

Abstract
Myocardial ischemia–reperfusion injury (MIRI) was often observed after surgeries, causing a lot of suffering to patients. Inflammation and apoptosis were critical determinants during MIRI. We conveyed experiments to reveal the regulatory functions of circHECTD1 in MIRI development. The Rat MIRI model was established and determined by 2,3,5‐triphenyl tetrazolium chloride (TTC) staining. We analyzed cell apoptosis using TUNEL and flow cytometry. Proteins expression was evaluated by western blot. The RNA level was determined by qRT‐PCR. Secreted inflammatory factors were analyzed by ELISA assay. To predict the interaction sequences on circHECTD1, miR‐138‐5p, and ROCK2, bioinformatics analysis was performed. Dual‐luciferase assay was used to confirm these interaction sequences. CircHECTD1 and ROCK2 were upregulated in the rat MIRI model, while miR‐138‐5p was decreased. CircHECTD1 knockdown alleviated H/R‐induced inflammation in H9c2 cells. Direct interaction and regulation of circHECTD1/miR‐138‐5p and miR‐138‐5p/ROCK2 were confirmed by dual‐luciferase assay. CircHECTD1 promoted H/R‐induced inflammation and cell apoptosis by inhibiting miR‐138‐5p. miR‐138‐5p alleviated H/R‐induced inflammation, while ectopic ROCK2 antagonized such effect of miR‐138‐5p. Our research suggested that the circHECTD1‐modulated miR‐138‐5p suppressing is responsible for ROCK2 activation during H/R‐induced inflammatory response, providing a novel insight into MIRI‐associated inflammation.
Keywords: circHECTD1, inflammation, ischemia/reperfusion injury, miR‐138‐5p, ROCK2
Abbreviations
- 2,3,5‐Triphenyl tetrazolium chloride
TTC
- ceRNA
competitive endogenous RNA
- circHECTD1
circular RNA HECTD1
- circRNA
circular RNA
- I/R injury
ischemia–reperfusion injury
- I/R
ischemia–reperfusion
- LncRNAs
Long noncoding RNAs
- MIRI
myocardial ischemia–reperfusion injury
- miRNA
microRNA
1. INTRODUCTION
The wildly accepted concept of myocardial ischemia–reperfusion injury (MIRI) refers to ischemic myocardium‐associated tissue damage. Inflammation and apoptosis along with microvascular obstruction, reactive oxygen species, and myocardial stunning were all considered important factors during MIRI development. 1 Importantly, inflammation and apoptosis were involved in tissue recovery or necrosis in I/R injuries not only myocardial but also in cerebral (e.g. in acute ischemic stroke patients), renal, and other tissues. 2 , 3 They might share the same regulatory mechanism which was not completely understood. Therefore, investigating the regulatory mechanism of inflammation and apoptosis during MIRI bears great importance.
Circular RNA (circRNA) included a unique closed ring structure that contained miRNA binding sequences and could act as a “miRNA sponge,” capturing its target miRNAs. 4 CircRNA played important regulatory roles in many diseases including MIRI through regulating diseases associated with miRNAs. 5 , 6 For example, Sun and colleagues constructed a ceRNA network involving multiple long non‐coding RNAs (lncRNA) during the regulation of the APMK pathway, which was associated with inflammation and apoptosis regulation during MIRI. 7 Recently, it was reported that circHECTD1 was increased in populations with acute ischemic stroke (AIS) and a higher cellular level of circHECTD1 could lead to increased disease severity, higher disease risk, and elevated recurrence of AIS. 8 circHECTD1 was much higher in ischemic brain tissues of AIS patients, as well as in mice models and circHECTD1 knockdown reduced infarct areas and neuronal deficits. 9 It was also observed that down‐regulation of circHECTD1 induced neuroprotection effects. 10 Due to the large similarities between neuronal and myocardial IRI, and considering that integrated network analysis of myocardial infarction also indicated circHECTD1 as a potential regulator of MIRI, 11 we speculated that circHECTD1 bears great regulatory potential in MIRI.
MicroRNA (miRNA) could regulate gene expression on the posttranscriptional level. Emerging studies in the last decades had implicated the fundamental role of miRNA in MIRI regulation and miRNA was even considered a potential therapeutic target for preventing MIRI. 12 , 13 , 14 For instance, Rno‐microRNA‐30c‐5p promoted myocardial ischemia–reperfusion injury in rats, 15 miR‐885 mediated cardioprotection against hypoxia/reoxygenation‐induced apoptosis in human cardiomyocytes, 16 miR‐206 could mitigate epithelial cell activation and neutrophil infiltration in the lungs after IR injury. 17 It was noteworthy that miR‐138 was downregulated in MIRI. 18 Besides, upregulation of miR‐138‐5p could alleviate I/R‐induced apoptosis of rat cardiac myoblast cells H9c2. 19 miR‐138‐5p was a fundamental regulatory factor during MIRI, however, the mechanism was poorly described. Revealing its regulation mechanism would greatly promote our understanding of MIRI regulation.
ROCK2 was a Rho‐associated serine/threonine kinase, which was downstream of Rho GTPase. ROCK2 was involved in various cellular activities such as cell adhesion, proliferation, apoptosis, and extracellular remodeling. It was also implicated in cardiovascular diseases including stroke. 20 The function of ROCK2 was identified by Dominik and colleagues in rat renal acute ischemia–reperfusion injury model. 21 In their study, inhibition of ROCK2 improved kidney function in rat MIRI models, suggesting that ROCK2 could aggravate MIRI. Although renal and myocytes were different tissues, we believe they might share several basic molecular pathways, especially in ischemic reperfusion injury. The opposite functions of ROCK1/2 in cardiomyocytes were also described in mice model, 22 whereas ROCK1 inhibited postcapillary pulmonary hypertension and cardiac dysfunction while ROCK2 exerted the opposite function. Taken together, ROCK2 could aggravate MIRI and lead to cardiac dysfunction, and might also contribute to MIRI.
The interaction between circHECTD1/miR‐138‐5p and between miR‐138‐5p/ROCK2 was predicted by starBase, respectively. We thus conveyed this research hoping to elucidate the hidden function of circHECTD1 in MIRI and to further explore its molecular mechanism. This study would provide a new perspective on MIRI research and treatment.
2. MATERIALS AND METHODS
2.1. Rat MIRI model
We obtained approval for animal experiments from the ethical committee of the First Affiliated Hospital of Xi'an Jiaotong University. All experiments were performed according to the Guide for the Care and Use of Laboratory Animals according to the regulation of the People's Republic of China. The number of the animal care committee approval was No. 2018‐044. Sprague–Dawley rats (6 weeks old, weighing 200–250 g) were obtained from the SLAC animal center. Myocardial I/R injury models were induced following the previous publication 23 (MIRI group n = 6, sham group n = 6). Briefly, rats were weighed and anesthetized with 1% sodium pentobarbital (1 mL/kg, i.p.). The thoracic cavity was opened in the intercostal space after linking the small animal ventilator and the heart was exposed. The left anterior descending coronary artery was ligated with a 4‐0 silk thread to ischemia for 30 mins, then the ligature was released and reperfused for 2 h. Ischemia was successfully performed based on the significant S‐T segment elevation of the II lead electrocardiogram. The elevated S‐T segment dropped half and above the recovery of the T wave indicating successful reperfusion. The sham group did not receive LAD ligation. In this research, blood samples were collected from caudal veins.
2.2. Establish of cellular MIRI model and cell culture
We purchased H9c2 cells (rat cardiac myoblast) from the American Type Culture Collection (ATCC, USA). We maintained H9c2 using DMEM+ 10% FBS in a humidified incubator containing 5% of CO2. For the MIRI model, we treated H9c2 cells using hypoxia‐reoxygenation (H/R). Briefly, H9c2 cells were treated under the hypoxia statement (94% N2, 5% CO2, 1% O2). Four hours later, cells were reoxygenated (by 21% O2, 5% CO2) for another 6 h. All media were obtained from Gibco, USA.
2.3. Plasmid construction and transfection
Plasmids of ROCK2 overexpression were constructed as follows: the cDNA of ROCK2 was cloned into pcDNA3.1. Luciferase plasmid systems for wild‐type and mutant circHECTD1 were established by cloning wild‐type and mutant circHECTD1 into the pRL luciferase reporter system (Promega, USA). The shRNA and control of circHECTD1 and the mimics and inhibitor of miR‐138‐5p were synthesized by Genepharm (Shanghai). We used lipo2000 to convey transfection following manufacturers' instructions.
2.4. qRT‐PCR
We extracted RNAs from prepared samples by Trizol and synthesized cDNA with a cDNA Reverse Transcription Kit (Thermo Fisher, USA). The reaction system of qPCR was prepared using SYBR green purchased from Takara. The qRT‐PCR reaction was conveyed on ABI Q7 (ABI, USA). To analyze the relative expression of each gene, we normalized their expression to GAPDH or U6. The primer sequence is as follows:
circHECTD1 forward: 5′‐ACTCCGTCACCTCGATTAGC‐3′.
circHECTD1 reverse: 5′‐ATCATCCCATGTTCTCCGGC‐3′.
miR‐138‐5p forward: 5′‐GGCCGGACTAAGTGTTGT‐3′.
miR‐138‐5p reverse: 5′‐GCAGGGTCCGAGGTATTC‐3′.
ROCK2 forward: 5′‐GTTCGTCATAAGGCATCACAGA‐3′.
ROCK2 reverse: 5′‐TGTTGGCAAAGGCCATAATATCT‐3′.
IL‐6 forward: 5′‐CCAGTATATACCACTTCACAAGTCGGA‐3′.
IL‐6 reverse: 5′‐CAAGATGAGTTGGATGGTCTTGGTC‐3′.
TNF‐α forward: 5′‐GCCTCTTCTCATTCCTGCTT‐3′.
TNF‐α reverse: 5′‐TGGGAACTTCTCATCCCTTTG‐3′.
IL‐1β forward: 5′‐CTCTGCCCTCTGGATGGCGG‐3′.
IL‐1βreverse: 5′‐CAGGTCATTCTCCTGGAAGG‐3′.
U6 forward: 5′‐CTCGCTTCGGCAGCACA‐3′.
U6 reverse: 5′‐AACGCTTCACGAATTTGCGT‐3′.
GAPDH forward: 5′‐CTGACTTCAACAGCGACACC‐3′.
GAPDH reverse: 5′‐GTGGTCCAGGGGTCTTACTC‐3′.
2.5. Western blot
After the required treatment, we collected cells and rinsed cells 1 time using PBS. Cells were collected and put into a centrifuge at 4°C. After centrifugation for 5 min at 1000 rpm, the pellets were resuspended and lysed by RIPA buffer (Millipore, USA) on ice for 20 min. For every 107 cells, 1 mL RIPA buffer was applied. Samples were subjected to centrifugation for 15 min (12,000 rpm) at 4°C and supernatants were collected. The concentration of proteins in the supernatant was quantified using a BCA kit (Thermo Fisher, USA) following the manufacturer's instructions. For the western blot, 20 μg protein was loaded into each well of SDS‐PAGE gel, separated, and transferred to the NC membrane. Membranes were incubated in 5% BSA. After 1 h, the blocking buffer was removed and membranes were incubated with primary antibodies at 4°C overnights. Membranes were washed with tris‐buffered saline tween‐20 (TBST) 3 times. Secondary antibodies were added onto membranes on a mild shaker avoiding light for 1 h. To remove extra antibodies, membranes were rinsed with TBST 3 times. The signal was detected by enhanced chemiluminescence (GE, USA).
2.6. ELISA
Supernatants of treated cells were collected. Secreted inflammatory factors were then detected by a kit of IL‐6 (ab100772), TNF‐α (ab100785), and IL‐1β (ab100768) following the manufacturer's instructions, respectively. All ELISA kits were obtained from Abcam. Briefly, standards and samples were added into appropriate wells. Samples were put on a shaker at RT. Two hours later, the solutions were discarded and each well was washed using the wash buffer (provided by the kit) 4 times. Detection antibodies (provided by kit) were added into each well and plates were put on a shaker at RT. After 1 h, the wells were washed 4 times. HRP‐streptavidin was added to these wells. Plates were put on a shaker at RT. Forty‐five minutes later, the wells were washed 4 times. The reaction was then proceeded by 30‐min incubation in dark with TMB One‐Step Substrate Reagent (provided by kit). Stop solution were applied to each well, and signals were read using a plate reader under a wavelength of 450 nm (Tecan, USA).
2.7. Flow cytometry analysis
We detected Apoptosis using Annexin V‐FITC (Sigma, USA) by flow cytometry. Experiments were performed under the guideline of the kit. Cells were digested with trypsin, and subjected to centrifugation for 5 min at 4°C at 1000 rpm. Supernatants were removed. Pellets were washed with PBS and centrifuged again. Pellets were then mixed thoroughly with 100 μL binding buffer. Then, mix Annexin V‐FITC and propidium iodide with the cell solution and put the samples stand avoiding light. Ten minutes later, the reaction was stopped. The apoptosis was then detected using flow cytometry Calibur (BD).
2.8. Luciferase assay
To perform a dual luciferase assay, the reporter plasmid system was transfected into H9c2 cells using lipo2000. A dual Luciferase Reporter Assay kit from Promega was used to conduct the experiments. Briefly, H9c2 cells transfected with wt‐circHECTD1, mut‐circHECTD1, and miR‐138‐5p mimics or inhibitor were collected and lysed with lysis buffer provided by the kit. Supernatants were collected and subjected to luciferase intensity detection by a luminometer. Renilla served as internal control.
2.9. RNA immunoprecipitation assay
We performed the RNA immunoprecipitation (RIP) assay using a RIP Kit (Millipore) following the manufacturers' instructions. Briefly, cells under the ninetieth confluence were harvested and treated with lysis buffer. Meanwhile, beads and Ago2 antibody (Abcam)were mixed and incubated for 1 h at RT. This step would conjugate the antibody to the beads. After conjugation, we collected beads using a magnetic separator, and the beads were resuspended using the Immunoprecipitation buffer. Take 100 μL of the lysates and mix with 900 μL resuspended beads and incubated the mixture for 4 h at 4°C. Beads were collected using a magnetic separator. Proteins were detached from beads by digesting using proteinase K. The negative control was rabbit IgG. We then detected RNA by qRT‐PCR to analyze our target genes.
2.10. RNA pulldown assay
RNA probe of CircRNA HECTD1 was synthesized by Genepharm (Shanghai, China). Pierce RNA 3′ End Desthiobiotinylation Kit was used for RNA pull down. Briefly, 20 μL RNA probe was incubated with prepared magnet beads in 400 μL binding buffer and 380 μL DEPC water and incubated at room temperature for 20 min. Then we used a magnetic separator to collect beads, and beads were washed with 800 μL binding buffer 3 times and with lysis buffer once. Conjugated beads with an RNA probe were then incubated with extracted RNA for 2 h at 4°C and captured RNA was analyzed using qPCR.
2.11. TUNEL assay
Collect and rinse cells with PBS once, then treated for 30 min with 4% PFA and washed again with PBS. Then, incubate samples with 0.3% Triton X‐100 cells for 5 min. Samples were rinsed again and incubated with 0.3% H2O2 dissolved in PBS followed by 3 times of washing. Then, we incubated samples for 30 min in Streptavidin‐HRP buffer at RT. After 3 times washing, the samples were mixed with 0.2 mL DAB solution, incubated for 20 min at RT, and washed 3 times. Results were analyzed using a microscope.
2.12. 2,3,5‐Triphenyl tetrazolium chloride staining
Blood in the aorta of rats was washed away by saline. Then the aorta was injected with 1 mL of 0.3% Evans Blue staining solution. Slices of the cardiac ventricle were then prepared by transect at 2 mm. Incubate the slice with 2,3,5‐triphenyl tetrazolium chloride (TTC) for 15 min at 37°C. The infarct area was then observed under a microscope and analyzed. The infarcted area is the white or light color area stained by TTC, and the noninfarcted ischemic‐free area is the color of normal myocardial tissue with no change after TTC staining. The junction of these two areas is determined as the infarct boundary area or peri‐infarct area.
2.13. Statistical analysis
Data were analyzed using GraphPad Prism 8.0 software (GraphPad Software, USA). All experiments were repeated at least 3 times, and results were presented as mean ± standard deviation (SD). The data were tested to conform to a normal distribution by Shapiro–Wilk. Differences between or among groups were analyzed using student's t‐test or one‐way ANOVA with the Tukey HSD post‐hoc test, respectively. Statistical significance was defined as a p value < 0.05.
3. RESULTS
3.1. Enhanced CircHECTD1 and ROCK2 and decreased miR‐138‐5p were detected in the rat MIRI model
First, we successfully constructed the MIRI rat model and investigated the expression of circHECTD1, miR‐138‐5p, and ROCK2 in this model. TTC staining showed that the myocardial infarction area of rats in the MIRI group was significantly larger than that in the sham group (Figure 1A). The apoptosis of the MIRI model also increased significantly in the MIRI model compared with the sham group (Figure 1B). The qRT‐PCR results showed that both circHECTD1 and ROCK2 were upregulated (Figure 1C,E), while miR‐138‐5p was reduced (Figure 1D) in the MIRI group. Then we treated H9c2 cells under the H/R condition, aiming to develop the MIRI cell model. circHECTD1 and ROCK2 were both upregulated, while miR‐138‐5p was downregulated in H9c2 cells after H/R exposure (Figure 1F–I). To further determine whether HECTD1 was greatest unregulated in the ischemia zone, we examined the expression level of circHECTD1 and its distribution in different tissues of the heart (for each group, n = 6). The expression of circHECTD1 was found to be the highest in the ischemic region, followed by the ischemic surrounding tissues, and the lowest in the nonischemic region (Figure S1A). In addition, the expression of circHECTD1 in the blood was detected and it was significantly increased in the MIRI model group (Figure S1B). The duration of circHECTD1 in the blood circulation was also measured, which was at a high level from 2.5 to 3 h, and returned to the normal level after 10 hours. These results indicated that circHECTD1, miR‐138‐5p, and ROCK2 participated in the development of MIRI. Besides, the results of Figure S1 showed that the expression of circHECTD1 was increased in myocardial infarction tissue and blood, so it should be knocked down in subsequent experiments to verify its function.
FIGURE 1.

Enhanced CircHECTD1 and ROCK2 and decreased miR‐138‐5p were detected in the rat MIRI model. (A) TTC staining of sham and MIRI rat model. Infarction size was analyzed and displayed in columns. (B) TUNEL assay of sham and MIRI rat model. The percentage of apoptosis myocytes was analyzed and displayed in columns. Tissues of the hearts were subjected to assays in C, D, E: (C) CircHECTD1 level was detected by qRT‐PCR. (D) The miR‐138‐5p level was detected by qRT‐PCR. (E) qRT‐PCR analysis of ROCK2 in sham and MIRI rat model. (F) qRT‐PCR of circHECTD1 in control or H/R‐treated H9c2 cells. (G) MiR‐138‐5p level in H9c2 cells was analyzed using qRT‐PCR. (H) qRT‐PCR analysis of ROCK2 in control or H/R‐treated H9c2 cells. (I) Western blot analysis of ROCK2 in control or H/R‐treated H9c2 cells. Data were analyzed using student's t‐test, *p < 0.05, **p < 0.01.
3.2. CircHECTD1 knockdown reduced H/R‐induced cell inflammatory response
Next, we tried to investigate whether circHECTD1 was involved in H/R‐induced inflammatory response in the H9c2 MIRI model. We established sh‐circHECTD1 and control H9c2 cell lines, and treated them by H/R treatment. Sh‐circHECTD1 successfully decreased circHECTD1 level and significantly attenuated H/R‐induced apoptosis compared with the control group (Figure 2A,D). H/R attenuated the level of miR‐138‐5p and enhanced both the mRNA and protein level of ROCK2, while knockdown of circHECTD1 reversed such effects and restored miR‐138‐5p and ROCK2 to a normal level as compared with the control group (Figure 2B,C). Moreover, expression and secretion of pro‐inflammatory factors (TNF‐α, IL‐6, and IL‐1 β) were all suppressed after transfection with sh‐circHECTD1 in the H/R‐induced H9c2 cells model (Figure 2E,F). In conclusion, the knockdown of circHECTD1 reduced H/R‐induced cell inflammation.
FIGURE 2.

Knockdown of circHECTD1 reduced H/R‐induced inflammatory response of cardiomyocytes. H9c2 cell was treated with H/R and transfected with sh‐NC or sh‐circHECTD1. (A) qRT‐PCR analysis of circHECTD1 in H9c2 cells. (B) qRT‐PCR analysis of miR‐138‐5p and ROCK2. (C) Western blot analysis of ROCK2 expression. (D) Flow cytometry analysis of apoptosis. Apoptosis rates were analyzed and displayed in columns. (E) RNA levels of IL‐6, TNF‐α, and IL‐1β were analyzed by qRT‐PCR. (F) ELISA analysis of IL‐6, TNF‐α, and IL‐1β in H9c2 cells. Data were analyzed using one‐way ANOVA with the Tukey HSD post‐hoc test, *p < 0.05, **p < 0.01, ***p < 0.001.
3.3. CircHECTD1 acted as a sponge of miR‐138‐5p
To investigate whether circHECTD1 could interact with miR‐138‐5p, we conveyed bioinformatics analysis. The binding sites of circHECTD1 and miR‐138‐5p were analyzed by starBase (http://starbase.sysu.edu.cn) and a complementary binding sequence was revealed (Figure 3A). To verify the correlation between miR‐138‐5p and circHECTD1, luciferase reporter plasmids containing wildtype (WT‐) and mutant (MUT‐) circHECTD1 binding sites were designed. The relative luciferase activity was significantly decreased by ectopic miR‐138‐5p mimics compared with mimics NC and was significantly increased by miR‐138‐5p inhibitor compared with inhibitor NC in the WT‐circHECTD1 group, while in MUT‐circHECTD1 group no such differences were observed (Figure 3B). RIP and RNA pulldown assays were then performed between circHECTD1 and miR‐138‐5p. Results demonstrated the direct interaction between circHECTD1 and miR‐138‐5p, where the enrichment between circHECTD1 and miR‐138‐5p were significantly higher than IgG groups (Figure 3C). In the RNA pull‐down experiment, miR‐138‐5p was remarkably enriched in the bio‐circHECTD1 group (Figure 3D). The level of miR‐138‐5p was significantly increased in sh‐circHECTD1 H9c2 cells (Figure 3E). The above results indicated that circHECTD1 could directly bind with miR‐138‐5p and regulated its expression as a sponge.
FIGURE 3.

CircHECTD1 acted as a sponge of miR‐138‐5p. (A) The interaction region of circHECTD1 and miR‐s138‐5p was analyzed by starBase and found that there were complementary binding sites between them. (B) H9c2 cells were transfected with circHECTD1 mutants and miR‐138‐5p mimics, inhibitors, or NC as indicated. A dual luciferase reporter assay was then performed. (C) Cells were harvested by RIP lysis buffer and incubated with human anti‐Ago2 antibody or normal IgG for RIP assay. RNA was detected by qRT‐PR and the relative enrichment level was analyzed. (D) The CircHECTD1 probe was labeled by biotin and incubated with RNA extracted from H9c2 cells. MiR‐138‐5p was detected by qRT‐PCR and the relative enrichment level was analyzed. (E) qRT‐PCR analysis of miR‐138‐5p in H9c2 cells transfected with sh‐circHECTD1 or sh‐NC. Data were analyzed using one‐way ANOVA with the Tukey HSD post‐hoc test, **p < 0.01, ***p < 0.001.
3.4. CircHECTD1‐regulated H/R‐induced inflammation through miR‐138‐5p
Next, we wondered whether miR‐138‐5p played a critical role in circHECTD1‐associated apoptosis and inflammation regulation during MIRI. Sh‐circHECTD1 and miR‐138‐5p inhibitor were thus obtained and subjected to H9c2 cells. MiR‐138‐5p inhibitor decreased the expression of miR‐138‐5p, which was counteracted by sh‐circHECTD1, while miR‐138‐5p inhibitor did not affect circHECTD1 expression (Figure 4A). Subsequently, we treated H9c2 cells under the H/R condition. According to flow cytometry analysis, the MiR‐138‐5p inhibitor aggravated H/R‐induced apoptosis. These effects of miR‐138‐5p inhibitor were nearly abolished by sh‐circHECTD1 (Figure 4B). When we tried to detect inflammatory factors, both qRT‐PCR and ELISA results showed that miR‐138‐5p inhibitor increased H/R‐induced pro‐inflammatory effect, while circHECTD1 knockdown reversed this phenomenon (Figure 4C,D). Through these results, we could conclude that circHECTD1 regulated H/R‐induced inflammation via miR‐138‐5p.
FIGURE 4.

CircHECTD1 regulates H/R‐induced inflammation through miR‐138‐5p. H9c2 cells were transfected with sh‐circHECTD1, sh‐NC, miR‐138‐5p inhibitor or NC inhibitor (A) qRT‐PCR analysis of circHECTD1 and miR‐138‐5p as indicated. (B) Apoptosis analysis of cells in H9c2 cells. (C) qRT‐PCR analysis of TNF‐α, IL‐6 and IL‐1β in H9c2 cells. (D) ELISA analysis of TNF‐α, IL‐6, and IL‐1β in H9c2 cells. Data were analyzed using one‐way ANOVA with the Tukey HSD post‐hoc test, *p < 0.05, **p < 0.01, ***p < 0.001.
3.5. MiR‐138‐5p interacted with the mRNA of ROCK2 and decreased its protein expression
To investigate the detailed regulation mechanism between miR‐138‐5p and ROCK2, the sequences of miR‐138‐5p and ROCK2 were analyzed by starBase. The binding sequence was found in the 3′‐UTR region of ROCK2 (Figure 5A). Dual luciferase reporter plasmid system containing Wild type (WT‐) or Mutant (MUT‐) ROCK2 3′‐UTR was then constructed and luciferase reporter assay was conveyed using this system. In H9c2 cells, the relative luciferase activity of WT‐ROCK2 was decreased by overexpression of miR‐138‐5p. This signal was enhanced by miR‐138‐5p inhibitor, but neither miR‐138‐5p mimics nor inhibitor significantly affected the luciferase activity of MUT‐ROCK2 (Figure 5B). We then successfully transfected inhibitors or mimics of miR‐138‐5p into H9c2 cells (Figure 5C), and detected ROCK2 expression by qRT‐PCR and WB assay. MiR‐138‐5p mimics significantly inhibited ROCK2 expression, while miR‐138‐5p inhibitors significantly promoted the expression of ROCK2 (Figure 5D,E). Furthermore, we also detected whether circHECTD1 knockdown could alter the expression of ROCK2. Results indicated that CircHECTD1 knockdown significantly inhibited the expression of ROCK2 (Figure 5F,G). According to these data, we could deduce that miR‐138‐5p could interact with the mRNA of ROCK2 and inhibit its protein expression.
FIGURE 5.

MiR‐138‐5p interacted with the mRNA of ROCK2 and decreased its protein expression. (A) Interaction region between miR‐138‐5p and ROCK2 was analyzed by starBase. (B) H9c2 cells transfected with ROCK2 mutants and miR‐138‐5p mimics, inhibitors, or NC as indicated. Dual luciferase analysis was performed 48 hours after transfection. (C) qRT‐PCR analysis of miR‐138‐5p in H9c2 cells. (D) qRT‐PCR analysis of ROCK2 mRNA in H9c2 cells transfected with miR‐138‐5p mimics, inhibitor, or NC as indicated. (E) Western blot analysis of ROCK2 in H9c2 cells. (F) qRT‐PCR analysis of ROCK2 mRNA in H9c2 cells. (G) Western blot analysis of ROCK2 in H9c2 cells. Data were analyzed using one‐way ANOVA with the Tukey HSD post‐hoc test, *p < 0.05, **p < 0.01, ***p < 0.001.
3.6. miR‐138‐5p attenuated H/R‐induced myocardial inflammatory response through inhibiting ROCK2
In the next step, we aimed to reveal whether miR‐138‐5p participated in H/R‐induced inflammatory response. MiR‐138‐5p mimics and pcDNA3.1‐ROCK2 were transfected to H9c2 cells and cells were treated with H/R conditions. We then analyzed the apoptosis of these cells by flow cytometry. MiR‐138‐5p mimics alleviated H/R‐induced apoptosis, while overexpression of ROCK2 counteracted such an effect (Figure 6A). Also, both expression and secretion of pro‐inflammatory factors were attenuated by miR‐138‐5p mimics, whereas ROCK2 overexpression antagonized such effects too (Figure 6B,C). Taken together, we concluded that miR‐138‐5p alleviated H/R‐induced inflammatory response by inhibiting ROCK2.
FIGURE 6.
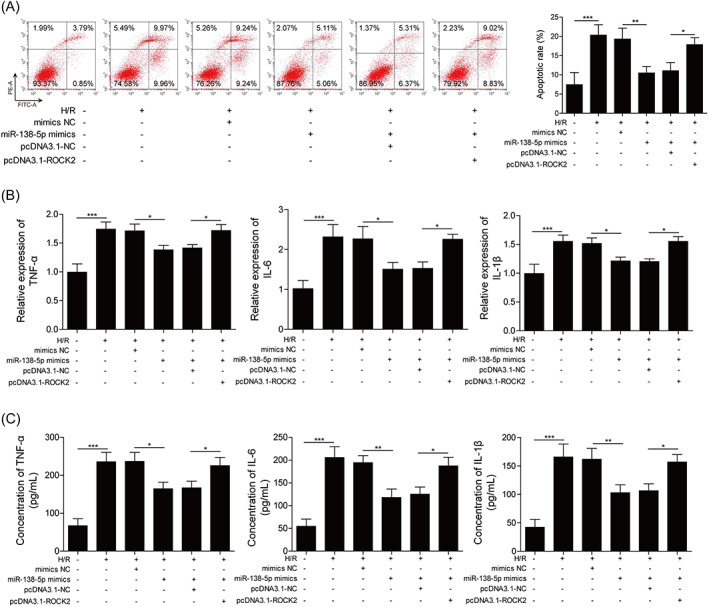
miR‐138‐5p attenuated H/R‐induced myocardial inflammatory response through ROCK2. (A) Apoptosis analysis of H9c2 cells transfected with miR‐138‐5p, pcDNA3.1‐ROCK2, or pcDNA3.1‐NC. (B) Expression of IL‐6, TNF‐α, and IL‐1β in H9c2 cells were detected by qRT‐PCR. (C) ELISA analysis of TNF‐α, IL‐6, and IL‐1β in H9c2 cells. Data were analyzed using one‐way ANOVA with the Tukey HSD post‐hoc test, *p < 0.05, **p < 0.01, ***p < 0.001.
4. DISCUSSION
MIRI, which usually occurred after surgeries such as open‐heart surgery, coronary artery bypass, coronary angioplasty, and thrombolysis, emerged as serious complications in recent decades. Severe MIRI might even cause sudden death due to serious arrhythmia, myocardial stunning or no‐reflow phenomenon, and lethal reperfusion injury. 24 MIRI, therefore, drew much attention. In this study, we first revealed that circHECTD1 played important role in MIRI regulation by sponging miR‐138‐5p. We also proved ROCK2 as a direct target of miR‐138‐5p and proved that miR‐138‐5p alleviated H/R‐induced inflammatory response by inhibiting ROCK2. Hence, circHECTD1 promoted MIRI‐associated inflammatory damage through miR‐138‐5p/ROCK2 axis.
We revealed that circHECTD1 was upregulated during rat MIRI. Similar abnormal expression of circHECTD1 was previously reported in patients with acute ischemic stroke. 8 Our results were in agreement with previous reports, indicating that the elevation of circHECTD1 is a common phenomenon in myocardial ischemic injury. While we identified miR‐138‐5p/ROCK2 as a key signal axis in rat MIRI, it was noteworthy that in similar ischemia‐induced injuries in brain tissue, the downstream signal pathways of circHECTD1 involved miR‐142 or miR‐133, which were different from our study. 9 , 10 This discrepancy might be explained partially by the difference in genomic and expression profiles among different tissues. The abundance and functions of miRNAs and proteins varied between the brain and myocytes. Besides, downstream signals and functions also varied between the brain and myocardial tissues. Meanwhile, circRNA and miRNA could have multiple targets. 25 For instance, circHIPK3 was reported to contain 18 potential binding sites and could sponge to at least 9 miRNAs. 26 In the bioinformatic analysis, we predicted 472 records of miRNAs binding to circHECTD1 through stareBase, and 306 miRNAs were obtained by screening and merging the same miRNAs. CircBank predicted 151 miRNAs binding to circHECTD1, and 44 miRNAs were predicted by both stareBase and circBank. Through a literature search, we found that in addition to the miR‐138‐5p we studied, five more miRNAs (miR‐98‐5p, miR‐338‐5p, miR‐25‐3p, miR‐200a‐3p, miR‐324‐5p) were reported to have low expression during myocardial ischemia and reperfusion, and improving their expression can alleviate MIRI. MiR‐98‐5p exerted such function by reducing TLR4 and activating PI3K/AKT signal pathway, 27 miR‐338‐5p alleviated cerebral I/R injury through rapamycin pathway, 28 miR‐25‐3p alleviated myocardial infarction by targeting EZH2 and pro‐apoptotic proteins, 29 miR‐200a‐3p regulated MIRI by PDCD4 signal axis, 30 miR‐324‐5p regulated MIRI by TRAF3 and Mtfr1 signal. 31 , 32 Based on these notions and evidence, miR‐138‐5p could be one of the key targets of circHECTD1 during MIRI regulation and reflect a facet of the complex signal network of circHECTD1 in MIRI.
Both in our study and previous report, knockdown of circHECTD1 attenuated ischemic reperfusion injury. 8 , 9 , 10 We believe that circHECTD1 was a master regulator in MIRI among different tissues. However, a comprehensive understanding of the regulation network of circHECTD1 requires more intensive studies. Directly upregulating miR‐138‐5p also attenuated ischemic reperfusion injury, indicating that miR‐138‐5p might be a potential therapeutic target in the future. So far, there were many articles investigating the application of miRNA to intervene or treat MIRI. For instance, microRNA‐29a inhibition protected MIRI, miR‐10b‐5p reduced infarct border zone, and they were all considered as potential clinical targets. 33 , 34 In our results, MiR‐138‐5p exerted a similar MIRI protection function.
Identifying miR‐138‐5p as a direct target of circHECTD1 was another highlight of our study. CircRNA was initially considered a widespread “junk” RNA produced from precursor mRNA. Its regulatory mechanism on miRNA was only recently discovered within a decade. The central notion is that circRNA contains multiple binding sites of its target miRNAs and therefore functions as a “sponge” to control the concentration of soluble cellular miRNAs. For example, miRNAs from the miR‐29 family could all be sponged by circRNA_100290, 35 and miR‐422a could be sponged by circNT5E. 36 This notion was now accepted by many scientists, and our finding added another evidence to this “sponge” theory. However, it was noteworthy that another potential function of circRNA was to interact with protein directly, as predicted by a dynamic circRNA‐protein interaction model. 37 Our understanding of circRNA was still limited and needs more investigation.
ROCK2 was implicated in cardiovascular diseases by aggravating MIRI and contributing to myocardial dysfunction. 20 The results of Dominik and Sunnamura 21 , 22 were consistent with our finding that ROCK2 overexpression enhanced apoptosis and inflammation in the MIRI cell model, and was fundamental in MIRI regulation. Such function of ROCK2 was also supported by other studies. For instance, ROCK2 inhibitor suppressed LPS‐induced apoptosis and inflammation in Pulmonary Microvascular Endothelial Cells. 38 In T cells, ROCK2 could induce an inflammatory response in inflammatory bowel disease. 39 Hence, we believe that ROCK2 was a key regulator promoting apoptosis and inflammation in a wide spectrum of diseases and was a key regulator in MIRI.
In this study, we first reported the function of circHECTD1 in MIRI and discovered miR‐138‐5p as a direct target of circHECTD1. We also proved that miR‐138‐5p regulated apoptosis and inflammation in MIRI via inhibiting ROCK2. Our study established a novel regulation signal axis that circHECTD1 promoted myocardial ischemia/reperfusion injury‐associated inflammatory damage through sponging miR‐138‐5p and upregulating ROCK2. This finding could deepen our understanding of the regulatory mechanism of MIRI, expand the potential therapeutic targets of MIRI, and provide theoretical support for follow‐up studies. For instance, delivering miRNA‐138‐5p into MIRI tissue might be a potential direction to develop new treatment options. Besides, inhibiting downstream signals of ROCK2 could also be a potential option. It was noteworthy that the control group of the MIRI model did not undergo ischemia. Although most studies about MIRI followed the same study design, there was still a possibility that the results were not caused by reperfusion damage after ischemia, but may also be caused by ischemia alone. Besides, we chose 30 min as ischemia time according to a previous publication, 23 we did notice that Chang et al (Acta Cardiol Sin 2016) reported that 30 min was not an ideal option for MIRI induction. We could not explain why these two groups generated contradictive results, but researchers must optimize methods from other publications and make sure desired results could be generated in their lab. This study only revealed the tip of the iceberg of the complex regulatory network of MIRI, and more research is still needed.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
Supporting information
Figure S1. (A) qRT‐PCR analysis of circHECTD1 in ischemic‐free, peri‐ischemic, ischemic zone. (B) qRT‐PCR analysis of circHECTD1 in Serum. (C) qRT‐PCR analysis of circHECTD1 in Serum at different times. Data were analyzed using one‐way ANOVA with the Tukey HSD post‐hoc test, *p < 0.05, **p < 0.01, ***p < 0.001. For each group of the animal experiment, n = 6.
{kind=link}
ACKNOWLEDGMENTS
The authors thank the members of our laboratory for their contributions.
Yang Y‐N, Luo Y‐B, Xu G, Li K, Ma R‐L, Yuan W. CircHECTD1 promoted MIRI‐associated inflammation via inhibiting miR‐138‐5p and upregulating ROCK2 . Kaohsiung J Med Sci. 2023;39(7):675–687. 10.1002/kjm2.12686
REFERENCES
- 1. Li J, Sun D, Li Y. Novel findings and therapeutic targets on cardioprotection of ischemia/ reperfusion injury in STEMI. Curr Pharm Des. 2019;25(35):3726–39. [DOI] [PubMed] [Google Scholar]
- 2. Carbone F, Bonaventura A, Montecucco F. Neutrophil‐related oxidants drive heart and brain remodeling after ischemia/reperfusion injury. Front Physiol. 2020;10:1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carney EF. Inflammation: activated protein C inhibits inflammasome activation in IRI. Nat Rev Nephrol. 2017;13(11):662. [DOI] [PubMed] [Google Scholar]
- 4. Chen L‐L, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12(4):381–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bai XF, Niu RZ, Liu J, Pan XD, Wang F, Yang W, et al. Roles of noncoding RNAs in the initiation and progression of myocardial ischemia‐reperfusion injury. Epigenomics. 2021;13(9):715–43. [DOI] [PubMed] [Google Scholar]
- 6. Zhang H‐d, Jiang L‐H, Sun D‐W, Hou J‐C, Ji Z‐L. CircRNA: a novel type of biomarker for cancer. Breast Cancer. 2018;25(1):1–7. [DOI] [PubMed] [Google Scholar]
- 7. Sun H, Wang J, Que J, Peng Y, Yu Y, Wang L, et al. RNA sequencing revealing the role of AMP‐activated protein kinase signaling in mice myocardial ischemia reperfusion injury. Gene. 2019;703:91–101. [DOI] [PubMed] [Google Scholar]
- 8. Peng X, Jing P, Chen J, Xu L. The role of circular RNA HECTD1 expression in disease risk, disease severity, inflammation, and recurrence of acute ischemic stroke. J Clin Lab Anal. 2019;33(7):e22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han B, Zhang Y, Zhang Y, Bai Y, Chen X, Huang R, et al. Novel insight into circular RNA HECTD1 in astrocyte activation via autophagy by targeting MIR142‐TIPARP: implications for cerebral ischemic stroke. Autophagy. 2018;14(7):1164–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai Q, Ma Y, Xu Z, Zhang L, Yang H, Liu Q, et al. Downregulation of circular RNA HECTD1 induces neuroprotection against ischemic stroke through the microRNA‐133b/TRAF3 pathway. Life Sci. 2021;264:118626. [DOI] [PubMed] [Google Scholar]
- 11. Shen LS, Hu XF, Chen T, Shen GL, Cheng D. Integrated network analysis to explore the key mRNAs and lncRNAs in acute myocardial infarction. Math Biosci Eng. 2019;16(6):6426–37. [DOI] [PubMed] [Google Scholar]
- 12. Ong S‐B, Katwadi K, Kwek X‐Y, Ismail NI, Chinda K, Ong S‐G, et al. Non‐coding RNAs as therapeutic targets for preventing myocardial ischemia‐reperfusion injury. Expert Opin Ther Targets. 2018;22(3):247–61. [DOI] [PubMed] [Google Scholar]
- 13. Morelli MB, Shu J, Sardu C, Matarese A, Santulli G. Cardiosomal microRNAs are essential in post‐infarction myofibroblast Phenoconversion. Int J Mol Sci. 2019;21(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kura B, Szeiffova Bacova B, Kalocayova B, Sykora M, Slezak J. Oxidative stress‐responsive MicroRNAs in heart injury. Int J Mol Sci. 2020;21(1):358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J, Zhang M, Zhang S, Wu J, Xue S. Rno‐microRNA‐30c‐5p promotes myocardial ischemia reperfusion injury in rats through activating NF‐κB pathway and targeting SIRT1. BMC Cardiovasc Disord. 2020;20(1):240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meng X, Mei L, Zhao C, Chen W, Zhang N. miR‐885 mediated cardioprotection against hypoxia/reoxygenation‐induced apoptosis in human cardiomyocytes via inhibition of PTEN and BCL2L11 and modulation of AKT/mTOR signaling. J Cell Physiol. 2020;235(11):8048–57. [DOI] [PubMed] [Google Scholar]
- 17. Cai J, Gehrau R, Tu Z, Leroy V, Su G, Shang J, et al. MicroRNA‐206 antagomiR–enriched extracellular vesicles attenuate lung ischemia–reperfusion injury through CXCL1 regulation in alveolar epithelial cells. J Heart Lung Transplant. 2020;39(12):1476–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fu Y, Liu M, Li F, Qian L, Zhang P, Lv F, et al. MiR‐221 promotes hepatocellular carcinoma cells migration via targeting PHF2. Biomed Res Int. 2019;2019:4371405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mao Q, Liang XL, Zhang CL, Pang YH, Lu YX. LncRNA KLF3‐AS1 in human mesenchymal stem cell‐derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR‐138‐5p/Sirt1 axis. Stem Cell Res Ther. 2019;10(1):393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hartmann S, Ridley AJ, Lutz S. The function of rho‐associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front Pharmacol. 2015;6:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kentrup D, Reuter S, Schnöckel U, Grabner A, Edemir B, Pavenstädt H, et al. Hydroxyfasudil‐mediated inhibition of ROCK1 and ROCK2 improves kidney function in rat renal acute ischemia‐reperfusion injury. PLoS One. 2011;6(10):e26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sunamura S, Satoh K, Kurosawa R, Ohtsuki T, Kikuchi N, Elias‐Al‐Mamun M, et al. Different roles of myocardial ROCK1 and ROCK2 in cardiac dysfunction and postcapillary pulmonary hypertension in mice. Proc Natl Acad Sci U S A. 2018;115(30):E7129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li D, Wang X, Huang Q, Li S, Zhou Y, Li Z. Cardioprotection of CAPE‐oNO(2) against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF‐κB pathway in vivo and in vitro. Redox Biol. 2018;15:62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Q, Li Z, Fan Z, Yang Y, Lu C. Involvement of non‐coding RNAs in the pathogenesis of myocardial ischemia/reperfusion injury (review). Int J Mol Med. 2021;47(4):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li X, Yang L, Chen L‐L. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018;71(3):428–42. [DOI] [PubMed] [Google Scholar]
- 26. Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang L, Wei Q, Liu X, Zhang T, Wang S, Zhou L, et al. Exosomal microRNA‐98‐5p from hypoxic bone marrow mesenchymal stem cells inhibits myocardial ischemia‐reperfusion injury by reducing TLR4 and activating the PI3K/Akt signaling pathway. Int Immunopharmacol. 2021;101(Pt B):107592. [DOI] [PubMed] [Google Scholar]
- 28. Yi X, Fang Q, Li L. MicroRNA‐338‐5p alleviates cerebral ischemia/reperfusion injury by targeting connective tissue growth factor through the adenosine 5′‐monophosphate‐activated protein kinase/mammalian target of rapamycin signaling pathway. Neuroreport. 2020;31(3):256–64. [DOI] [PubMed] [Google Scholar]
- 29. Peng Y, Zhao JL, Peng ZY, Xu WF, Yu GL. Exosomal miR‐25‐3p from mesenchymal stem cells alleviates myocardial infarction by targeting pro‐apoptotic proteins and EZH2. Cell Death Dis. 2020;11(5):317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun R, Zhang L. Long non‐coding RNA MALAT1 regulates cardiomyocytes apoptosis after hypoxia/reperfusion injury via modulating miR‐200a‐3p/PDCD4 axis. Biomed Pharmacother. 2019;111:1036–45. [DOI] [PubMed] [Google Scholar]
- 31. Huang L, Guo B, Liu S, Miao C, Li Y. Inhibition of the LncRNA Gpr19 attenuates ischemia‐reperfusion injury after acute myocardial infarction by inhibiting apoptosis and oxidative stress via the miR‐324‐5p/Mtfr1 axis. IUBMB Life. 2020;72(3):373–83. [DOI] [PubMed] [Google Scholar]
- 32. Jin A, Zhang Q, Cheng H, Yang C, Wang X. Circ_0050908 up‐regulates TRAF3 by sponging miR‐324‐5p to aggravate myocardial ischemia‐reperfusion injury. Int Immunopharmacol. 2022;108:108740. [DOI] [PubMed] [Google Scholar]
- 33. Wu L, Chen Y, Chen Y, Yang W, Han Y, Lu L, et al. Effect of HIF‐1α/miR‐10b‐5p/PTEN on hypoxia‐induced cardiomyocyte apoptosis. J Am Heart Assoc. 2019;8(18):e011948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ding S, Liu D, Wang L, Wang G, Zhu Y. Inhibiting MicroRNA‐29a protects myocardial ischemia‐reperfusion injury by targeting SIRT1 and suppressing oxidative stress and NLRP3‐mediated Pyroptosis pathway. J Pharmacol Exp Ther. 2020;372(1):128–35. [DOI] [PubMed] [Google Scholar]
- 35. Chen L, Zhang S, Wu J, Cui J, Zhong L, Zeng L, et al. circRNA_100290 plays a role in oral cancer by functioning as a sponge of the miR‐29 family. Oncogene. 2017;36(32):4551–61. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Wang R, Zhang S, Chen X, Li N, Li J, Jia R, et al. CircNT5E acts as a sponge of miR‐422a to promote glioblastoma tumorigenesis. Cancer Res. 2018;78(17):4812–25. [DOI] [PubMed] [Google Scholar]
- 37. Du WW, Zhang C, Yang W, Yong T, Awan FM, Yang BB. Identifying and characterizing circRNA‐protein interaction. Theranostics. 2017;7(17):4183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang J, Ruan F, Zheng Z. Ripasudil attenuates lipopolysaccharide (LPS)‐mediated apoptosis and inflammation in pulmonary microvascular endothelial cells via ROCK2/eNOS signaling. Med Sci Monit. 2018;24:3212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang W, Zhou G, Yu T, Chen L, Yu L, Guo Y, et al. Critical role of ROCK2 activity in facilitating mucosal CD4(+) T cell activation in inflammatory bowel disease. J Autoimmun. 2018;89:125–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) qRT‐PCR analysis of circHECTD1 in ischemic‐free, peri‐ischemic, ischemic zone. (B) qRT‐PCR analysis of circHECTD1 in Serum. (C) qRT‐PCR analysis of circHECTD1 in Serum at different times. Data were analyzed using one‐way ANOVA with the Tukey HSD post‐hoc test, *p < 0.05, **p < 0.01, ***p < 0.001. For each group of the animal experiment, n = 6.