391. 10.1371/journal.pcbi.1006391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ho YY, Parmigiani G, Louis TA. et al. Modeling liquid association. Biometrics 2011;67:133–41. 10.1111/j.1541-0420.2010.01440.x [DOI] [PubMed] [Google Scholar]
- 10. Efron B. Large-scale simultaneous hypothesis testing: the choice of a null hypothesis. J Am Stat Assoc 2004;99:96–104. 10.1198/016214504000000089 [DOI] [Google Scholar]
- 11. Loh P-R, Tucker G, Bulik-Sullivan BK. et al. Efficient bayesian mixed-model analysis increases association power in large cohorts. Nat Genet 2015;47:284–90. 10.1038/ng.3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Devlin B, Roeder K. Genomic control for association studies. Biometrics 1999;55:997–1004. 10.1111/j.0006-341X.1999.00997.x [DOI] [PubMed] [Google Scholar]
- 13. Reich DE, Goldstein DB. Detecting association in a case? Control study while correcting for population stratification. Genet Epidemiol 2001;20:4–16. [DOI] [PubMed] [Google Scholar]
- 14. Lonsdale J, Thomas J, Salvatore M. et al. The genotype-tissue expression (gtex) project. Nat Genet 2013;45:580–5. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Teslovich TM, Kim DS, Yin X. et al. Identification of seven novel loci associated with amino acid levels using single-variant and gene-based tests in 8545 finnish men from the metsim study. Hum Mol Genet 2018;27:1664–74. 10.1093/hmg/ddy067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Little RJA, Rubin DB. Statistical Analysis With Missing Data. Hoboken, NJ: John Wiley & Sons, 2002. [Google Scholar]
- 17. Hrydziuszko O, Viant MR. Missing values in mass spectrometry based metabolomics: an undervalued step in the data processing pipeline. Metabolomics 2012;8:161–74. 10.1007/s11306-011-0366-4 [DOI] [Google Scholar]
- 18. Di Guida R, Jasper Engel J, Allwood W. et al. Non-targeted uhplc-ms metabolomic data processing methods: a comparative investigation of normalisation, missing value imputation, transformation and scaling. Metabolomics 2016;12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wei R, Wang J, Mingming S. et al. Missing value imputation approach for mass spectrometry-based metabolomics data. Nat Sci Rep 2018;8:1–10. 10.1038/s41598-017-19120-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bliss CI. Statistics in Biology; Statistical Methods for Research in the Natural Sciences. New York: McGraw-Hill Book Company, 1967. [Google Scholar]
- 21. Stelzer G, Rosen N, Plaschkes I. et al. The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics 2016;54:1–1. 10.1002/cpbi.5 [DOI] [PubMed] [Google Scholar]
- 22. Moszak M, Szulińska M, Bogdański P. You are what you eat—the relationship between diet, microbiota, and metabolic disorders—a review. Nutrients 2020;12:1096. 10.3390/nu12041096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Botchlett R, Lawler JM, Guoyao W. L-arginine and l-citrulline in sports nutrition and health. In: Bagchi D, Nair S, Sen CK (eds.), Nutrition and Enhanced Sports Performance. Academic Press, London, 2019, 645–652. 10.1016/B978-0-12-813922-6.00055-2. [DOI] [Google Scholar]
- 24. Platten M, Nollen EAA, Röhrig UF. et al. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 2019;18:379–401. 10.1038/s41573-019-0016-5 [DOI] [PubMed] [Google Scholar]
- 25. Petrovic A, Vukadin S, Sikora R. et al. Anabolic androgenic steroid-induced liver injury: an update. World J Gastroenterol 2022;28:3071–80. 10.3748/wjg.v28.i26.3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garrido F, Aptsiauri N. Cancer immune escape: Mhc expression in primary tumours versus metastases. Immunology 2019;158:255–66. 10.1111/imm.13114 [DOI] [PMC free article] [PubMed] [Google Scholar]
-
27.
Šestan M, Marinović S, Kavazović I. et al. Virus-induced interferon-
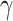 causes insulin resistance in skeletal muscle and derails Glycemic control in obesity. Immunity 2018;49:164–177.e6. 10.1016/j.immuni.2018.05.005
[DOI] [PubMed] [Google Scholar]
causes insulin resistance in skeletal muscle and derails Glycemic control in obesity. Immunity 2018;49:164–177.e6. 10.1016/j.immuni.2018.05.005
[DOI] [PubMed] [Google Scholar] - 28. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell 2015;160:816–27. 10.1016/j.cell.2015.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muller LMAJ, Gorter KJ, Hak E. et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis 2005;41:281–8. 10.1086/431587 [DOI] [PubMed] [Google Scholar]
- 30. Pearson-Stuttard J, Blundell S, Harris T. et al. Diabetes and infection: assessing the association with glycaemic control in population-based studies. Lancet Diabetes Endocrinol 2016;4:148–58. 10.1016/S2213-8587(15)00379-4 [DOI] [PubMed] [Google Scholar]
- 31. Fang M, Li P, Xiaoyan W. et al. Class II transactivator (CIITA) mediates transcriptional repression of pdk4 gene by interacting with hypermethylated in cancer 1 (HIC1). J Biomed Res 2015;29:308–15. 10.7555/JBR.29.20150055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hasler D, Meister G, Fischer U. Stabilize and connect: The role of LARP7 in nuclear non-coding RNA metabolism. RNA Biol 2021;18:290–303. 10.1080/15476286.2020.1767952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu H, Zhang F, Yan P. et al. LARP7 protects against heart failure by enhancing mitochondrial biogenesis. Circulation 2021;143:2007–22. 10.1161/CIRCULATIONAHA.120.050812 [DOI] [PubMed] [Google Scholar]
- 34. Qingyu W, Chen S. Proprotein convertase subtilisin/kexin 6 in cardiovascular biology and disease. Int J Mol Sci 2022;23:13429. 10.3390/ijms232113429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rykaczewska U, Suur BE, Röhl S. et al. PCSK6 is a key protease in the control of smooth muscle cell function in vascular remodeling. Circ Res 2020;126:571–85. 10.1161/CIRCRESAHA.119.316063 [DOI] [PubMed] [Google Scholar]
- 36. Testa G, Staurenghi E, Giannelli S. et al. Up-regulation of PCSK6 by lipid oxidation products: a possible role in atherosclerosis. Biochimie 2021;181:191–203. 10.1016/j.biochi.2020.12.012 [DOI] [PubMed] [Google Scholar]
- 37. Graber R, Sumida C, Nunez EA. Fatty acids and cell signal transduction. J Lipid Mediat Cell Signal 1994;9:91–116. [PubMed] [Google Scholar]
- 38. De Carvalho CCCR, Caramujo MJ. The various roles of fatty acids. Molecules 2018;23:2583. 10.3390/molecules23102583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lange H, Kispal G, Lill R. Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria. J Biol Chem 1999;274:18989–96. 10.1074/jbc.274.27.18989 [DOI] [PubMed] [Google Scholar]
- 40. Katunuma N, Okada M, Nishii Y. Regulation of the urea cycle and tca cycle by ammonia. Adv Enzyme Regul 1966;4:317–35. 10.1016/0065-2571(66)90025-2 [DOI] [PubMed] [Google Scholar]
- 41. Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 2005;6:826–35. 10.1038/nrg1710 [DOI] [PubMed] [Google Scholar]
- 42. Towbin JA, Jefferies JL. Cardiomyopathies due to left ventricular noncompaction, mitochondrial and storage diseases, and inborn errors of metabolism. Circ Res 2017;121:838–54. 10.1161/CIRCRESAHA.117.310987 [DOI] [PubMed] [Google Scholar]
- 43. Ballard VLT, Edelberg JM. Stem cells and the regeneration of the aging cardiovascular system. Circ Res 2007;100:1116–27. 10.1161/01.RES.0000261964.19115.e3 [DOI] [PubMed] [Google Scholar]
- 44. Bouten CVC, Dankers PYW, Driessen-Mol A. et al. Substrates for cardiovascular tissue engineering. Adv Drug Deliv Rev 2011;63:221–41. 10.1016/j.addr.2011.01.007 [DOI] [PubMed] [Google Scholar]
- 45. Atehortúa L, Rojas M, Vásquez GM. et al. Endothelial alterations in systemic lupus erythematosus and rheumatoid arthritis: Potential effect of monocyte interaction. Mediators Inflamm 2017;2017:1–12. 10.1155/2017/9680729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lleo A, Invernizzi P, Gao B. et al. Definition of human autoimmunity–autoantibodies versus autoimmune disease. Autoimmun Rev 2010;9:A259–66. 10.1016/j.autrev.2009.12.002 [DOI] [PubMed] [Google Scholar]
- 47. Olson NC, Sallam R, Doyle MF. et al. T helper cell polarization in healthy people: Implications for cardiovascular disease. J Cardiovasc Transl Res 2013;6:772–86. 10.1007/s12265-013-9496-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Misu T, Onodera H, Fujihara K. et al. Chemokine receptor expression on t cells in blood and cerebrospinal fluid at relapse and remission of multiple sclerosis: imbalance of th1/th2-associated chemokine signaling. J Neuroimmunol 2001;114:207–12. 10.1016/S0165-5728(00)00456-2 [DOI] [PubMed] [Google Scholar]
- 49. Qian S, Tang Y, Tang QQ. Adipose tissue plasticity and the pleiotropic roles of BMP signaling. J Biol Chem 2021;296:100678. 10.1016/j.jbc.2021.100678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev 2021;101:1371–426. 10.1152/physrev.00026.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019;19:477–89. 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Koh A, De Vadder F, Kovatcheva-Datchary P. et al. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 2016;165:1332–45. 10.1016/j.cell.2016.05.041 [DOI] [PubMed] [Google Scholar]
- 53. Zhou W, Cheng Y, Ping Zhu MI. et al. Implication of gut microbiota in cardiovascular diseases. Oxid Med Cell Longev 2020;2020:1–14. 10.1155/2020/5394096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Peter J, Slotte. Biological functions of sphingomyelins. Prog Lipid Res 2013;52:424–37. 10.1016/j.plipres.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 55. Badenhorst CPS, Erasmus E, Van der Sluis R. et al. A new perspective on the importance of glycine conjugation in the metabolism of aromatic acids. Drug Metab Rev 2014;46:343–61. 10.3109/03602532.2014.908903 [DOI] [PubMed] [Google Scholar]
- 56. Schiffer L, Barnard L, Baranowski ES. et al. Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: a comprehensive review. J Steroid Biochem Mol Biol 2019;194:105439. 10.1016/j.jsbmb.2019.105439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ng TW, Khan AA, Meikle PJ. Investigating the pathogenesis and risk of type 2 diabetes: clinical applications of metabolomics. Clinical Lipidology 2012;7:641–59. 10.2217/clp.12.75 [DOI] [Google Scholar]
-
58.
Bluher M, Kratzsch J, Paschke R. Plasma levels of tumor necrosis factor-
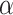 , angiotensin II, growth hormone, and IGF-I are not elevated in insulin-resistant obese individuals with impaired glucose tolerance. Diabetes Care 2001;24:328–34. 10.2337/diacare.24.2.328
[DOI] [PubMed] [Google Scholar]
, angiotensin II, growth hormone, and IGF-I are not elevated in insulin-resistant obese individuals with impaired glucose tolerance. Diabetes Care 2001;24:328–34. 10.2337/diacare.24.2.328
[DOI] [PubMed] [Google Scholar] - 59. Tramunt B, Smati S, Grandgeorge N. et al. Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 2020;63:453–61. 10.1007/s00125-019-05040-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yassin A, Haider A, Haider KS. et al. Testosterone therapy in men with hypogonadism prevents progression from prediabetes to type 2 diabetes: eight-year data from a registry study. Diabetes Care 2019;42:1104–11. 10.2337/dc18-2388 [DOI] [PubMed] [Google Scholar]
- 61. Obrist PA. Cardiovascular Psychophysiology: A Perspective. New York: Plenum Press, 1981. [Google Scholar]
- 62. Yousri NA, Suhre K, Yassin E. et al. Metabolic and metabo-clinical signatures of type 2 diabetes, obesity, retinopathy, and dyslipidemia. Diabetes 2022;71:184–205. 10.2337/db21-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang K, Cui L, Zhang G. et al. The jidong eye cohort study: objectives, design, and baseline characteristics. Eye Vis 2020;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stehouwer CDA. Microvascular dysfunction and hyperglycemia: a vicious cycle with widespread consequences. Diabetes 2018;67:1729–41. 10.2337/dbi17-0044 [DOI] [PubMed] [Google Scholar]
- 65. Østergaard L, Finnerup NB, Terkelsen AJ. et al. The effects of capillary dysfunction on oxygen and glucose extraction in diabetic neuropathy. Diabetologia 2015;58:666–77. 10.1007/s00125-014-3461-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang L, Peng Q, Xie Y. et al. Cell–cell contact-driven EphB1 cis- and trans-signalings regulate cancer stem cells enrichment after chemotherapy. Cell Death Dis 2022;13:980. 10.1038/s41419-022-05385-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McNeill EM, Roos KP, Moechars D. et al. Nav2 is necessary for cranial nerve development and blood pressure regulation. Neural Dev 2010;5:6. 10.1186/1749-8104-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang R, Li M, Ding Q. et al. Neuron navigator 2 is a novel mediator of rheumatoid arthritis. Cell Mol Immunol 2021;18:2288–9. 10.1038/s41423-021-00696-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tao W, Moore R, Meng Y. et al. Endocytic adaptors Arh and Dab2 control homeostasis of circulatory cholesterol. J Lipid Res 2016;57:809–17. 10.1194/jlr.M063065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Struik D, Dommerholt MB, Jonker JW. Fibroblast growth factors in control of lipid metabolism: from biological function to clinical application. Curr Opin Lipidol 2019;30:235–43. 10.1097/MOL.0000000000000599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gasser E, Sancar G, Downes M. et al. Metabolic messengers: fibroblast growth factor 1. Nat Metab 2022;4:663–71. 10.1038/s42255-022-00580-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rahimi R, Malek I, Lerrer-Goldshtein T. et al. TMF1 is upregulated by insulin and is required for a sustained glucose homeostasis. FASEB J 2021;35:e21295. 10.1096/fj.202001995R [DOI] [PubMed] [Google Scholar]
- 73. Narayanan M, Huynh JL, Wang K. et al. Common dysregulation network in the human prefrontal cortex underlies two neurodegenerative diseases. Mol Syst Biol 2014;10:743. 10.15252/msb.20145304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lai Y, Wu B, Chen L. et al. A statistical method for identifying differential gene-gene co-expression patterns. Bioinformatics 2004;20:3146–55. 10.1093/bioinformatics/bth379 [DOI] [PubMed] [Google Scholar]
- 75. McKenzie AT, Katsyv I, Song WM. et al. DGCA: a comprehensive R package for differential gene correlation analysis. BMC Syst Biol 2016;10:106. 10.1186/s12918-016-0349-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Li KC. Genome-wide coexpression dynamics: theory and application. Proc Natl Acad Sci U S A 2002;99:16875–80. 10.1073/pnas.252466999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Suhara T, Baba Y, Shimada BK. et al. The mtor signaling pathway in myocardial dysfunction in type 2 diabetes mellitus. Curr Diab Rep 2017;17:1–10. 10.1007/s11892-017-0865-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yuan T, Rafizadeh S, Gorrepati KDD. et al. Reciprocal regulation of mtor complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia 2017;60:668–78. 10.1007/s00125-016-4188-9 [DOI] [PubMed] [Google Scholar]
- 79. Panarina M, Kisand K, Alnek K. et al. Interferon and interferon-inducible gene activation in patients with type 1 diabetes. Scand J Immunol 2014;80:283–92. 10.1111/sji.12204 [DOI] [PubMed] [Google Scholar]
- 80. Jia-Ning G, Yang C-X, Ding Y-Y. et al. Identification of bmp10 as a novel gene contributing to dilated cardiomyopathy. Diagnostics 2023;13:242. 10.3390/diagnostics13020242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Besseling J, Kastelein JJP, Defesche JC. et al. Association between familial hypercholesterolemia and prevalence of type 2 diabetes mellitus. JAMA 2015;313:1029–36. 10.1001/jama.2015.1206 [DOI] [PubMed] [Google Scholar]
- 82. Fall T, Xie W, Poon W. et al. Using genetic variants to assess the relationship between circulating lipids and type 2 diabetes. Diabetes 2015;64:2676–84. 10.2337/db14-1710 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.