

Abstract
Acute lung injury (ALI) is an adverse disease of the respiratory system, and one of its prevalent causes is sepsis induction. Cell pyroptosis facilitates the progression of ALI and lncRNAs play critical roles in ALI. Thus, this research seeks to investigate the specific mechanism of NEAT1 in sepsis‐ALI.BEAS‐2B cells were exposed to lipopolysaccharide (LPS) to construct a cell model of sepsis‐induced ALI. The gene and protein expression were assessed using qRT‐PCR and western blot. Cell viability was identified by CCK‐8. Cell death was discovered using PI staining. The secretion of IL‐1β and IL‐18 was examined using ELISA. The interconnections among NEAT1, miR‐26a‐5p, and ROCK1 were confirmed using starbase, luciferase assay, and RIP.LPS treatment augmented NEAT1 and ROCK1 levels while mitigating miR‐26a‐5p level in BEAS‐2B cells. Additionally, LPS treatment facilitated cell death and cell pyroptosis, whereas NEAT1 silencing could reverse these effects in BEAS‐2B cells. Mechanistically, NEAT1 positively mediated ROCK1 expression by targeting miR‐26a‐5p. Furthermore, miR‐26a‐5p inhibitor offset NEAT1 depletion‐mediated suppressive effects on cell death and cell pyroptosis. ROCK1 upregulation decreased the inhibitory impacts produced by miR‐26a‐5p overexpression on cell death and cell pyroptosis. Our outcomes demonstrated NEAT1 could reinforce LPS‐induced cell death and cell pyroptosis by repressing the miR‐26a‐5p/ROCK1 axis, thereby worsening ALI caused by sepsis. Our data indicated NEAT1, miR‐26a‐5p, and ROCK1 might be biomarkers and target genes for relieving sepsis‐induced ALI.
Keywords: acute lung injury, lncRNA NEAT1, miR‐26a‐5p, ROCK1, sepsis
Abbreviation
- ALI
acute lung injury
- LPS
lipopolysaccharide
- ROCK1
Rho kinase 1
- RIP
RNA immunoprecipitation
- CTGF
connective tissue growth factor
1. INTRODUCTION
Sepsis is a life‐threatening disease accompanied by fatal multiorgan dysfunction because of the imbalance of infection response. 1 Strong inflammatory reaction promotes further damage to the body, which may induce cardiovascular paralysis and shock, multiple organ injury, and even death. 2 The lung is the most vulnerable organ. 3 Then, sepsis typically induces acute lung injury (ALI) characterized by cell death of lung epithelial cells and excessively inflammatory response, eventually evolving into respiratory failure. 4 Cell pyroptosis, also known as inflammatory necrosis, is a type of programmed cell death, which has a well‐established close association with sepsis‐induced ALI. 5 , 6 Therefore, we focus on enhancing sepsis‐induced ALI by seeking techniques for suppressing cell pyroptosis.
MicroRNAs are an indispensable component of non‐coding RNAs involving various biological regulatory processes in eukaryotes. 7 Several microRNAs such as miR‐223 and miR‐30d‐5p were found to be engaged in relieving or worsening the advancement of ALI by mediating cell pyroptosis. 8 , 9 The miR‐26a‐5p has been found to play protective roles in sepsis‐induced ALI. 10 , 11 However, whether miR‐26a‐5p participates in sepsis‐induced ALI by impacting pyroptosis and its underlying molecular regulation mechanism remains to be further clarified.
Generally, lncRNAs can sponge microRNAs to modulate the progression of diseases from the viewpoint of epigenetics. 12 lncRNA NEAT1 as a remarkable lncRNA has been studied and played important regulatory roles in many diseases including ALI caused by sepsis. 13 , 14 Starbase 15 forecasted binding sequences between NEAT1 and miR‐26a‐5p. Currently, these two molecules for the regulation of sepsis‐induced ALI have not been evaluated.
Rho kinase 1 (ROCK1) is a momentous downstream effector of RhoA, which was found to be closely linked with ALI. 16 , 17 As previously described, ROCK1 controlled by miR‐539‐5p worsened sepsis‐induced ALI by promoting apoptosis and inflammation. 16 Notably, ROCK1 could regulate NLRP3‐mediated pyroptosis to influence sepsis‐induced acute kidney injury and ventilator‐lung injury, 18 , 19 implying that ROCK1 might impact cell pyroptosis to mediate sepsis‐induced ALI. Furthermore, starbase forecasted miR‐26a‐5p bind to ROCK1. Therefore, we presume that the miR‐26a‐5p/ROCK1 axis might regulate cell pyroptosis to influence sepsis‐induced ALI, currently, which has not been demonstrated.
Based on these backgrounds, we present our hypothesis that miR‐26a‐5p, regulated by NEAT1, could target ROCK1 to suppress LPS‐induced cell pyroptosis of BEAS‐2B cells, further relieving the advancement of sepsis‐induced ALI.
2. METHODS
2.1. Cell culture and treatment
Human lung bronchus epithelial cells (BEAS‐2B) and 293 T cells were obtained from ATCC (USA). Under the condition of 37°C and 5% CO2, these cells were cultured in DMEM (Thermo Fisher Scientific, USA) which had 10% FBS (Gibco, USA), 1% penicillin, and streptomycin (Sigma). To simulate sepsis‐induced ALI in vitro, BEAS‐2B cells were given 1 μg/mL LPS for 12 h referring to a previous study. 20 This cell model was employed for subsequent experiments.
2.2. Cell transfection
si‐NEAT1, miR‐26a‐5p mimic/inhibitor, pcDNA3.1‐ROCK1, and corresponding control groups (si‐NC, miR‐NC, pcDNA3.1) were obtained from GenePharma (China). Following the manual, BEAS‐2B cells and 293 T cells were subjected to transfection with the above sequences and plasmids based on our experimental design using Lipofectamine™ 3000 (Invitrogen, CA).
2.3. RNA isolation, reverse transcription, and RT‐qPCR
The RNA was collected from BEAS‐2B cells with LPS treatment and transfection by TRIzol reagent (Invitrogen, USA). Then, using Prime Script Reverse Transcription Reagent Kit (TaKaRa, China) synthesized cDNA based on RNA. SYBR Premix Ex Taq II Kit (TaKaRa) was processed in qPCR. Below are the primer sequences:
NEAT1 (F): GTGGCTGTTGGAGTCGGTAT.
NEAT1 (R): TAACAAACCACGGTCCATGA.
miR‐26a‐5p (F): GCCCGTTCAAGTAATCCAGGA.
miR‐26a‐5p (R): GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCCTA.
ROCK1(F): TGCAACTGGAACTCAACCAA.
ROCK1 (R): GTTTAGCACGCAATTGCTCA.
U6 (F): CTCGCTTCGGCAGCACA.
U6 (R): AACGCTTCACGAATTTGCGT.
GAPDH (F): AGGTCGGAGTCAACGGATTT.
GAPDH (R): TGACGGTGCCATGGAATTTG.
The relative quantification was estimated with the 2−ΔΔCt formula. U6 or GAPDH functioned as the reference gene.
2.4. The evaluation of cell viability
Cell viability of BEAS‐2B cells with indicated treatment was assessed using Cell Counting Kit‐8 (CCK‐8, Beyotime, China). BEAS‐2B cells with indicated treatment were embedded onto 96‐well plates (3 × 103 cells/well) and cultured overnight. CCK‐8 solution was added to the plates. Then, the absorbance was analyzed at 450 nm after 4 h incubation with CCK‐8 solution.
2.5. Propidium iodide staining
BEAS‐2B cells were seeded onto a 24‐well plate (8 × 104 cells/well) and were cultured overnight. DAPI solution (5 μg/mL) and propidium iodide (PI) staining solution (2 μg/mL) were integrated into each well. After incubation at 4°C for 20 min, all cells were monitored under a fluorescence microscope.
2.6. Western blot
BEAS‐2B cells with indicated treatment were lysed with RIPA buffer (Beyotime) to obtain proteins. The concentration of total protein in multiple groups was identified using a BCA Assay Kit (Beyotime). After the quantitative, equivalent proteins (30 μg) were separated by SDS–PAGE and then were transferred onto PVDF membranes. Blocked by non‐fat milk, anti‐ROCK1 (abcam, 1:1000, ab134181, UK), anti‐NLRP3 (abcam, 1:1000, ab263899), anti‐ASC (1:1000, ab283684), anti‐Cleaved caspase1 (1:1000, ab207802), anti‐Cleaved Gasdermin‐D (1:1000, ab215203), and anti‐GAPDH (1:4000, ab8245) incubated the PVDF membranes for 12 h at 4°C. Then, an HRP‐labeled secondary antibody (1:10000, Absin, no. abs20039, China) incubated the membranes for 1 h. ECL chemiluminescent reagent (Beyotime) was conducted to react with the proteins on PVDF membranes and an ECL chemiluminescence detection system (Thermo Fisher Scientific) was carried out to visualize these protein bands. The densitometry analysis was assessed using ImageJ (National Institutes of Health, USA).
2.7. ELISA
BEAS‐2B cells with indicated treatment were lysed onto a 6‐well plate (5 × 105 cells/well) and were cultured overnight. The detection kits of IL‐1β and IL‐18 (Biolegend, China) were applied to identify the concentration of inflammatory cytokines in supernatants of BEAS‐2B cells after the corresponding protocol. Afterward, the data was documented at an absorbance value of 450 nm.
2.8. Luciferase assay
The starbase (https://starbase.sysu.edu.cn) forecasted the binding sites of NEAT1/miR‐26a‐5p and miR‐26a‐5p/ROCK1. The binding sequences of NEAT1/ROCK1 which bind with miR‐26a‐5p were cloned into psiChECK2vectors (Ke Lei Biological Technology Co., Ltd, China) for constructing reporter vectors NEAT1/ROCK1 (WT/MUT). MiR‐26a‐5p mimics in combination with NEAT1/ROCK1 (WT/MUT) were transfected into the 293 T cells by Lipofectamine™ 3000 (Invitrogen, USA) following the protocol. After transfection for 48 h, the luciferase activity was discovered using a dual‐luciferase reporter assay system (Promega, Madison, WI).
2.9. RNA immunoprecipitation assay
Following the manual, Magna RIP RNA‐Binding Protein Immunoprecipitation Kit (Millipore, USA) was employed for RNA immunoprecipitation (RIP) analysis. Briefly, BEAS‐2B cells (5 × 106 cells/mL) were lysed and the extractive was subjected to incubation with magnetic beads conjugated with antibodies against Argonaute2 (Ago2, ab32381, abcam) and immunoglobulin G (IgG, ab109489, abcam). Afterward, Immunoprecipitated RNA (NEAT1 and miR‐26a‐5p) was determined utilizing qRT‐PCR analysis.
2.10. Data analysis
Data were expressed as mean ± SD and all data were obtained from at least three replicate experiments. Statistical analysis was prepared by GraphPad Prism 6 (San Diego, USA) using a Student's paired T‐test between two groups and a one‐way analysis of variance followed by Tukey's post hoc test among three or more three groups. The p‐values <0.05 was deemed statistically significant.
3. RESULTS
3.1. NEAT1 knockdown attenuated LPS‐induced pyroptosis in BEAS‐2B cells
To imitate sepsis‐induced cell injury, BEAS‐2B cells were given 1 μg/mL LPS treatment for 12 h. Compared with control, LPS induced NEAT1 expression (Figure 1A). To evaluate the role of NEAT1 in ALI caused by sepsis, si‐NEAT1 was introduced into BEAS‐2B cells and underwent LPS induction. Our results displayed that NEAT1 silencing largely mitigated NEAT1 expression when compared with the LPS + si‐NC group (Figure 1B). Furthermore, LPS administration suppressed cell viability, while NEAT1 knockdown abrogated this impact (Figure 1C). PI staining exhibited LPS stimulation resulted in elevated cell death, which were attenuated by NETA1 downregulation (Figure 1D). Furthermore, the expression pyroptosis‐related proteins including NLRP3, ASC, Cleaved caspase1, and Cleaved Gasdermin‐D were evidently LPS stimulation, however, these alterations influenced by LPS treatment were antagonized by NEAT1 knockdown (Figure 1E). LPS treatment could notably elevate the levels of IL‐1β and IL‐18, which were antagonized by NEAT1 deficiency (Figure 1F). Taken together, NEAT1 deficiency could mitigate cell injury caused by LPS‐treatment in BEAS‐2B cells.
FIGURE 1.
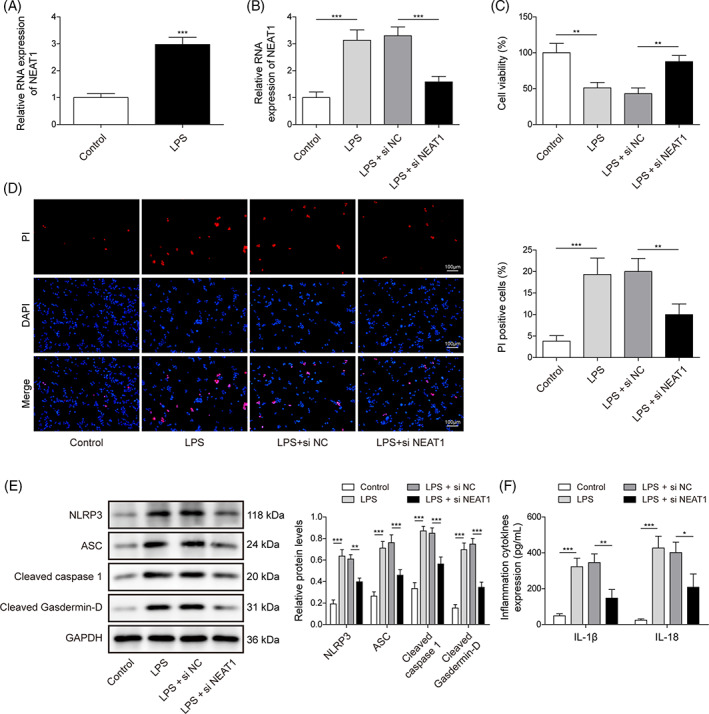
NEAT1 knockdown attenuated LPS‐induced pyroptosis in BEAS‐2B cells. (A) NEAT1 was analyzed using qRT‐PCR in BEAS‐2B cells with/without LPS treatment. BEAS‐2B cells suffered si‐NEAT1 or si‐NC transfection upon LPS administration. (B) NEAT1 was evaluated using qRT‐PCR. (C) Cell viability was analyzed using CCK8. (D) Cell death was determined by PI staining. (E) The protein levels of pyroptosis‐related proteins were investigated using a western blot. (F) The levels of IL‐1β and IL‐18 were determined using ELISA. All data collected from at least three replicate experiments were indicated as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
3.2. NEAT1 restricted miR‐26a‐5p expression
Next, we determined the key miRNA that could be modulated by NEAT1 and is involved in sepsis‐induced ALI. Consequently, we found miR‐26a‐5p level was apparently mitigated by LPS treatment in BEAS‐2B cells (Figure 2A). Furthermore, starbase forecasted miR‐26a‐5p could bind to NEAT1 (Figure 2B). Luciferase activity in the NEAT1‐WT group was overtly subdued by miR‐26a‐5p mimics; however, miR‐26a‐5p mimics acquired failure to change luciferase activity in NEAT1‐MUT group (Figure 2C). Furthermore, the RIP assay demonstrated Ago2 enriched NEAT1 and miR‐26a‐5p, but IgG failed to enrich those (Figure 2D). Additionally, NEAT1 silencing could recover the miR‐26a‐5p level which reduced by LPS‐treatment in BEAS‐2B cells (Figure 2E). As mentioned above, NEAT1 negatively mastered miR‐26a‐5p expression via interplaying with miR‐26a‐5p.
FIGURE 2.
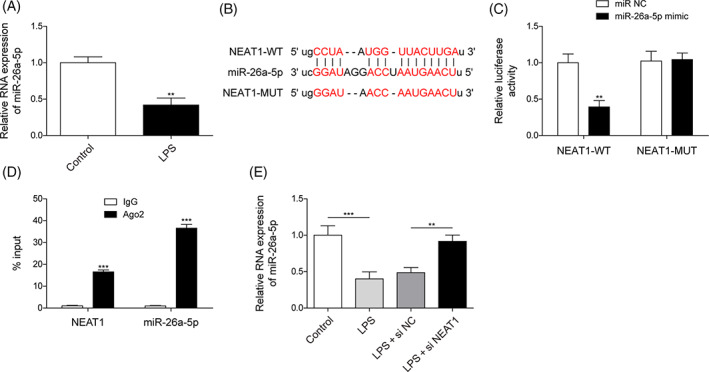
NEAT1 restricted miR‐26a‐5p expression. (A) MiR‐26a‐5p was calculated using qRT‐PCR in BEAS‐2B cells with/without LPS treatment. (B–D) The intercorrelation of NEAT1 and miR‐26a‐5p was forecasted and validated using Starbase, dual luciferase assay, and RIP. (E) miR‐26a‐5p was identified using qRT‐PCR in BEAS‐2B cells with si‐NEAT1 or si‐NC transfection upon LPS treatment. All data collected from at least three replicate experiments were expressed as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
3.3. miR‐26a‐5p inhibition abolished the suppression of NEAT1 knockdown on cell pyroptosis
To further unearth whether NEAT1 promotes cell pyroptosis caused by LPS induction via targeting miR‐26a‐5p, BEAS‐2B cells were delivered with si‐NEAT1 and miR‐26a‐5p inhibitor and followed LPS treatment. NEAT1 silencing substantially elevated LPS‐reduced miR‐26a‐5p level, while miR‐26a‐5p inhibitor suppressed NEAT1 silencing‐enhanced miR‐26a‐5p but failed to alter NEAT1 expression (Figure 3A). For biological function analysis, NEAT1 knockdown increased cell viability, and decreased cell death in LPS‐treated BEAS‐2B cells, whereas these phenomena were impaired by miR‐26a‐5p depletion (Figure 3B,C). Furthermore, NEAT1‐mediated inhibition of pyroptosis‐related proteins including NLRP3, ASC, Cleaved caspase1, and Cleaved Gasdermin‐D, and levels of IL‐1β and IL‐18 were recovered by loss of miR‐26a‐5p (Figure 3D,E). In total, NEAT1 knockdown relieved cell injury with LPS treatment by boosting miR‐26a‐5p.
FIGURE 3.
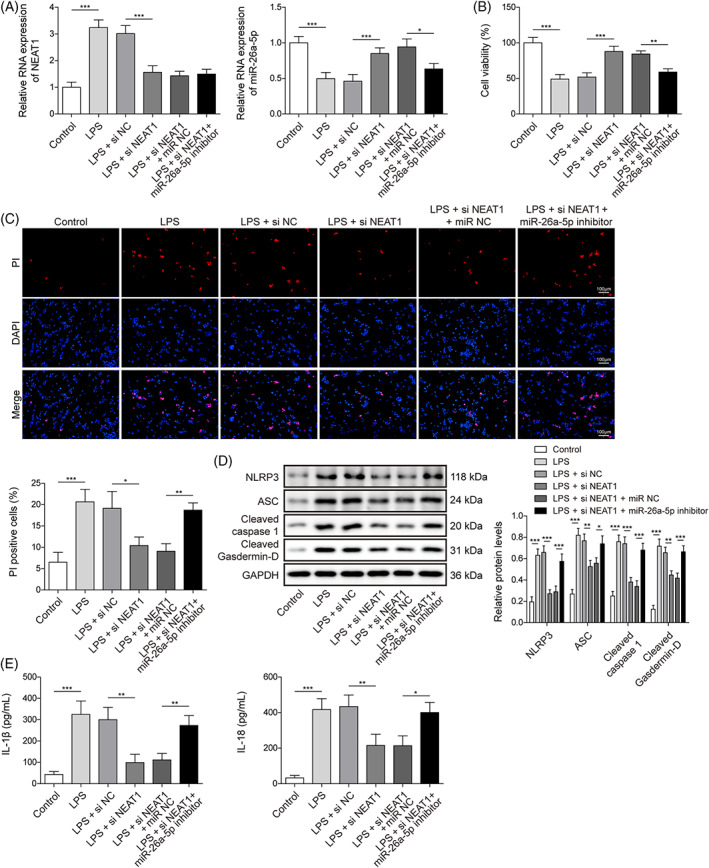
miR‐26a‐5p inhibition abolished the suppression of NEAT1 knockdown on cell pyroptosis. si‐NEAT1 or in conjunction with miR‐26a‐5p inhibitor was transferred into BEAS‐2B cells and followed LPS treatment. (A) NEAT1 and miR‐26a‐5p were evaluated using qRT‐PCR. (B) Cell viability was calculated using CCK8. (C) Cell death was assessed by PI staining. (D) The protein levels of pyroptosis‐related proteins including NLRP3, ASC, Cleaved caspase1, and Cleaved Gasdermin‐D were identified using western blot. (E) The levels of IL‐1β and IL‐18 were tested using ELISA. All data collected from at least three replicate experiments were denoted as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
3.4. miR‐26a‐5p targeted ROCK1 and mediated ROCK1 expression
We further excavated the targeted molecule of miR‐26a‐5p. Primarily, in LPS‐treated BEAS‐2B cells, the ROCK1 level was memorably enhanced (Figure 4A,B). Subsequently, starbase predicted miR‐26a‐5p could bond to ROCK1 (Figure 4C). Furthermore, miR‐26a‐5p mimic dramatically constrained luciferase activity in 293 T cells containing ROCK1‐WT, while had no alteration in the ROCK‐MUT group (Figure 4D). As anticipated, miR‐26a‐5p overexpression eliminated LPS‐induced ROCK1 level (Figure 4E,F). Therefore, we deduced the miR‐26a‐5p bond to ROCK1 and suppressed its expression.
FIGURE 4.
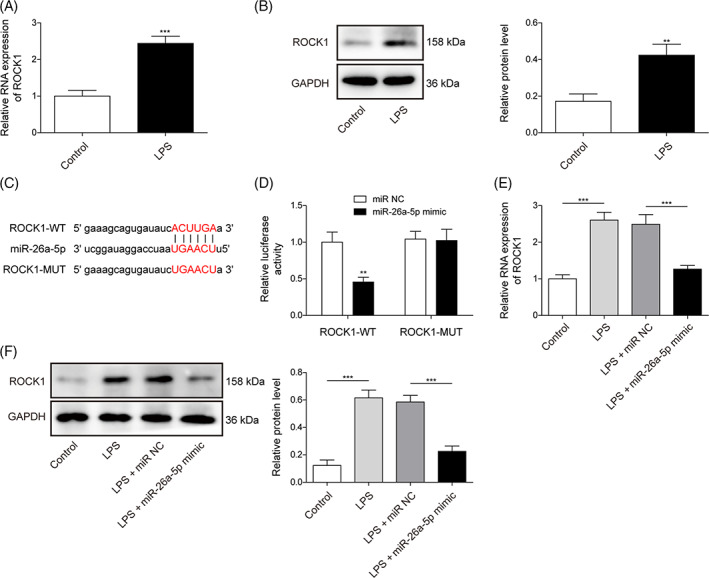
miR‐26a‐5p targeted ROCK1 and mediated ROCK1 expression. (A,B) ROCK1 was evaluated using qRT‐PCR and western blot in BEAS‐2B cells with/without LPS treatment. (C,D) the connection between ROCK1 and miR‐26a‐5p was forecasted and validated using Starbase and dual luciferase assay. (E,F) ROCK1 was analyzed using qRT‐PCR and western blot in BEAS‐2B cells with miR‐26a‐5p mimic or miR‐NC transfection upon LPS administration. All data procured from at least three replicate experiments were denoted as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
3.5. ROCK1 upregulation compromised the suppression of miR‐26a‐5p mimic on cell pyroptosis
Here, the roles of the miR‐26a‐5p/ROCK1 axis in cell pyroptosis with LPS treatment were thoroughly assessed. First, in BEAS‐2B cells, we upregulated miR‐26a‐5p or/and ROCK1 expression via miR‐26a‐5p mimic or/and pcDNA3.1‐ROCK1 transfection. MiR‐26a‐5p mimic significantly increased miR‐26a‐5p and substantially lowered ROCK1 in BEAS‐2B cells with LPS induction. pcDNA3.1‐ROCK1 evidently enhanced ROCK1 expression and eliminated miR‐26a‐5p upregulation effects on ROCK1 expression, but failed to alter miR‐26a‐5p (Figure 5A,B). Furthermore, miR‐26a‐5p overexpression could abrogate LPS‐induced repression of cell viability and upregulation of cell death, but these effects were eliminated by ROCK1 overexpression (Figure 5C,D). Besides, miR‐26a‐5p mimic inhibited the protein levels of NLRP3, ASC, Cleaved caspase1, and Cleaved Gasdermin‐D and the level of IL‐1β and IL‐18, which were abrogated by ROCK1 overexpression (Figure 5E,F). Conclusively, miR‐26a‐5p mitigated LPS‐induced cell pyroptosis by reducing the ROCK1 level.
FIGURE 5.
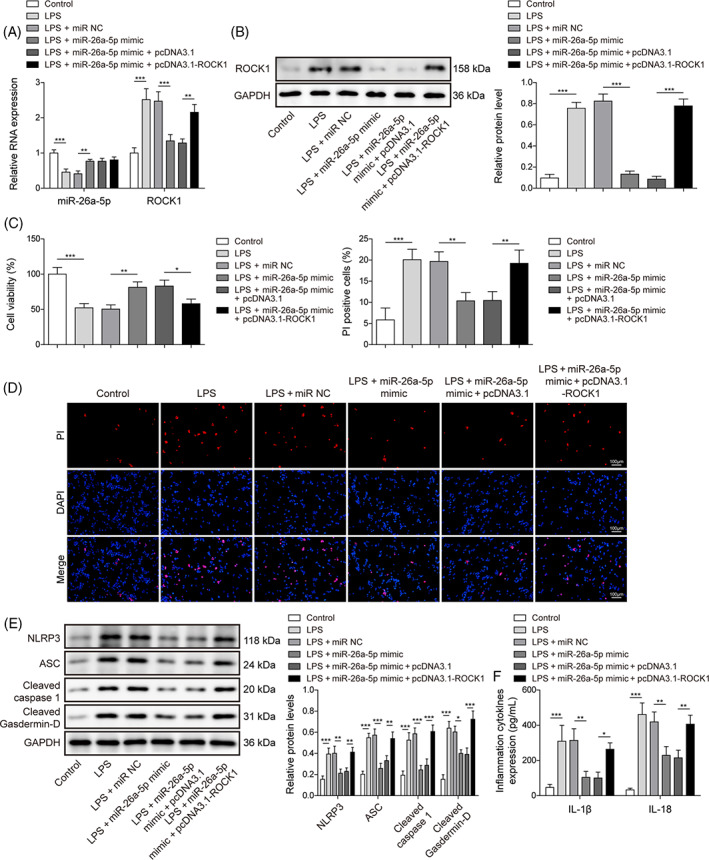
ROCK1 upregulation compromised the suppression of miR‐26a‐5p mimic on pyroptosis. miR‐26a‐5p mimic or in conjunction with pcDNA3.1‐ROCK1 was transfected into BEAS‐2B cells and followed LPS treatment. (A) ROCK1 and miR‐26a‐5p were evaluated using qRT‐PCR. (B) ROCK1 was determined using a western blot. (C) cell viability was identified using CCK8. (D) cell death was determined with PI staining. (E) protein levels of pyroptosis‐related proteins including NLRP3, ASC, Cleaved caspase1, and Cleaved Gasdermin‐D were assessed by western blot. (F) The levels of IL‐1β and IL‐18 were tested using ELISA. All data procured from at least three replicate experiments were implied as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001.
4. DISCUSSION
Pyroptosis mediates cell death defined by producing holes in the cell membrane, influencing the release of proinflammatory cytokines, and eventually occurring cell lysis. 21 NLRP3 inflammasome is one of triggers to cell pyroptosis, which combines with ASC to promote caspase‐1 activation which further cleaves GSDMD and pro‐IL‐1β and pro‐IL‐18 to contribute to a cascade amplification of inflammation. 22 Numerous studies have indicated that cell pyroptosis is responsible for damage to a large extent in sepsis‐induced ALI. 23 , 24 Therefore, curbing cell pyroptosis is a direction of relieving sepsis‐induced ALI. In our research, we highlighted NEAT1 knockdown regulated miR‐26a‐5p/ROCK1 axis to lower LPS‐induced cell pyroptosis, indicating that targeting NEAT1/miR‐26a‐5p/ROCK1 axis might mitigate sepsis‐induced ALI.
NEAT1 was widely evaluated in multi‐diseases and was shown to contribute to regulating several biological functions including cell pyroptosis. 25 , 26 As previously described, NEAT1 has been determined to have a close correlation with cell pyroptosis in cerebral ischemia/reperfusion injury, diabetic nephropathy, and so on. 25 , 27 However, whether NEAT1 controls cell pyroptosis in sepsis‐induced ALI has not been determined. Previous analyses have indicated that NEAT1 could worsen ALI. 14 , 28 , 29 For instance, NEAT1 suppression could inactivate HMGB1/RAGE pathway to stimulate cell viability and constrain cell apoptosis and inflammatory response, eventually improving LPS‐induced ALI. 29 Besides, NEAT1 targeted miR‐944/TRIM37 axis or miR‐98‐5p/TLR4 axis or miR‐16‐5p/BRD4 axis to worsen ALI influenced by LPS treatment via increasing cell apoptosis and inflammatory response. 14 , 28 , 30 Herein, we discovered LPS administration elevated NEAT1 expression, inhibition of cell viability and enhancement of cell death and cell pyroptosis. However, NEAT1 downregulation compromised LPS‐induced cell viability inhibition and cell pyroptosis promotion.
Moreover, we determined that miR‐26a‐5p was a downstream gene of NEAT1. MiR‐26a‐5p was widely assessed in diverse diseases such as cardiac hypertrophy, gastric cancer, and sepsis‐induced ALI. 10 , 31 , 32 For example, in LPS‐induced lung cells, miR‐26a‐5p upregulation could evidently hinder cell apoptosis and inflammatory response. 10 , 11 A prior study indicated miR‐26a‐5p was mitigated by LPS administration in mice and human lung cells or lung tissues of mice, 11 suggesting miR‐26a‐5p was involved in sepsis‐induced ALI. Here, the miR‐26a‐5p level was lowered in BEAS‐2B cells upon LPS induction. Moreover, LPS treatment‐decreased miR‐26a‐5p level was compromised by NEAT1 knockdown. Besides, miR‐26a‐5p inhibitor could neutralize NEAT1 knockdown‐mediated restrained influences on cell death and cell pyroptosis. In total, we initially propose that NEAT1 suppressed miR‐26a‐5p level via interaction with miR‐26a‐5p to promote cell death and cell pyroptosis in LPS‐induced ALI.
It is well‐known that one microRNA could bind to multi‐genes via complementary sequences between miRNA and targeted mRNAs, thereby partaking in biological regulation. 33 , 34 For example, miR‐326 could target TLR4 to mitigate sepsis‐induced ALI. 35 MiR‐129/TAK1 axis plays a protective role in sepsis‐induced ALI by regulating the NF‐κB pathway. 36 Here, we initially confirmed ROCK1 was a downstream molecule of miR‐26a‐5p to mediate sepsis‐induced ALI via influencing cell death and cell pyroptosis. ROCK1 is a critical molecule in the regulation of inflammatory response. 18 , 37 Extracellular vesicles carrying Caspase‐3 in lung epithelial cells with hyperoxia treatment worsened inflammatory lung responses via activating the ROCK1 pathway. 37 Additionally, ROCK1 could regulate cell viability, apoptosis, and inflammation in ALI. 16 , 38 , 39 , 40 Furthermore, Wang et al. showed that ROCK1 could regulate cell pyroptosis to influence sepsis‐induced acute kidney injury. 19 In our research, we determined that miR‐26a‐5p was directly combined with ROCK1 and suppressed ROCK1 expression. Furthermore, ROCK1 upregulation reversed miR‐26a‐5p mimic‐mediated inhibition of cell death and cell pyroptosis in LPS‐induced BEAS‐2B cells.
Conclusively, we revealed that NEAT1 is competitively bound to miR‐26a‐5p to hinder the targeted inhibition of ROCK1 by miR‐26a‐5p, thereby influencing LPS‐induced ALI by worsening cell pyroptosis. Our findings might present crucial molecules to impede the progression of sepsis‐induced ALI. Although the functions and inter‐correlations of NEAT1/miR‐26a‐5p/ROCK1 axis on cell pyroptosis in LPS‐induced ALI were demonstrated by in vitro experiments, however, the experimental data from clinical is still inconclusive. We will further validate our outcomes at the clinical level.
CONFLICT OF INTEREST STATEMENT
These authors declared no competing interests in this work.
Fan X‐Y, Ma Z‐X, Tang L‐B, Shen H‐Z, Qi F, Xia J‐W. lncRNA NEAT1 mediates LPS‐induced pyroptosis of BEAS‐2B cells via targeting miR‐26a‐5p/ROCK1 axis. Kaohsiung J Med Sci. 2023;39(7):665–674. 10.1002/kjm2.12681
Xiu‐Ying Fan and Zhong‐Xu Ma are the co‐first authors.
REFERENCES
- 1. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet. 2018;392(10141):75–87. [DOI] [PubMed] [Google Scholar]
- 2. Wu C, Li H, Zhang P, Tian C, Luo J, Zhang W, et al. Lymphatic flow: a potential target in sepsis‐associated acute lung injury. J Inflamm Res. 2020;13:961–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park I, Kim M, Choe K, Song E, Seo H, Hwang Y, et al. Neutrophils disturb pulmonary microcirculation in sepsis‐induced acute lung injury. Eur Respir J. 2019;53(3):1800786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao H, Chen H, Xiaoyin M, Yang G, Hu Y, Xie K, et al. Autophagy activation improves lung injury and inflammation in sepsis. Inflammation. 2019;42(2):426–39. [DOI] [PubMed] [Google Scholar]
- 5. Sun J, Li Y. Pyroptosis and respiratory diseases: a review of current knowledge. Front Immunol. 2022;13:920464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu B, He R, Zhang L, Hao B, Jiang W, Wang W, et al. Inflammatory caspases drive pyroptosis in acute lung injury. Front Pharmacol. 2021;12:631256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beermann J, Piccoli MT, Viereck J, Thum T. Non‐coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96(4):1297–325. [DOI] [PubMed] [Google Scholar]
- 8. Ji J, Ye W, Sun G. lncRNA OIP5‐AS1 knockdown or miR‐223 overexpression can alleviate LPS‐induced ALI/ARDS by interfering with miR‐223/NLRP3‐mediated pyroptosis. J Gene Med. 2022;24(4):e3385. [DOI] [PubMed] [Google Scholar]
- 9. Jiao Y, Zhang T, Zhang C, Ji H, Tong X, Xia R, et al. Exosomal miR‐30d‐5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis‐related acute lung injury. Crit Care. 2021;25(1):356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li H, Yang T, Fei Z. miR‐26a‐5p alleviates lipopolysaccharide‐induced acute lung injury by targeting the connective tissue growth factor. Mol Med Rep. 2021;23(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun Q, Luo M, Gao Z, Han X, Wu W, Zhao H. Long non‐coding RNA OIP5‐AS1 aggravates acute lung injury by promoting inflammation and cell apoptosis via regulating the miR‐26a‐5p/TLR4 axis. BMC Pulm Med. 2021;21(1):236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen L, Zhou Y, Li H. LncRNA, miRNA and lncRNA‐miRNA interaction in viral infection. Virus Res. 2018;257:25–32. [DOI] [PubMed] [Google Scholar]
- 13. Bu FT, Wang A, Zhu Y, You HM, Zhang YF, Meng XM, et al. lncRNA NEAT1: shedding light on mechanisms and opportunities in liver diseases. Liver Int. 2020;40(11):2612–26. [DOI] [PubMed] [Google Scholar]
- 14. Chen J, Liu Q, Ding Z, Wang Y, Zhou L, Zheng Y, et al. lncRNA NEAT1 aggravates lipopolysaccharide‐induced acute lung injury by regulating the miR‐98‐5p/TLR4 axis. J Biochem Mol Toxicol. 2021;35(12):e22927. [DOI] [PubMed] [Google Scholar]
- 15. Shang A, Gu C, Wang W, Wang X, Sun J, Zeng B, et al. Exosomal circPACRGL promotes progression of colorectal cancer via the miR‐142‐3p/miR‐506‐3p‐ TGF‐β1 axis. Mol Cancer. 2020;19(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meng L, Cao H, Wan C, Jiang L. MiR‐539‐5p alleviates sepsis‐induced acute lung injury by targeting ROCK1. Folia Histochem Cytobiol. 2019;57(4):168–78. [DOI] [PubMed] [Google Scholar]
- 17. Yin J, Lv L, Zhai P, Long T, Zhou Q, Pan H, et al. Connexin 40 regulates lung endothelial permeability in acute lung injury via the ROCK1‐MYPT1‐ MLC20 pathway. Am J Physiol Lung Cell Mol Physiol. 2019;316(1):L35–l44. [DOI] [PubMed] [Google Scholar]
- 18. Zhang S, Zhu L, Dai H, Pan L. Silencing ROCK1 ameliorates ventilator‐induced lung injury in mice by inhibiting macrophages' NLRP3 signaling. Int Immunopharmacol. 2021;101:108208. [DOI] [PubMed] [Google Scholar]
- 19. Wang QL, Xing W, Yu C, Gao M, Deng LT. ROCK1 regulates sepsis‐induced acute kidney injury via TLR2‐mediated endoplasmic reticulum stress/pyroptosis axis. Mol Immunol. 2021;138:99–109. [DOI] [PubMed] [Google Scholar]
- 20. Lv X, Zhou X, Yan J, Jiang J, Jiang H. Propofol inhibits LPS‐induced apoptosis in lung epithelial cell line, BEAS‐2B. Biomed Pharmacother. 2017;87:180–7. [DOI] [PubMed] [Google Scholar]
- 21. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He G, Chen K, Wang H, Li X, Li W, Liu L, et al. Fudosteine attenuates acute lung injury in septic mice by inhibiting pyroptosis via the TXNIP/NLRP3/GSDMD pathway. Eur J Pharmacol. 2022;926:175047. [DOI] [PubMed] [Google Scholar]
- 24. Wang W, Xiong Y, Zhao H, Xu R. CircEXOC5 facilitates cell pyroptosis via epigenetic suppression of Nrf2 in septic acute lung injury. Mol Cell Biochem. 2022;478(4):743–54. [DOI] [PubMed] [Google Scholar]
- 25. Zhang HS, Ouyang B, Ji XY, Liu MF. Gastrodin alleviates cerebral ischaemia/reperfusion injury by inhibiting pyroptosis by regulating the lncRNA NEAT1/miR‐22‐3p axis. Neurochem Res. 2021;46(7):1747–58. [DOI] [PubMed] [Google Scholar]
- 26. Prinz F, Kapeller A, Pichler M, Klec C. The implications of the long non‐coding RNA NEAT1 in non‐cancerous diseases. Int J Mol Sci. 2019;20(3):627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhan JF, Huang HW, Huang C, Hu LL, Xu WW. Long non‐coding RNA NEAT1 regulates pyroptosis in diabetic nephropathy via mediating the miR‐34c/NLRP3 axis. Kidney Blood Press Res. 2020;45(4):589–602. [DOI] [PubMed] [Google Scholar]
- 28. Chen C, Zhang H, Ge M, Ye J, Li R, Wang D. LncRNA NEAT1 acts as a key regulator of cell apoptosis and inflammatory response by the miR‐944/TRIM37 axis in acute lung injury. J Pharmacol Sci. 2021;145(2):202–12. [DOI] [PubMed] [Google Scholar]
- 29. Zhou H, Wang X, Zhang B. Depression of lncRNA NEAT1 antagonizes LPS‐evoked acute injury and inflammatory response in alveolar epithelial cells via HMGB1‐RAGE signaling. Mediators Inflamm. 2020;2020:8019467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yin J, Han B, Shen Y. LncRNA NEAT1 inhibition upregulates miR‐16‐5p to restrain the progression of sepsis‐induced lung injury via suppressing BRD4 in a mouse model. Int Immunopharmacol. 2021;97:107691. [DOI] [PubMed] [Google Scholar]
- 31. Li Y, Wang P, Wu LL, Yan J, Pang XY, Liu SJ. miR‐26a‐5p inhibit gastric cancer cell proliferation and invasion through mediated Wnt5a. Onco Targets Ther. 2020;13:2537–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi H, Li H, Zhang F, Xue H, Zhang Y, Han Q. MiR‐26a‐5p alleviates cardiac hypertrophy and dysfunction via targeting ADAM17. Cell Biol Int. 2021;45(11):2357–67. [DOI] [PubMed] [Google Scholar]
- 33. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Króliczewski J, Sobolewska A, Lejnowski D, Collawn JF, Bartoszewski R. microRNA single polynucleotide polymorphism influences on microRNA biogenesis and mRNA target specificity. Gene. 2018;640:66–72. [DOI] [PubMed] [Google Scholar]
- 35. Wang Z, Yan J, Yang F, Wang D, Lu Y, Liu L. MicroRNA‐326 prevents sepsis‐induced acute lung injury via targeting TLR4. Free Radic Res. 2020;54(6):408–18. [DOI] [PubMed] [Google Scholar]
- 36. Yao W, Xu L, Jia X, Li S, Wei L. MicroRNA‐129 plays a protective role in sepsis‐induced acute lung injury through the suppression of pulmonary inflammation via the modulation of the TAK1/NF‐κB pathway. Int J Mol Med. 2021;48(1):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moon HG, Cao Y, Yang J, Lee JH, Choi HS, Jin Y. Lung epithelial cell‐derived extracellular vesicles activate macrophage‐mediated inflammatory responses via ROCK1 pathway. Cell Death Dis. 2015;6(12):e2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin Q, Liang Q, Qin C, Li Y. CircANKRD36 knockdown suppressed cell viability and migration of LPS‐stimulated RAW264.7 cells by sponging miR‐330. Inflammation. 2021;44(5):2044–53. [DOI] [PubMed] [Google Scholar]
- 39. Siddiqui MR, Akhtar S, Shahid M, Tauseef M, McDonough K, Shanley TP. miR‐144‐mediated inhibition of ROCK1 protects against LPS‐induced lung endothelial hyperpermeability. Am J Respir Cell Mol Biol. 2019;61(2):257–65. [DOI] [PubMed] [Google Scholar]
- 40. Zhang J, Xiang J, Liu T, Wang X, Tang Y, Liang Y. miR‐495 targets ROCK1 to inhibit lipopolysaccharides‐induced WI‐38 cells apoptosis and inflammation. Kaohsiung J Med Sci. 2020;36(8):607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]