

Abstract
The prevalence of post-translational modification in nature suggests that expanding beyond the suite of functional groups found in the 20 canonical amino acids can impart specialized utility to peptide-based biomolecules. Thioamides, amidines, and heterocycles are three classes of such modifications that can act as peptide bond isosteres to alter the peptide backbone. Thioimidate protecting groups can address many of the problematic synthetic issues surrounding installation of these groups. Historically, amidines have received considerably little attention in peptides due to limitations in methods to access them. The first robust and general procedure for the introduction of amidines into peptide backbones exploits the utility of thioimidate protecting groups as a means to side-step reactivity that ultimately renders existing methods unsuitable for the installation of amidines along the main-chain of peptides. Further, amidines formed on-resin can be reacted to form (4H)-imidazolone heteorcycles which have recently been shown to act as amide isosteres. General methods for heterocyclic installation capable of geometrically restricting peptide conformation are also under-developed. This work is significant because it describes a generally applicable and divergent approach to access unexplored peptide designs and architectures.
Keywords: Thioimidate, Thioamide, Amidine, Imidazolone Peptide, Isostere, Solid-phase, Backbone
1. Introduction
The installation of peptide bond isosteres in place of natural amide bonds is one avenue to interrogate interactions along the peptide backbone.[1] Aside from the ester functional group in depsipeptides, thioamides and amidines are the only isosteres that attempt to replicate, in an altered fashion, the hydrogen-bond donating and accepting ability of the amide. The thioamide has been known for some time, but synthetic access to this isostere is far from general. In contrast, the amidine has received almost no attention in peptides. Additionally, Nature has mimicked the electronics of the amide bond through the enzymatic installation of heterocycles along the peptide backbone which are less prone to proteolytic degradation. Imidazolones have recently emerged as a non-aromatic heterocycle that mimics the electronics of the amide bond with more favorable pharmacological properties.[2] The procedures below document how three isosteres of the amide bond—thioamides, amidines, and imidazolone heterocycles—can be accessed through the thioimidate functional group. Critically, these methods are compatible and readily implemented within the work-flow of standard solid-phase peptide synthesis (SPPS), which should facilitate their implementation.
2. Thioamide Peptide-Bond Isosteres
Thioamides have emerged as versatile tools for probing interactions in peptides including hydrogen-bonding,[3–6] hydrophobicity,[7, 8] and stereoelectronic effects.[9–14] Thioamides also display photophysical behavior that allows them to operate as conformational photoswitches[15, 16] or as efficient quenchers of fluorescent dyes in experiments designed to study proteolysis and elucidate peptide conformation.[17–25] Additionally, they can be exploited for a variety of site-specific reactions within peptide-based substrates.[26–31] Collectively, these reports create a perception that current methodologies for thioamide incorporation into peptides have reached a state of adequacy, but thioamides are far from a plug-and-play probe that can be inserted into any peptide sequence of interest.[32] While thionopeptides have been known for some time, their synthesis is far from straight forward and many syntheses fail due to sequence dependent side reactions. Indeed, the idiosyncratic nature of thionopeptide synthesis can lead many researchers to abandon such projects and any information these structural probes would provide.
Below, we document side reactions of thioamides at different stages of solid-phase peptide synthesis and demonstrate how protection of thioamides as thioimidates can resolve synthetic pitfalls. Importantly, thioimidates can be easily incorporated with little modification of the standard SPPS work-flow, and do not require implementation of non-standard (and non-commercially available) N-terminal and side chain protection chemistries,[15, 33, 34] providing a comprehensive solution to the vagaries of thionopeptide synthesis.
2.1. Thioamide during Fmoc-deprotection
The benefits of thioimidate protection of thioamides can be most readily understood by considering the Fmoc-deprotection step in the SPPS cycle (Figure 2). Thioamides can be coupled into peptides using pre-formed thioacyl reagents (residue 5 in Figure 2, which were originally developed by Rapaport and coworkers[35] and later refined by Chatterjee and coworkers.[36] This chemistry has been reviewed in this journal within the last few years,[37] and procedures for their use are described in section 6.2 below. Following installation of the thioamide, peptides are elongated, one amino-acid residue at a time, through the sequential coupling of Fmoc-protected amino acids to the N-terminal amine of the resin-bound peptide, followed by removal of the Fmoc protecting group to reveal the newly coupled amine to repeat the cycle again.
Figure 2:
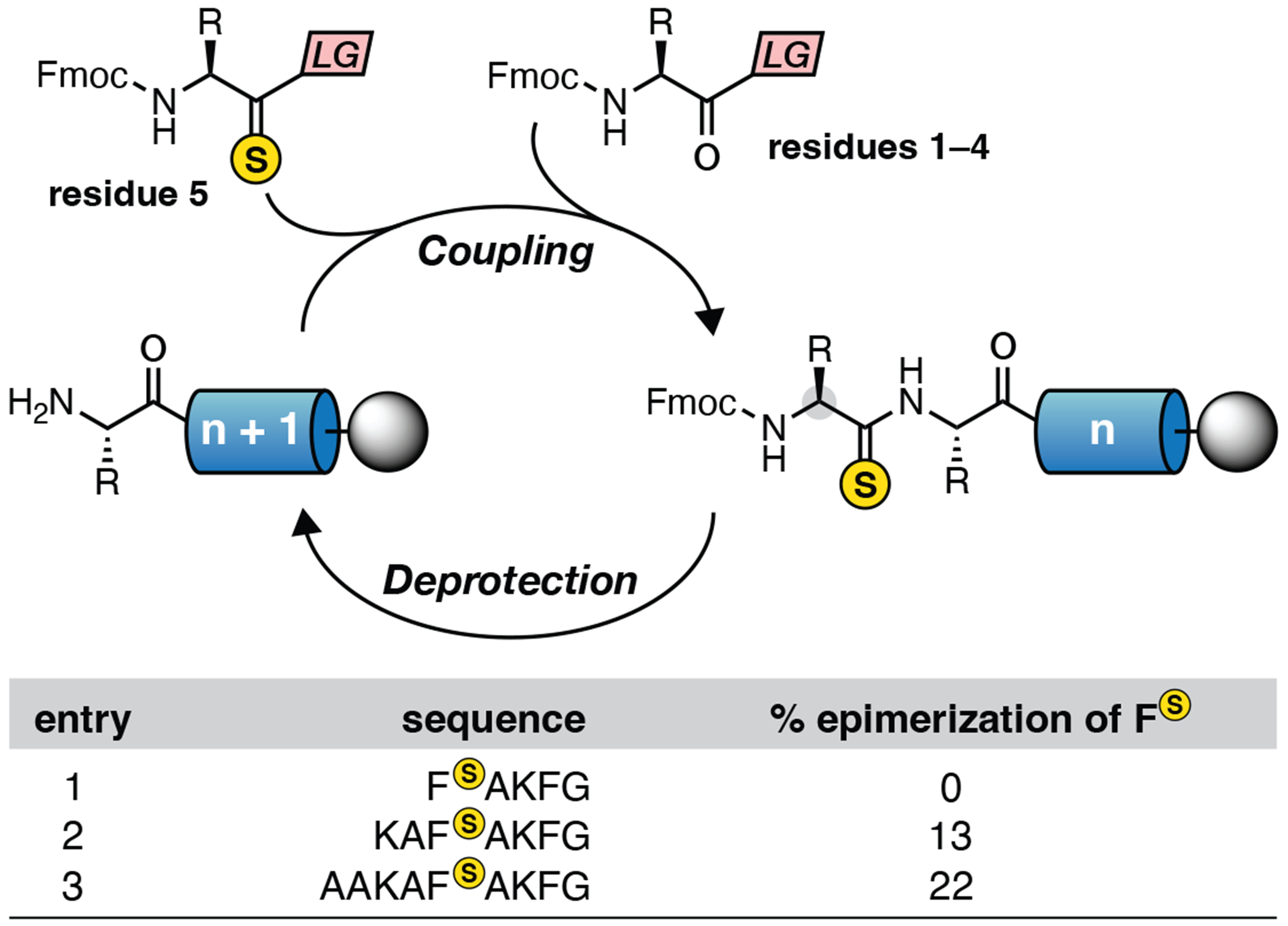
Stereochemical fragility of thioamide residues during SPPS
In the context of oxoamides, stereochemically pure peptides between 50–100 amino acids can be obtain via SPPS. With thioamides, however, the strong piperidine base used for Fmoc deprotection leads to epimerization of the α-carbon stereochemistry of the thioamide residue (entry 2, Figure 2), which continues to erode as the peptide is elongated (entry 3).[38–41]
To resolve this synthetic pitfall, we sought to directly address the mechanism of racemization. Namely, the acidity of the thioamide α-CH leads to deprotonation by piperidine and loss of stereochemical integrity. The increased α-CH acidity is due to the weaker C=S bond and lower energy π*C=S orbital that is able to stablize the α-anion of the conjugate base.[40] While different basic conditions have been developed to circumvent racemization,[38, 39] abbreviated deprotection times increase the risk of incomplete Fmoc deprotection and amino acid deletions upon subsequent coupling cycles. Alternatively, reversible protection of the thioamide as a thioimidate alters the key orbital that determines the acidity of the α-CH (a higher energy π*C=N), and provides robust protection of stereochemistry during peptide elongation.[40] As an example, the thioimidate protection-deprotection strategy in Figure 3 was used to synthesize the stereochemically pure peptide AAKAFSAKFG, where significant epimerization (22%) was observed if the thioamide was left unprotected (entry 3).[40, 41]
Figure 3:
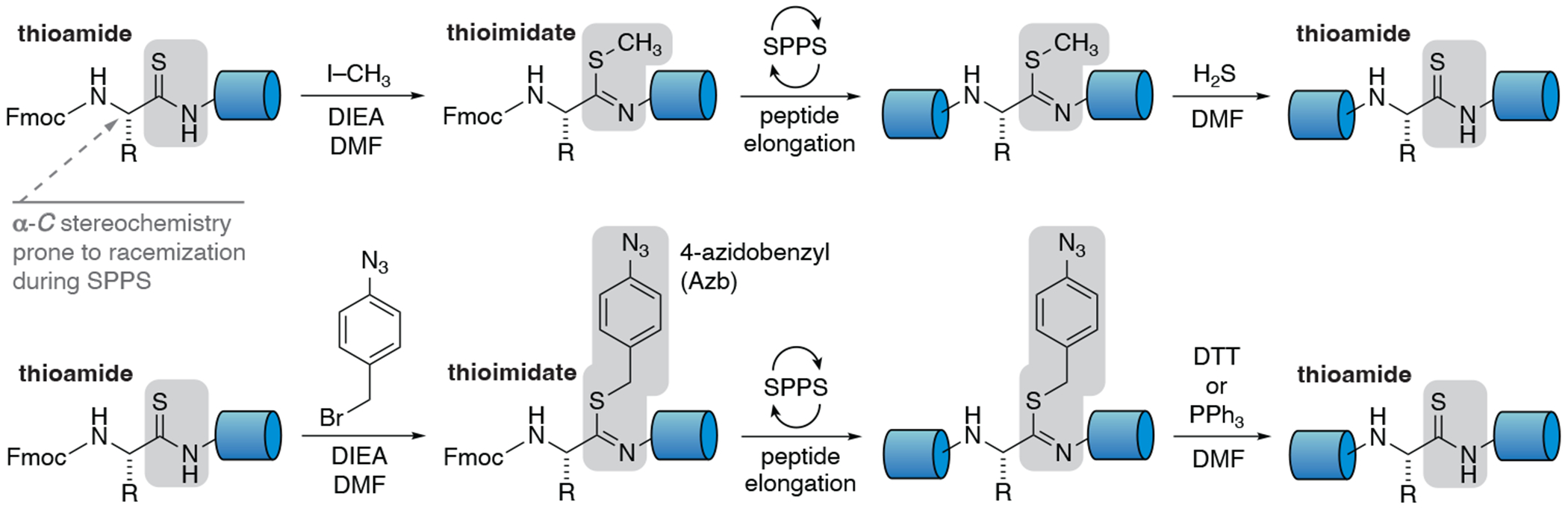
Reversible Protection of Thioamide Residue Stereochemistry with Thioimidate[40, 41]
Methods to employ thioimidates in this manner to circumvent the stereochemical fragility of thioamides during peptide elongation can be found in section 6.3 below. After a thioamide has been coupled into the peptide, prior to subsequent elongation, the thioamide can be protected as the thioimidate through alkylation (Figure 3). The process is compatible with standard SPPS conditions and apparatus. Once protected, peptide elongation can proceed as normal. After the sequence is complete, the thioamide can either be be restored prior to cleavage and deprotection from resin or after liberation from the resin. Section 2.3 discusses reactivity that suggests deprotection after cleavage can be beneficial in certain instances. Protection is completely reversible and conditions for deprotection depend on the identity of the alkyl group of the thioimidate (Figure 3).
2.2. Thioamide during peptide cleavage
The strong acid conditions (typically TFA) necessary for side-chain deprotection and cleavage of the final peptide can induce thioamides to attack the adjacent amide, n+1 amino acids along the chain, resulting in scission of the final peptide strand (Figure 4).[37, 42] These chain-scission products can often dominate unless significant optimization of the cleavage cocktail is undertaken.[42]
Figure 4:

Acid-induced chain scission of thionopeptides during cleavage
The propensity of the nucleophilic thioamide to cyclize with the n+1 residue is also problematic when a thioamide is the second residue from the C-terminus (Figure 5).[37] Protonation of the amide/acid attached to the resin is a pre-requisite for liberation from the resin. This event would also entices attack by the preceding thioamide, leading to epimerization of the C-terminal residue (Figure 5).
Figure 5:

Epimerization of the C-terminal residue is a particular risk when a thioamide is present in the pentultimate residue.
Protection of the thioamide as a thiomidate can circumvent these processes because the sulfur atom of the thioimidate is not nucleophilic. Indeed, the thioimidate is more basic than the thioamide (because of the =N– site) leading to protonation under the acidic conditions of cleavage and eliminating any nucleophilic behavior.[43] Thus, isolation of thionopeptides can be improved by cleaving the peptide (and side-chain protecting groups) from the resin with TFA while leaving the thioimidate protecting group in place (Figure 6). Once TFA is removed, deprotection to reveal the final thioamide can be accomplished using the deprotection reagents discussed as part of section 2.1 and Figure 2 above.
Figure 6:

Thioimidate protection preserves thioamides through elongation and resin cleavage
The only concern with thioimidates for these purposes is that they are prone to hydrolysis if water is present in the TFA cleavage cocktail.[44, 45] Mixtures of 3:1 TFA:DCM were found to provide the best yield of side-chain deprotection and cleavage from resin (>90%). Thioimidates protected with benzyl groups gave the highest yields of peptide with minimal hydrolysis products.[46]
Methods to protect thioamides during cleavage from the solid support can be found in section 6 below.
3. Amidine Peptide-Bond Isosteres
The amidine peptide-bond isostere, in which the oxygen of the amide is swapped for an NH group, has received considerably less attention relative to other amide-bond mimics.[1] The amidine can introduce substantial changes to the properties of the peptide (e.g. basicity, charge, hydrogen-bonding). Indeed, the mutable hydrogen-bonding characteristics of amidines were exploited by Boger and coworkers to combat the key mechanism of resistance that gram-positive bacteria exploit to counter antibiotics based on vancomycin.[30, 47–51] The unique properties of amidines may be a reason why nature evolved biosynthetic pathways for their installation,[52, 53] and amidines have even been proposed as prebiotic building blocks.[54, 55]
There is a surprising lack of examples of amidines in peptides, however, perhaps due to a lack of general methods to access them.[56, 57] Although the thioamide can itself serve as a reactive handle for amidine installation,[30, 31], it is prone to side reactions that make amidine insertion directly from the thioamide impossible for linear peptides.[26, 58] Driven by this gap in knowledge, we developed the first general method to install an amidine in place of the traditional amide functional group along the peptide backbone.[26] The new method exploits the site-selective installation of thioimidates, in which the protonation of the thioimidate provides a reactive site suitable for conversion into amidine. Because thioimidates are already a desirable protecting group for thioamides during peptide coupling and elongation (see section 2 above), further chemistry to elaborate these groups into a host of diverse amidine peptides expands the synthetic utility of thioimidates.
The amidine can be installed on-resin and is compatible with the strong acid conditions associated with removal of side-chain protecting groups and cleavage from resin. Methods to protect thioamides during cleavage from the solid support can be found in section 7 below.
4. 4H-Imidazolone Peptide-Bond Isosteres
Single-atom isosteres are not the only substitutions capable of mimicking the electronics and activity of the native amide bond.[59] Nature has evolved methods to install heterocycles, commonly oxazoles and thiazoles (derived from Ser/Thr and Cys residues respectively) into the backbone of peptides.[60] These modifications are capable of tuning the conformational behavior and enhancing the proteolytic stability of the peptide products.[60–62] Indeed, synthetic chemists have mimicked this approach in order to stabilize peptide-based therapeutics. Nearly all substitution patterns of 5-membered ring heterocyles found in Nature are in the form of 1,3-substitutions, which mimic the prototypical trans-amide conformation (Figure 8 left).[60, 63, 64]
Figure 8:

Heterocyclic motifs along the peptide backbone can restrict the amide bonds into either all-trans-amide or all-cis-amide conformations. Nature utilizes enzymatic cyclization to yield all-trans heterocycles, while naturally-occurring all-cis-inducing heterocycles remain unknown with unreliable methods for their synthesis.
Interestingly, methods to synthesize heterocycles capable of geometrically locking the cis-amide conformation (which exists only 0.2% of the time in peptides[65] at room temperature) have been under-developed, isolated to triazole and tetrazole examples synthesized as dipeptides and tripeptides in solution phase (Figure 8 right) .[66–68] Attempts to install these cis-amide locked heterocycles during the course of SPPS provides unreliable yields[69, 70], which has greatly limited their utility and application in the study amide bond conformation in peptides.[71]
Recently, 4(H)-imidazolones have been shown to be potent non-aromatic bioisosteres of the traditional amide bond[2], capable of retaining and potentially improving pharmacological properties. Additionally, imidazolones are featured in a variety of bioactive and agrochemical compounds.[72–78] Thioimidates provide an avenue to access these important heterocycles in peptides and, notably, permit unique access to α-chiral imidazolones, which are generally not possible by traditional imidazolone formation methods.[79]
Thioimidates in peptides can be converted into imidazolones through a cyclization cascade initiated by amidine formation. If the n+1 residue to the amidine is an ester, subsequent 5-exo-trig cyclization onto the ester leads to the final imidazolone, (see Figure 9) and locking in a cis-amide-type conformation.[79]
Figure 9:

Activation of thioimidate-precursor with acid enables formation of a transient amidine capable of cyclization to 4(H)-imidazolone locked cis-amide bioisostere during SPPS.[79]
The thioimidate necessary for the transformation can be derived from any commercial Fmoc-protected amino acid in 3 steps (1 chromatographic step) enabling ’plug-and-play’ insertion at any amide position in the peptide of interest (see Section 9.1 below). Disubstitution at the 4-position of the imidazolone is required due to the aromatic imidazole tautomer which epimerizes any stereochemistry at this position. Imidazolones are known to epimerize at the exocyclic α-position (position highlighted in red in Figure 9) under acidic conditions[80], however have been shown to be stable to repetitive Fmoc- deprotections using 20% piperidine. Optimized conditions for installation and acidiolytic resin cleavage (see Section 9.2 and 9.3 below) ensure that epimerization of the α-C stereochemistry is minimized (<10%).[79]
5. Materials
Fmoc-protected amino acids, all reagents, and solvents were commercially available and used without further purification. Fmoc-protected thioaminoanilides and thiobenzotriazolides (thioacylation reagents) were synthesized according to literature procedures.[35–37] Solvents indicated as dry were prepared by exposure to either activated molecular sieves (4Å) or dried via a Glass Contours Inc. solvent purification system.
General Supplies for Solid-Phase Peptide Chemistry:
2-Chlorotrityl resin (CTC; 200-400 mesh; Chem-Impex, Cat No. 33310)
Rink amide MBHA resin (Supplier Protein Technology, Cat No. RAM-25-LL)
Wang resin (100-200 mesh; Chem-Impex, Cat No. 01927)
Peptide Synthesis Vessel (Chemglass, Cat No. CG-1866)
Fmoc- protected amino acids (Chem-Impex)
α-Aminoisobutyric acid methyl ester hydrochloride (HCl●NH2-Aib-OMe;Chem-Impex, Cat No. 14869)
O-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU; Chem-Impex, Cat No. 12881)
3-(3-Dimethylaminopropyl)-1-ethyl-carbodiimide hydrochloride (EDC●HCl; Chem-Impex, Cat No. 00050)
N-methylmorpholine (NMM; Chem-Impex, Cat No. 02333)
N,N’-Diisopropylethylamine (DIEA; Chem-Impex, Cat No. 00141)
Piperidine (Chem-Impex, Cat No. 02351)
Triethylamine (TEA; Sigma-Aldrich, Cat No. 471283)
Acetic anhydride (Ac2O; Fisher Scientific, Cat No. A10-500)
General Supplies for Solution-Phase Chemistry:
Lawesson’s reagent (LR; Chem-Impex, Cat No. 26927)
Iodomethane (MeI; Fisher Scientific, Cat No. AC122375000)
Tetrabutylammonium iodide (TBAI; Sigma-Aldrich, Cat No. 140775)
p-Toluidine (Sigma-Aldrich, Cat No. 461121)
Sodium nitrite (NaNO3; Fisher Scientific, Cat No. S347-250)
Sodium azide (NaN3; Chem-Impex, Cat No. 29961)
2,2′-Azobis(2-methylpropionitrile) (AIBN; Sigma-Aldrich, Cat No. 441090)
N-Bromosuccinimide (NBS; Sigma-Aldrich, Cat No. B81255)
Peptide Cleavage and Thioimidate Deprotection:
Trifluoroacetic acid (TFA; Chem-Impex, Cat No. 02883)
Triisopropylsilane (TIPS; Chem-Impex, Cat No. 01966)
1,4-Dithio-DL-threitol (DTT; Chem-Impex, Cat No. 00127)
Solvents:
Diethyl ether (Et2O; Fisher Scientific, Cat No. AC615080040)
Methylene chloride (DCM; Fisher Scientific, Cat No. D37-4)
N,N-Dimethylformamide (DMF; Fisher Scientific, Cat No. D119-4)
Acetonitrile (MeCN; Fisher Scientific, Cat No. A998-4)
Tetrahydrofuran (THF; Fisher Scientific, Cat No. T397-500)
2,2,2-Trifluoroethanol (TFE; Fisher Scientific, Cat No. AC139755000)
Acetic acid (AcOH; Fisher Scientific, Cat No. A38C-212)
General Supplies:
Analytical Thin-Layer Chromatography Silica Plates, Aluminum-Backed (200 μm, 20 x 20 cm, F254; Silicycle, Cat No. TLA-R10011B-323)
Micro Bio-Spin Columns (Small Cleavage Funnels, Bio-Rad, Cat. No. 732-6204)
Poly-Prep Chromatography Columns (Large Cleavage Funnels, Bio-Rad, Cat. No. 731-1550)
Eppendorf 50 mL Conical Tubes (Falcon/Centrifuge Tube, Eppendorf, Cat. No. 0030122178)
Eppendorf Safe-Lock Tubes 2.0 mL (microcentrifuge tubes; Eppendorf, Cat. No. 0030123620)
6. Protocol for Synthesis of Protected Thionopeptides
In general, the thioimidate should be in place prior to any treatment of the peptide with Fmoc-deprotection conditions (i.e. piperidine) in order to preserve the α-CH stereochemistry of the amino acid bearing the intended thioamide.[40] Thioimidates derived from 4-azidobenzyl are the preferred method for the synthesis of thionopeptides because of the ease and safety of deprotection.[41] Methyl thioimidates require H2S (a potential safety concern) to unveil the final thioamide and are generally less stable to cleavage conditions with higher amounts of TFA. However, methyl thioimidates are useful for insertion of amidines via on-resin functionalization (see section 7). Note: Thioimidates cannot be formed preceding a proline (XSR-Pro) residue because the tertiary amide group can not be alkylated.
6.1. General Solid-Phase Peptide Synthesis Protocol
Transfer resin of choice (CTC, Wang, or Rink Amide) into a Chemglass peptide synthesis vessel, and swell resin in dry DMF (5-10 mL) with N2 agitation (bubbling) for 10 minutes. Drain solution from vessel.
Fmoc deprotection is affected by agitating the resin in deprotection cocktail containing 20% piperidine in DMF (v/v). Initial deprotection is performed for 2 minutes, drained, and a second deprotection for 8 minutes before draining and rinsing the resin (5 x 2 mL).
A pre-activated solution containing Fmoc amino acid (Fmoc-Xaa-OH, 5 eq. to resin loading) with HATU (4.9 eq. to resin loading) and NMM (10 eq. to resin loading in dry DMF is added to the synthesis vessel. The coupling reaction is agitated for 30 minutes with N2 before draining and rinsing the resin (3 x 2 mL)
All following Fmoc deprotections, couplings, and washes were performed in the same manner.
Note: Automated SPPS using commercial microwave peptide synthesizers can be used to synthesize the oxoamide fragment up to the thioamide residue installation. Incorporation of thioamides/thioimidates on resin using microwave synthesis has not been tested and their stability to microwave heating is unknown.
6.2. Synthesis & Resin Installation of Thionopeptides via Thiobenzotriazolides

Fmoc-protected thioaminoanilides and thiobenzotriazolides (thioacylation reagents) can be synthesized according to literature procedures. Such syntheses and subsequent installation on-resin during peptide chain-growth has been explained in greater detail by Chatterjee and co-workers in separate publications.[36, 37]
6.3. Protection of Thionopeptides via Resin Alkylation

Acylation with acetic anhydride prior to alkylation is suggested to truncate un-reacted −NH2 groups on the resin from incomplete thioacylation.
Resin is transferred to a polypropylene cleavage funnel.
-
A fresh alkylation cocktail (2 mL) is prepared.
For p-Azidobenzyl thioimidates: DIEA (0.1 M), p-Azidobenzyl bromide (see synthesis below, 0.5 M), and TBAI (0.01 M) in dry DMF.
For Methyl thioimidates: DIEA (0.1 M), and MeI (0.5 M) in dry DMF.
The resin is swelled in the alkylation cocktail of choice, capped, and agitated on a rotary mixer for 6 hours.
Alkylation mixture is drained and another fresh alkylation cocktail was prepared and added.
Resin is capped again and agitated via rotary mixer for an additional 12 hours.
To validate complete conversion of thioamide, a small amount of resin (ca. 10 mg resin) is added to a small polypropylene cleavage funnel, and subjected test cleavage in TFA/DCM/TIPS (75:20:5) for 30 minutes.
Cleavage cocktail is drained into a microcentrifuge tube, dried to a thin film under a stream of N2, and resuspended in minimal amount of H2O/MeCN (v/v) and analyzed via HPLC-MS. Presence of any thioamide in the LC trace is indicative of un-reacted thioamide on resin and the resin should be subjected to steps 5-7 until no thioamide presence is detected.
Note: Significant thioester or thiazolone–indicative of the presence of thioimidate or thioamide respectively–may be detected during test cleavage. These side products are addressed in final cleavage protocol (see below).
6.4. Synthesis of p-Azidobenzyl Bromide

p-Azidobenzyl bromide was prepared according to previous literature procedures.[81]
p-Toluidine (3.00 g, 1 eq., 28.0 mmol) is dissolved in 5 M HCl (20 mL) at 0°C with stirring.
A solution of sodium nitrite (2.12 g, 1.1 eq., 30.8 mmol) in H2O (5 mL) is added very slowly with stirring at 0°C for 30 min.
A solution of sodium azide (2.00 g, 1.1 eq., 30.8 mmol) in H2O (5 mL) is then added very slowly. Reaction was allowed to warm to room temperature and is stirred for 1 hour.
Reaction is quenched via careful addition of saturated aqueous NaHCO3.
DCM (20 mL) is then added, the mixture poured into a separatory funnel, and layers separated and extracted with DCM (2 x 25 mL). Combined organic layers are then washed with water (20 mL), dried over Na2SO4, and concentrated in vacuo to afford 1-azido-4-methylbenzene (3.50 g, 26.3 mmol, 94%) as a brown oil which is used without further purification.
To an oven-dried round bottom flask equipped with a water-cooled reflux condenser and stirring under N2, 1-azido-4-methylbenzene (3.50 g, 1 eq., 26.3 mmol) is dissolved in benzene (30 mL).
AIBN (1.29 g, 0.3 eq., 7.89 mmol) and freshly recrystallized NBS (5.61 g, 1.2 eq., 31.5 mmol) is added under N2 blanket. Reaction vessel should be covered in aluminum foil to prevent photodegradation.
Reaction is heated at reflux with stirring for 18 hours with monitoring by TLC. In the event of incomplete conversion, a small amount of AIBN (100 mg) is added and reaction heated at reflux for an additional 4 hours.
The mixture is then cooled to room temperature and poured over 30 mL water, then extracted with diethyl ether (2 x 20 mL).
Combined organics are dried over Na2SO4, and concentrated in vacuo. The crude residue is purified via column chromatography in 100% hexanes to yield 1-azido-4-(bromomethyl)benzene (3.71 g, 17.5 mmol, 66.6 %) as a light brown oil which is stored in a dark freezer (for up to 3 months) to prevent degradation of the azide.
7. Protocol for Cleavage & Deprotection of Protected Thionopeptides
After a thioimidate peptide has been installed on-resin, either by alkylation of thioamides or direct thioimidate dipeptide coupling, Fmoc SPPS can be continued as usual until completion of the peptide. Due to thioimidate protection, no modifications of the Fmoc-deprotection conditions are required (standard 20% piperidine in DMF (v/v)) regardless of thioimidate identity.[40, 41]
Methyl thioimidates have variable–often extremely poor–resistance to hydrolysis during high % TFA cleavage and are best utilized for on-resin amidine transformations (see protocol for formation of amidinopeptides).[26, 40] 4-azidobenzyl thioimidates should be cleaved from resin taking special care to preserve the thioimidate as much as possible, and then subsequently deprotected (in solution) to the thionopeptide using the protocol below.
7.1. Resin Cleavage of Protected Thioimidate Peptides

Completed on-resin thioimidate peptides are washed thoroughly with dry DCM (3 x 3 mL) and dried with N2 flow, and transferred to a large cleavage funnel.
- Determination of a reasonable cleavage cocktail by analysis of side-chain protecting groups, thioimidate identity, and resin linker is critical to their isolation. Some specific considerations:
- 4-Azidobenzyl thioimidates: Utilize 75% TFA and anhydrous, non-reducing scavengers such as phenols, TIPS, anisole, or m-cresol.
- Cys/Met residues with 4-Azidobenzyl thioimdiate: Check for oxidation; DTT reduction (required for 4-Azidobenzyl deprotection, see below) may reduce Cys disulfides or Met(O). If a thiol scavenger is required, DTT is preferred for retention of azides on resin.[82]
- Methyl thioimidate: Not recommended – should be used for functional group transformations on-resin. In the event they must be used, special care should be taken to reduce TFA exposure due to high propensity to hydrolyze. Utilize low-acid cleaving resins (such as CTC or Sieber resins) if possible.
Prior to complete peptide cleavage, a test cleavage should be performed. A small amount of resin (ca. 10 mg resin) is added to a small polypropylene cleavage funnel, and subjected to cleavage conditions determined above for 1 hour.
Cleavage cocktail is drained into a microcentrifuge tube, dried to a thin film under a stream of N2, and resuspended in minimal amount of H2O/MeCN (v/v) and analyzed via HPLC-MS to determine the efficacy of the cleavage cocktail used.
Repeat previous steps if thioimidate does not survive cleavage conditions (indicated by large portion of corresponding thioester in the LC trace)
Once ideal resin conditions have been determined by test cleavage, the resin is swelled in enough cleavage cocktail that it is free-flowing, capped, and agitated for 1-2 hours with gentle rocking.
The solution is then drained into a centrifuge tube and evaporated under a stream of N2 gas.
The crude film is precipiated by addition of one 50 mL portion of very cold diethyl ether (cooled on dry ice for 30 minutes), and pelleted by centrifugation (10,000 rpm, 5 min).
The supernatant diethyl ether is discarded and the previous step repeated with another 50 mL portion of very cold diethyl ether.
The supernatant is again discarded, and the pelleted peptide is allowed to dry in air. Thioimidates generally may degrade on exposure to water, and 4-azidobenzyl thioimidates may degrade on exposure to light. Deprotection of the thioimidate should be immediately affected or the material should be stored in the freezer in the dark until deprotection.
7.2. Solution-Phase Deprotection of p-Azidobenzyl Thioimidates

Take care to use anhydrous reagents when manipulating cleaved thioimidate peptides. Residual TFA from resin cleavage can affect acidic hydrolysis of thioimdiates.
To the crude 4-azidobenzyl thioimidate peptide pellet is added DTT (0.5 M) and DIEA (0.1 M) in dry DMF (2 mL).
The mixture is allowed to stand for 2 hours with occasional agitation.
Deprotection progress is monitored using LC-MS.
The deprotected thionopeptide is precipitated again by addition of one 50 mL portion of very cold diethyl ether (cooled on dry ice for 30 minutes), and pelleted by centrifugation (10,000 rpm, 5 min).
The supernatant diethyl ether is discarded and the previous step repeated with another 50 mL portion of very cold diethyl ether.
The supernatant is again discarded, and the pelleted peptide is allowed to dry in air.
Crude thionopeptide is then purified using preparatory reverse-phase HPLC.
8. Protocol for Amidinopeptide Synthesis via Thioimidates

8.1. Resin Conversion of Thioimidates into Amidines
Transfer resin containing completed methyl thioimidate peptide into large cleavage funnel (DCM or DMF may be used to assist transfer).
-
A fresh amine cocktail containing amine of choice (2 M) and AcOH (0.5 M) in DMF/TFE (1:1, v/v) is prepared.
Note: DMF was selected as it swells polystyrene resins very effectively, preliminary results from our group show evidence that THF and DCM are also effective for this transformation.
Enough amine cocktail is then added to the resin to swell and ensuring the resin beads are free-flowing.
The cleavage funnel is capped, and the resin is then agitated with gentle rocking for 8 hours.
After 8 hours, the cleavage funnel is drained and a test cleavage is performed to validate complete consumption of the thioimidate on resin. A small amount of resin (ca. 10 mg resin) is added to a small polypropylene cleavage funnel, and subjected test cleavage in TFA/DCM/TIPS (75:20:5) for 30 minutes.
-
Cleavage cocktail is drained into a microcentrifuge tube, dried to a thin film under a stream of N2, and resuspended in minimal amount of H2O/MeCN (v/v) and analyzed via HPLC-MS. Presence of thioimidate in the LC trace is indicative of incomplete consumption of the thioimidate on resin and the resin should be subjected to steps 2-6 until no thioimidate is detected.
Note: Significant thioester–indicative of the presence of thioimidate–may be detected during test cleavage, this is indicative of thioimidate on the resin.
Upon completion of amidine formation, the cleavage funnel is drained and the resin rinsed with DMF (3 x 5 mL).
8.2. Resin Cleavage of Amidino Peptides

Resin cleavage is affected using a standard cleavage cocktail dictated by side-chains and type of resin used. Amidines are stable to high % TFA cleavage conditions and typical cleavages are performed using a cocktail of 95/2.5/2,5 v/v of TFA/TIPS/DCM.
The resin is swelled in enough cleavage cocktail that it is free-flowing, capped, and agitated for 1-5 hours with gentle rocking.
Upon cleavage, the cleavage cocktail is drained into a centrifuge tube and dried to a thin film under a stream of N2 gas.
The crude film is precipitated by addition of one 50 mL portion of very cold diethyl ether (cooled on dry ice for 30 minutes), and pelleted by centrifugation (10,000 rpm, 5 min).
The supernatant diethyl ether is discarded and the previous step repeated with another 50 mL portion of very cold diethyl ether.
The supernatant is again discarded, and the pelleted peptide is allowed to dry in air.
Crude amidinopeptide is then purified using preparatory reverse-phase HPLC.
Note: It is recommended that a 0.1% TFA additive is used in HPLC eluents to enhance peak resolution and ensure protonation of the amidine.
9. Protocol for Backbone Imidazolone Peptide Synthesis via Thioimidates
9.1. Synthesis of Thioimidate Precursor

Fmoc-Phe-OH (2.32 g, 1 eq., 6.00 mmol) and N-methylmorpholine (2.0 mL, 3 eq., 18.0 mmol) were dissolved in DMF (60 mL, 0.1 M) and cooled in an ice bath to 0°C.
HATU (2.28 g, 1 eq., 6.00 mmol) was added slowly to the reaction with stirring and allowed to pre-activate for 15 minutes.
HCl●NH2-Aib-OMe (1.01 g, 1.1 eq. 6.60 mmol) was added slowly to the reaction with stirring.
-
The reaction was allowed to stir for 4-6 hours with TLC monitoring until complete consumption of starting material.
Note: Aib is an α,α-disubstituted amino acid and hindered amino acid couplings may take longer to complete.
After the reaction was completed, the DMF solution was diluted with water (200 mL, 3-4 times the DMF solution volume). The aqueous layer was extracted with EtOAc (3 x 100 mL) and the organic layers combined.
Combined organic layers were washed with 1 M aq. HCl (2 x 100 mL), saturated NaHCO3 (2 x 100 mL), and brine (100 mL). The organic layer was then dried over Na2SO4, and concentrated in vacuo to afford Fmoc-Phe-Aib-OMe which was used without further purification.
Fmoc-Phe-Aib-OMe (ca. 6.00 mmol) and Lawesson’s reagent (3.64 g, 1.5 eq, 9.00 mmol) was dissolved in DME (20 mL, 0.3 M).
The reaction was heated for 4 hours at 85°C with TLC monitoring.
After 4 hours, any solid particulates in the reaction mixture were removed via vacuum filtration, and the solvent was concentrated in vacuo to a crude residue.
The crude residue was re-suspended in EtOAc (150 mL) and washed with water (200 mL), saturated NaHCO3 (4 x 200 mL), and brine (100 mL). The organic layer was then dried over Na2SO4, and concentrated in vacuo to afford Fmoc-PheS-Aib-OMe which was used without further purification.
Fmoc-PheS-Aib-OMe (ca. 6.00 mmol) was dissolved in acetone (20 mL, 0.3 M).
Anhydrous potassium carbonate (1.66 g, 2 eq., 12.0 mmol) and MeI (1.1 mL, 3 eq. 18.0 mmol) were added to the solution.
-
The reaction was sealed with a septa, and allowed to stir at 35circC for 18 hours with occasional TLC monitoring.
Note: If reaction appears sluggish, additional equivalents of MeI can be added to push reaction to completion.
Upon completion of the reaction, the potassium carbonate is removed using vaccuum filtration and the residue concentrated in vacuo.
Crude residue is purified via automated flash silica gel chromatography with gradient elution in 0-25% EtOAc in hexanes to yield Fmoc-PheSMe-Aib-OMe (1.10 g, 2.1 mmol, 36 % over 3 steps).
9.2. Resin Installation of Backbone Imidazolones via Thioimidate Precursor

Pre-swell resin for 10 minutes in dry THF then drain solution.
Transfer resin containing N-deprotected resin (either main peptide chain or side-chain amine) into large cleavage funnel (THF may be used to assist transfer). C-terminal imidazolones can be affected by direct reaction of Fmoc-deprotected Rink amide resin. Note: For thioimidate pre-cursors with no α-stereochemistry (such as Gly or Aib) heating can be used to accelerate reactivity by transfer of the dried resin to a microcentrifuge tube of appropriate size.
-
Depending on the amino acid following the imidazolone ring (α-position) the imidazolone installation cocktail below is freshly prepared:
For all residues other than Gly: Fmoc-Xa.a.SMe-Aib-OMe (see synthesis above, 0.2 M) and AcOH (0.1 M) in dry THF/TFE (v/v).
For residues with no α-stereochemistry such as Gly: Fmoc-GlySMe-Aib-OMe (see synthesis above, 0.1 M) and AcOH (0.1 M) in dry THF/TFE (v/v).
-
Enough amine cocktail is then added to the resin to swell and ensuring the resin beads are free-flowing. Depending on the amino acid following the imidazolone ring (α-position) the imidazolone installation conditions below are employed:
For all residues other than Gly: The cleavage funnel is capped, and the resin is then agitated with gentle rocking for 18 hours.
For residues with no α-stereochemistry such as Gly: A microscale stirbar is added to the microcentrifuge tube, sealed, and the resin is heated at 55°C with gentle stirring agitation for 18 hours.
After completion of reaction, the imidazolone installation cocktails are drained and the resin washed (3 x DMF).
Test cleavage in of a small portion of resin can be used to assess degree of un-reacted peptide remaining. If insufficient conversion occurs repeat the installation with a fresh cocktail. Note: Slow epimerization of α-stereochemistry of the imidazolone may occur under repeated or prolonged exposure to acid. Method is optimized to reduce epimerization (<10%), increase the concentration of thioimidate (to 0.3-0.5 M) in imidazolone cocktail if insufficient conversion occurs.
9.3. Resin Cleavage of Backbone Imidazolone Peptides

Resin cleavage is affected using a modified cleavage cocktail dictated by side-chains and type of resin used. Imidazolones with stereochemistry are stable to 75% TFA cleavage conditions and cleavages have been tested with a cocktail of 75/2.5/2.5/20 (v/v) of TFA/TIPS/H2O/DCM. Imidazolone peptides without stereochemistry can be cleaved using standard high % TFA cleavage conditions.
The resin is swelled in enough cleavage cocktail that it is free-flowing, capped, and agitated for 1 hour with gentle rocking.
Upon cleavage, the cleavage cocktail is drained into a centrifuge tube and dried to a thin film under a stream of N2 gas.
An excess (ca. 1 mL) of triethylamine in DCM (2-3 mL) is added to the peptide residue to ensure complete quenching of any acid which could slowly epimerize α-stereochemistry of the imidazolone peptides. Note: This step is not necessary for imidazolone peptides without stereochemistry.
The crude film is precipiated by addition of one 50 mL portion of very cold diethyl ether (cooled on dry ice for 30 minutes), and pelleted by centrifugation (10,000 rpm, 5 min).
The supernatant diethyl ether is discarded and the previous step repeated with another 50 mL portion of very cold diethyl ether.
The supernatant is again discarded, and the pelleted peptide is allowed to dry in air.
Crude imidazolone peptides are then purified using preparatory reverse-phase HPLC.
Note: It is recommended that a 0.1% TFA additive is used in HPLC eluents to enhance peak resolution.
10. Summary and Conclusions
Peptide-bond isosteres are important tools in drug design and the study of interactions in peptides. Biochemical methods for the installation of these structural probes into proteins are still in their infancy. Thus, synthetic organic chemistry is required to implement such isosteres into peptide-based biomolecules. For thioamides in particular, synthetic pitfalls exist at different stages of SPPS. The reversible protection of the thioamide as a thioimidate provides an avenue to navigate around each pitfall. The thioimidate motif also continues to expand the possibility of potential isosteric substitutions at the peptide backbone. The site-selective installation of thioimidates permits the first robust and general procedure for the introduction of amidines into the peptide backbone; there are limited examples of amidines in peptide scaffolds. Further, reactions of thioimidate precursor dipeptides have also enabled the formation of transient amidine species capable of ring-closing reactions which form (4H)-imidazolones on-resin, another known bioisostere of the amide bond. Conformational restriction imparted by the imidazolone substitution pattern also introduces a locked all-cis-amide geometry which may prove useful for stabilizing secondary structure, preventing proteolysis, and probing the importance of conformational rigidity in peptide-based ligand binding. Thus, thioimidates continue to be a versatile and general strategy to install new unnatural and bioisosteric modifications to the peptide without significantly altering the normal SPPS work-flow.
Figure 1:

Stereochemical fragility of thioamide residues during SPPS
Figure 7:

Activation of thioimidates with acid for insertion of amidines during SPPS.
11. Acknowledgements
The authors acknowledge the National Institutes of Health, National Institutes of General Medical Sciences under award number R35 GM142883. We also acknowledge Dr. Luis Camacho III and Dr. Bryan J. Lampkin for enabling conversations regarding this work.
12 References
- (1).Choudhary A; Raines RT An Evaluation of Peptide-Bond Isosteres. ChemBioChem 2011, 12, 1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Jackson JJ et al. Imidazolone as an Amide Bioisostere in the Development of β-1,3-N-Acetylglucosaminyltransferase 2 (B3GNT2) Inhibitors. J. Med. Chem 0000, 0, null. [DOI] [PubMed] [Google Scholar]
- (3).Lampkin BJ; VanVeller B Hydrogen Bond and Geometry Effects of Thioamide Backbone Modifications. J. Org. Chem 2021, 86, 18287–18291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Walters CR, Szantai-Kis DM, Zhang Y, Reinert ZE, Horne WS, Chenoweth DM, Petersson EJ The effects of thioamide backbone substitution on protein stability: a study in α-helical, β-sheet, and polyproline II helical contexts. Chem. Sci 2017, 8, 2868–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Newberry RW, VanVeller B, Raines RT Thioamides in the collagen triple helix. Chem. Commun 2015, 51, 9624–9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fiore KE, Patist MJ, Giannakoulias S, Huang C-H, Verma H, Khatri B, Cheng RP, Chatterjee J, Petersson EJ Structural impact of thioamide incorporation into a β-hairpin. RSC Chem. Biol 2022, 3, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Verma H, Khatri B, Chakraborti S, Chatterjee J Increasing the bioactive space of peptide macrocycles by thioamide substitution. Chem. Sci 2018, 9, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ghosh P, Raj N, Verma H, Patel M, Chakraborti S, Khatri B, Doreswamy CM, Anandakumar S, Seekallu S, Dinesh M, et al. An amide to thioamide substitution improves the permeability and bioavailability of macrocyclic peptides. Nat. Commun 2023, 14, 6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Newberry RW, Raines RT The n→π* Interaction. Acc. Chem. Res 2017, 50, 1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Newberry RW, VanVeller B, Guzei IA, Raines RT n→π* Interactions of Amides and Thioamides: Implications for Protein Stability. J. Am. Chem. Soc 2013, 135, 7843–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bartlett GJ, Newberry RW, VanVeller B, Raines RT, Woolfson DN Interplay of Hydrogen Bonds and n→π* Interactions in Proteins. J. Am. Chem. Soc 2013, 135, 18682–18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Newberry RW, Bartlett GJ, VanVeller B, Woolfson DN, Raines RT Signatures of n→π* interactions in proteins. Protein Science 2014, 23, 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Khatri B, Majumder P, Nagesh J, Penmatsa A, Chatterjee J Increasing protein stability by engineering the n → π* interaction at the β-turn. Chem. Sci 2020, 11, 9480–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Huang Y, Ferrie JJ, Chen X, Zhang Y, Szantai-Kis DM, Chenoweth DM, Petersson EJ Electronic interactions of i, i + 1 dithioamides: increased fluorescence quenching and evidence for n-to-π* interactions. Chem. Commun 2016, 52, 7798–7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wildemann D, Schiene-Fischer C, Aumüller T, Bachmann A, Kiefhaber T, Lücke C, Fischer G A Nearly Isosteric Photosensitive Amide-Backbone Substitution Allows Enzyme Activity Switching in Ribonuclease S. Journal of the American Chemical Society 2007, 129, 4910–4918. [DOI] [PubMed] [Google Scholar]
- (16).Huang Y, Cong Z, Yang L, Dong S A photoswitchable thioxopeptide bond facilitates the conformation-activity correlation study of insect kinin. Journal of Peptide Science 2008, 14, 1062–1068. [DOI] [PubMed] [Google Scholar]
- (17).Robkis DM, Hoang EM, Po P, Deutsch CJ, Petersson EJ Side-chain thioamides as fluorescence quenching probes. Biopolymers 2021, 112, e23384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Walters CR, Ferrie JJ, Petersson EJ Dithioamide substitutions in proteins: effects on thermostability, peptide binding, and fluorescence quenching in calmodulin. Chem. Commun 2018, 54, 1766–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Petersson EJ, Goldberg JM, Wissner RF On the use of thioamides as fluorescence quenching probes for tracking protein folding and stability. Phys. Chem. Chem. Phys 2014, 16, 6827–6837. [DOI] [PubMed] [Google Scholar]
- (20).Goldberg JM, Batjargal S, Chen BS, Petersson EJ Thioamide Quenching of Fluorescent Probes through Photoinduced Electron Transfer: Mechanistic Studies and Applications. J. Am. Chem. Soc 2013, 135, 18651–18658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Culik RM, Jo H, DeGrado WF, Gai F Using Thioamides To Site-Specifically Interrogate the Dynamics of Hydrogen Bond Formation in β-Sheet Folding. J. Am. Chem. Soc 2012, 134, 8026–8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Goldberg JM, Wissner RF, Klein AM, Petersson EJ Thioamide quenching of intrinsic protein fluorescence. Chem. Commun 2012, 48, 1550–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Goldberg JM, Batjargal S, Petersson EJ Thioamides as Fluorescence Quenching Probes: Minimalist Chromophores To Monitor Protein Dynamics. J. Am. Chem. Soc 2010, 132, 14718–14720. [DOI] [PubMed] [Google Scholar]
- (24).Chen X, Mietlicki-Baase EG, Barrett TM, McGrath LE, Koch-Laskowski K, Ferrie JJ, Hayes MR, Petersson EJ Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones. J. Am. Chem. Soc 2017, 139, 16688–16695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Barrett TM, Chen XS, Liu C, Giannakoulias S, Phan HAT, Wang J, Keenan EK, Karpowicz RJJ, Petersson EJ Studies of Thioamide Effects on Serine Protease Activity Enable Two-Site Stabilization of Cancer Imaging Peptides. ACS Chem. Biol 2020, 15, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).O’Brien EA, Sharma KK, Byerly-Duke J, Camacho LA 3rd, VanVeller B A general strategy to install amidine functional groups along the peptide backbone. J. Am. Chem. Soc 2022, 144, 22397–22402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thombare VJ, Hutton CA Rapid, Traceless, AgI-Promoted Macrocyclization of Peptides Possessing an N-Terminal Thioamide. Angew. Chem., Int. Ed 2019, 58, 4998–5002. [DOI] [PubMed] [Google Scholar]
- (28).Shabani S, Hutton CA Depsipeptide synthesis using a late-stage Ag(i)-promoted macrolactonisation of peptide thioamides. Chem. Commun 2021, 57, 2081–2084. [DOI] [PubMed] [Google Scholar]
- (29).Taresh AB, Hutton CA Backbone thioamide directed macrocyclisation: lactam stapling of peptides. Org. Biomol. Chem 2022, 20, 1488–1492. [DOI] [PubMed] [Google Scholar]
- (30).Okano A, James RC, Pierce JG, Xie J, Boger DL Silver(I)-Promoted Conversion of Thioamides to Amidines: Divergent Synthesis of a Key Series of Vancomycin Aglycon Residue 4 Amidines That Clarify Binding Behavior to Model Ligands. J. Am. Chem. Soc 2012, 134, 8790–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Huh S, Appavoo SD, Yudin AK Amidine Functionality As a Conformational Probe of Cyclic Peptides. Org. Lett 2020, 22, 9210–9214. [DOI] [PubMed] [Google Scholar]
- (32).Byerly-Duke J, Donovan A, Sharma K, Ibrahim R, VanVeller B Thioimidate Solutions to Thioamide Problems during Peptide Synthesis. ChemRxiv 2023, DOI: 10.26434/chemrxiv-2023-3823q. [DOI] [Google Scholar]
- (33).Wildemann D, Drewello M, Fischer G, Schutkowski M Extremely selective Mg(ClO4)2 mediated removal of Bpoc/Ddz moieties suitable for the solid phase peptide synthesis of thioxo peptides. Chem. Commun 1999, 1809–1810. [Google Scholar]
- (34).Bachmann A, Wildemann D, Praetorius F, Fischer G, Kiefhaber T Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proceedings of the National Academy of Sciences 2011, 108, 3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shalaby MA, Grote CW, Rapoport H Thiopeptide synthesis. Alpha-amino thionoacid derivatives of nitrobenzotriazole as thioacylating agents. J. Org. Chem 1996, 61, 9045–9048. [DOI] [PubMed] [Google Scholar]
- (36).Khatri B, Bhat P, Chatterjee J Convenient synthesis of thioamidated peptides and proteins. J. Pept. Sci 2020, 26, e3248. [DOI] [PubMed] [Google Scholar]
- (37).Khatri B, Raj N, Chatterjee J In Synthetic and Enzymatic Modifications of the Peptide Backbone, Petersson EJ, Ed., Methods in Enzymology, Vol. 656, Academic Press: 2021, pp 27–57. [DOI] [PubMed] [Google Scholar]
- (38).Mukherjee S, Chatterjee J Suppressing the epimerization of endothioamide peptides during Fmoc/t-Bu-based solid phase peptide synthesis. J. Pept. Sci 2016, 22, 664–672. [DOI] [PubMed] [Google Scholar]
- (39).Szantai-Kis DM, Walters CR, Barrett TM, Hoang EM, Petersson EJ Thieme Chemistry Journals Awardees–Where Are They Now? Improved Fmoc Deprotection Methods for the Synthesis of Thioamide-Containing Peptides and Proteins. Synlett 2017, 28, 1789–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Camacho LA, Lampkin BJ, VanVeller B A Bottom-Up Approach To Preserve Thioamide Residue Stereochemistry during Fmoc Solid-Phase Peptide Synthesis. Org. Lett 2019, 21, 7015–7018. [DOI] [PubMed] [Google Scholar]
- (41).Camacho LA 3rd, Nguyen YH, Turner J, VanVeller B Deprotection strategies for thioimidates during Fmoc solid-phase peptide synthesis: A safe route to thioamides. J. Org. Chem 2019, 84, 15309–15314. [DOI] [PubMed] [Google Scholar]
- (42).Miwa JH, Margarida LA, Meyer AE Improved acidolytic deprotection conditions for the Fmoc-based solidphase synthesis of thioxo peptides. Tetrahedron Lett. 2001, 42, 7189–7191. [Google Scholar]
- (43).Chaturvedi RK, Schmir GL Hydrolysis of thioimidate esters. II. Evidence for the formation of three species of the tetrahedral intermediate. J. Am. Chem. Soc 1969, 91, 737–746. [Google Scholar]
- (44).Chaturvedi RK, MacMahon AE, Schmir GL Hydrolysis of thioimidate esters. Tetrahedral intermediates and general acid catalysis. J. Am. Chem. Soc 1967, 89, 6984–6993. [Google Scholar]
- (45).Sharma I, Crich D Direct Fmoc-chemistry-based solid-phase synthesis of peptidyl thioesters. J. Org. Chem 2011, 76, 6518–6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Byerly-Duke J, VanVeller B Thioimidate Solutions to Thioamide Problems during Thionopeptide Deprotection. Organic Letters 2024, 26, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Boger DL The difference a single atom can make: Synthesis and design at the chemistry–biology interface. The Journal of organic chemistry 2017, 82, 11961–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Okano A, Nakayama A, Wu K, Lindsey EA, Schammel AW, Feng Y, Collins KC, Boger DL Total syntheses and initial evaluation of [Ψ [C (═ S) NH] Tpg4] vancomycin,[Ψ [C (═ NH) NH] Tpg4] vancomycin,[Ψ [CH═NH] Tpg4] vancomycin, and their (4-chlorobiphenyl) methyl derivatives: synergistic binding pocket and peripheral modifications for the glycopeptide antibiotics. Journal of the American Chemical Society 2015, 137, 3693–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL Total synthesis of [Ψ [C (═ S) NH] Tpg4] vancomycin aglycon,[Ψ [C (═ NH) NH] Tpg4] vancomycin aglycon, and related key compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. Journal of the American Chemical Society 2012, 134, 1284–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wu Z-C, Boger DL Maxamycins: durable antibiotics derived by rational redesign of vancomycin. Accounts of chemical research 2020, 53, 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Moore MJ, Qin P, Keith DJ, Wu Z-C, Jung S, Chatterjee S, Tan C, Qu S, Cai Y, Stanfield RL, et al. Divergent Total Synthesis and Characterization of Maxamycins. Journal of the American Chemical Society 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Clark KA, Seyedsayamdost MR Bioinformatic atlas of radical SAM enzyme-modified RiPP natural products reveals an isoleucine-tryptophan crosslink. J. Am. Chem. Soc 2022, 144, 17876–17888. [DOI] [PubMed] [Google Scholar]
- (53).Dong S-H, Liu A, Mahanta N, Mitchell DA, Nair SK Mechanistic basis for ribosomal peptide backbone modifications. ACS Central Science 2019, 5, 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Foden CS, Islam S, Fernández-García C, Maugeri L, Sheppard TD, Powner MW Prebiotic synthesis of cysteine peptides that catalyze peptide ligation in neutral water. Science 2020, 370, 865–869. [DOI] [PubMed] [Google Scholar]
- (55).Singh J, Whitaker D, Thoma B, Islam S, Foden CS, Aliev AE, Sheppard TD, Powner MW Prebiotic catalytic peptide ligation yields proteinogenic peptides by intramolecular amide catalyzed hydrolysis facilitating regioselective lysine ligation in neutral water. J. Am. Chem. Soc 2022, 144, 10151–10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Vastl J, Kartika R, Park K, Cho AE, Spiegel DA Peptidines: glycine-amidine-based oligomers for solution- and solid-phase synthesis. Chem. Sci 2016, 7, 3317–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Inokuchi E, Yamada A, Hozumi K, Tomita K, Oishi S, Ohno H, Nomizu M, Fujii N Design and synthesis of amidine-type peptide bond isosteres: application of nitrile oxide derivatives as active ester equivalents in peptide and peptidomimetics synthesis. Org. Biomol. Chem 2011, 9, 3421–3427. [DOI] [PubMed] [Google Scholar]
- (58).Hitotsuyanagi Y, Sasaki S-I, Matsumoto Y, Yamaguchi K, Itokawa H, Takeya K Synthesis of [L-Ala-1]RA-VII, [D-Ala-2]RA-VII, and [D-Ala-4]RA-VII by epimerization of RA-VII, an antitumor bicyclic hexapeptide from Rubia plants, through oxazoles. J. Am. Chem. Soc 2003, 125, 7284–7290. [DOI] [PubMed] [Google Scholar]
- (59).Kumari S, Carmona AV, Tiwari AK, Trippier PC Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. Journal of Medicinal Chemistry 2020, 63, 12290–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Smolyar IV, Yudin AK, Nenajdenko VG Heteroaryl Rings in Peptide Macrocycles. Chem. Rev 2019, 119, 10032–10240. [DOI] [PubMed] [Google Scholar]
- (61).Biron E, Chatterjee J, Kessler H Solid-Phase Synthesis of 1,3-Azole-Based Peptides and Peptidomimetics. Org. Lett. 2006, 8, 2417–2420. [DOI] [PubMed] [Google Scholar]
- (62).Kaldas SJ, Yudin AK Achieving Skeletal Diversity in Peptide Macrocycles through The Use of Heterocyclic Grafts. Chem. Eur. J 2018, 24, 7074–7082. [DOI] [PubMed] [Google Scholar]
- (63).Soor HS, Appavoo SD, Yudin AK Heterocycles: Versatile control elements in bioactive macrocycles. Bioorg. Med. Chem 2018, 26, Peptide Therapeutics, 2774–2779. [DOI] [PubMed] [Google Scholar]
- (64).Hamdan F, Tahoori F, Balalaie S Synthesis of novel cyclopeptides containing heterocyclic skeletons. RSC Adv. 2018, 8, 33893–33926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Zhang J, Germann MW Characterization of secondary amide peptide bond isomerization: Thermodynamics and kinetics from 2D NMR spectroscopy. Biopolymers 2011, 95, 755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Yu KL, Johnson RL Synthesis and chemical properties of tetrazole peptide analogs. The Journal of Organic Chemistry 1987, 52, 2051–2059. [Google Scholar]
- (67).Boeglin D, Cantel S, Heitz A, Martinez J, Fehrentz J-A Solution and Solid-Supported Synthesis of 3,4,5-Trisubstituted 1,2,4-Triazole-Based Peptidomimetics. Org. Lett 2003, 5, 4465–4468. [DOI] [PubMed] [Google Scholar]
- (68).Tam A, Arnold U, Soellner MB, Raines RT Protein Prosthesis: 1,5-Disubstituted[1,2,3]triazoles as cis-Peptide Bond Surrogates. J. Am. Chem. Soc 2007, 129, 12670–12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Tischler M, Nasu D, Empting M, Schmelz S, Heinz DW, Rottmann P, Kolmar H, Buntkowsky G, Tietze D, Avrutina O Braces for the Peptide Backbone: Insights into Structure–Activity Relationships of Protease Inhibitor Mimics with Locked Amide Conformations. Angewandte Chemie International Edition 2012, 51, 3708–3712. [DOI] [PubMed] [Google Scholar]
- (70).Empting M, Avrutina O, Meusinger R, Fabritz S, Reinwarth M, Biesalski M, Voigt S, Buntkowsky G, Kolmar H “Triazole Bridge”: Disulfide-Bond Replacement by Ruthenium-Catalyzed Formation of 1,5-Disubstituted 1,2,3-Triazoles. Angewandte Chemie International Edition 2011, 50, 5207–5211. [DOI] [PubMed] [Google Scholar]
- (71).Grob NM, Schibli R, Béhé M, Valverde IE, Mindt TL 1,5-Disubstituted 1,2,3-Triazoles as Amide Bond Isosteres Yield Novel Tumor-Targeting Minigastrin Analogs. ACS Medicinal Chemistry Letters 2021, 12, 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Hernandez-Olmos V, Heering J, Bischoff-Kont I, Kaps A, Rajkumar R, Liu T, Fürst R, Steinhilber D, Proschak E Discovery of Irbesartan Derivatives as BLT2 Agonists by Virtual Screening. ACS Med. Chem. Lett 2021, 12, 1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Mallinger A et al. Discovery of Potent, Selective, and Orally Bioavailable Small-Molecule Modulators of the Mediator Complex-Associated Kinases CDK8 and CDK19. J. Med. Chem 2016, 59, 1078–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Metwally KA, Dukat M, Egan CT, Smith C, DuPre A, Gauthier CB, Herrick-Davis K, Teitler M, Glennon RA Spiperone: Influence of Spiro Ring Substituents on 5-HT2A Serotonin Receptor Binding. J. Med. Chem 1998, 41, 5084–5093. [DOI] [PubMed] [Google Scholar]
- (75).Jones LH, Dupont T, Mowbray CE, Newman SD A Concise and Selective Synthesis of Novel 5-Aryloxyimidazole NNRTIs. Org. Lett 2006, 8, 1725–1727. [DOI] [PubMed] [Google Scholar]
- (76).Tan S, Evans RR, Dahmer ML, Singh BK, Shaner DL Imidazolinone-tolerant crops: history, current status and future. Pest Manag. Sci 2005, 61, 246–257. [DOI] [PubMed] [Google Scholar]
- (77).Mancini I, Guella G, Pietra F, Debitus C, Waikedre J From Inactive Nortopsentin D, a Novel Bis(indole) Alkaloid Isolated from the Axinellid Sponge Dragmacidon sp. from Deep Waters South of New Caledonia, to a Strongly Cytotoxic Derivative. Helv. Chim. Acta 1996, 79, 2075–2082. [Google Scholar]
- (78).Keel KL, Tepe JJ The preparation of (4H)-imidazol-4-ones and their application in the total synthesis of natural products. Org. Chem. Front 2020, 7, 3284–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Wall B, O’Brien E, Sharma K, Donovan A, VanVeller B General Installation of (4H)-Imidazolone cis-Amide Bioisosteres Along the Peptide Backbone. ChemRxiv 2023, DOI: 10.26434/chemrxiv-2023-9t1f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Panov I, Drabina P, Hanusek J, Sedlák M The synthesis and characterisation of N-(1-carbamoyl-1,1-dialkyl-methyl)-(S)-prolinamides and related pyrrolidin-2-yl-4,5-dihydro-1H-imidazol-5-ones as potential enantioselective organocatalysts. Tetrahedron: Asymm. 2011, 22, 215–221. [Google Scholar]
- (81).Gjonaj L, Roelfes G Selective chemical modification of DNA with alkoxy- and benzyloxyamines. Org. Biomol. Chem 2015, 13, 6059–6065. [DOI] [PubMed] [Google Scholar]
- (82).Schneggenburger PE, Worbs B, Diederichsen U Azide reduction during peptide cleavage from solid support—the choice of thioscavenger? J. Pept. Sci 2010, 16, 10–14. [DOI] [PubMed] [Google Scholar]